

Abstract
A highly efficient chemoenzymatic method for synthesizing glycosphingolipids using α-Gal pentasaccharyl ceramide as an example is reported here. Enzymatic extension of chemically synthesized lactosyl sphingosine using efficient sequential one-pot multienzyme (OPME) reactions allowed glycosylation be carried out in aqueous solution. Facile C18 cartridge-based quick (<30 minutes) purification proctols were established using minimal amounts of green solvents (CH3CN and H2O). Simple acylation in the last step led to the formation of the target glycosyl ceramide in 4 steps with an overall yield of 57%.
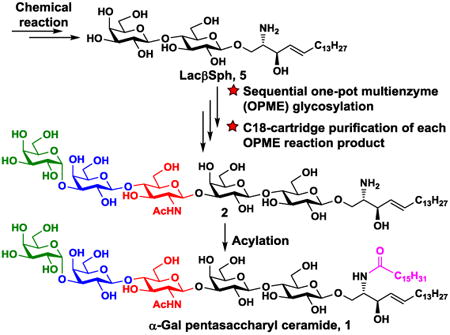
Graphical abstract
α-Gal pentasaccharyl ceramide was synthesized using sequential one-pot multienzyme (OPME) systems with facile purification by C18 cartridge followed by acylation.
Glycosphingolipids (GSLs) are glycoconjugates consisting of an oligosaccharide linked to a ceramide, a lipid consisting of a sphingoid base (sphingosine in mammalian glycosphingolipids). In ceramide, the amino group of the sphingoid base was coupled to a fatty acid via an amide bond.1 GSLs are ubiquitous components of mammalian cell membranes and are well known for their important roles in human health and diseases.2-6 De novo synthesis of glycosphingolipids in nature involves the formation of ceramide7 at the cytoplasmic face of the endoplasmic reticulum (ER), transfer of the ceramide to the cytoplasmic face of the Golgi, formation of glucosylceramide and translocation to the luminal face of the Golgi, and subsequent extension of the oligosaccharide chain by glycosyltransferases for the formation of more complex glycosphingolipids in the Golgi, followed by delivery to cell surface.2 All complex glycosphingolipids share a common lactosyl ceramide (LacβCer) core.8-10 LacβCer has a low solubility in water and is a poor acceptor for in vitro enzymatic reactions using glycosyltransferases in aqueous solutions.9, 10
GSLs used in functional studies and clinical applications have been commonly purified from mammalian cells, blood, and/or tissues.11-13 The inherited heterogeneity, the presence of other compounds with similar properties, and potential contamination by infectious agents11 make large scale purification of desired glycosphingolipids challenging, especially for low abundant compounds.
Complex GSLs are also challenging synthetic targets despite advances in the development of modern chemical, enzymatic, and chemoenzymatic methods. A general strategy for synthesizing GSLs has been using multistep chemical synthesis14-17 of a trichloroacetimidate glycosyl donor16, 18-21 for coupling with an azido derivative of the protected glycosphingosine followed by reduction of the azido group, coupling with an acyl chain, and deprotection. Unavoidably, the chemical synthetic approaches involve multiple tedious protection and deprotection processes which lead to extended preparation time and low overall yields. Alternatively, glycans synthesized enzymatically and chemoenzymatically have been protected and activated to generate glycosyl donors, such as trichloroacetimidate21 and more recently perbenzoylated glycosyl N-phenyltrifluoroacetimidate, for chemical glycosylation with a selectively benzoyl protected azido-sphingosine as the glycosylation acceptor. Another chemoenzymatic strategy is an endoglycoceramidase glycosynthase strategy using an oligosaccharyl fluoride as the donor substrate and a sphingoid base or its derivative as the acceptor substrate.11, 22 Both chemoenzymatic strategies require pre-assembly of non-protected oligosaccharides or oligosaccharyl fluorides in aqueous solution which involve time-consuming non-trivial purification of non-protected glycans after each glycosylation step. The products that can be obtained by glycosynthase-catalyzed direct glycosylation strategy are also limited by the substrate specificities of the enzyme mutants used towards both the glycan and the lipid components.
We propose an alternative chemoenzymatic strategy for synthesizing GSLs. As shown in Scheme 1, we envision that complex glycosphingosines can be readily obtained by sequential glycosyltransferase-dependent one-pot multienzyme (OPME) systems to extend the glycan chain in lactosyl sphingosine (LacβSph), a glycolipid that is readily soluble in aqueous solutions. The sphingosine (lipid) component in the glycosyl sphingosine products can be used as a hydrophobic tag, allowing facile purification of the product by C18 cartridge with simple green solvents such as acetonitrile and water. Acylation of the glycosyl sphingosine product with a fatty acid will form desired glycosphingolipids. The method allows the enzyme-catalyzed glycosylation reactions to occur in aqueous solutions and facile purifications by solid-phase extraction.23 An additional advantage is that intermediates for long chain complex glycosphingosines can be acylated to form other naturally occurring glycosphingolipids.
Scheme 1.

An efficient chemoenzymatic strategy for synthesizing complex glycosphingolipids by enzymatic extension of lactosyl sphingosine (LacβSph, 3) using sequential one-pot multienzyme (OPME) reactions with C18-cartridge purification after each glycosylation reaction followed by a simple acylation process. The structures of target α-Gal pentasaccharyl sphingosine Galα3nLc4βSph (2) and α-Gal pentasaccharyl ceramide Galα3nLc4βCer (1) are also shown.
For a proof-of-concept experiment, α-Gal pentasaccharyl ceramide (Galα3Galβ4GlcNAcβ3Galβ4GlcβCer) or α1–3-galactosyl-lacto-N-neotetraosyl β-ceramide Galα3nLc4βCer (1) (Scheme 1) was chosen as a target for synthesis. Galα3nLc4βCer (1) was initially identified from rabbit red blood cells.24 It was later found to be the major non-acid glycosphingolipid in pig kidney25 and the major α-Gal structure in pig aorta.26 Together with other α-Gal epitopes, it binds to naturally existing human anti-Gal antibodies and is a major cause for organ rejection in pig to human xenotransplantation.27-29
For economic and effective chemoenzymatic synthesis of glycosphingolipids using the proposed method, the first step is to identify an efficient chemical method for large-scale synthesis of lactosyl sphingosine (LacβSph) from commercially available inexpensive phytosphingosine (6) and lactose.
According to several previous reports30-32 for the synthesis of glucosyl, galactosyl, or lactosyl sphingosine, the azido-derivative of sphingosine was a better acceptor than other N-protected sphingosine derivatives for glycosylation with trichloroacetimidate glycosylation donors. For example, our attempts for glycosylation using N-tetrachlorophthaloyl (N-TCP) protected acceptor30 and lactosyl trichloroacetimidate led to very low yields. In addition to using the azido protecting group, benzoyl protection of the secondary alcohol in sphingosine was designed to improve the regioselectivity of glycosylation. Therefore, 2-azido-3-O-benzoyl sphingosine (12) (Scheme 2) was chosen as the glycosylation acceptor for the formation of lactosyl sphingosine. An efficient strategy33 was chosen to synthesize 12 from inexpensive D-erythro-sphingosine (or phytosphingosine, 6) by converting its amino group to an azido group by treating with freshly prepared triflic-azide in the presence of catalytic CuSO4 and triethylamine to form compound 7 in a quantitative yield without chromatographic purification. The primary hydroxyl of 7 was selectively protected by tert-butyldiphenylsilyl (TBDPS) group33 to produce 8 in 98% yield. Conversion of the 3,4-vicinal diol in 8 to its cyclic sulfate (9) was achieved in high yield (92%) by using thionyl chloride in the presence of triethylamine followed by oxidation with RuCl3/NaIO4. Selectively opening of the cyclic sulfate by tetrabutylammonium iodide and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), dehydrohalogenation to form alkene, followed by acidic hydrolysis to remove the allylic O-sulfate group were carried out in one pot33 to furnish compound 10 in 85% yield. Conventional benzoylation of compound 10 produced 11 in 95% yield. Removal of the O-sialyl ether of 11 was carried out using HF·pyridine to produce the glycosylation acceptor 12 (2.3 grams) in 97% yield. The total yield for the chemical synthesis of the compound 12 from phytosphingosine 6 was 61% insix steps.
Scheme 2.

Synthesis of sphingosine acceptor 12. Reagents and conditions: (a) TfN3, CuSO4, Et3N, MeOH, CH2Cl2, H2O, r.t., 6h, > 90%; (b) TBDPSCl, Et3N, DMAP, CH2Cl2, r.t., 8h, 98%; (c) SO2Cl2, Et3N, CH2Cl2, 0°C, 0.5–1 h; (d) RuCl3.3H2O, NaIO4, CCl4:CH3CN:H2O (1:1:1), r.t., 2 h; 89% in two steps (e) (i) Bu4NI, DBU, Toluene, reflux, 4 h; (ii) H2SO4/H2O/THF, r.t., 45 min; 85% in two steps (f) BzCl, DMAP, Et3N, CH2Cl2, 0°C to r.t., 12 h, 95%; (g) HF Pyridine, THF, 0°C to r.t., 12 h, 97%.
Glycosylation of 12 with per-O-benzoyl lactosyl trichloroacetimidate (13)34 in the presence of BF3·OEt2 in CH2Cl2 at -18 °C produced per-O-benzoyl lactoside (14)35 in 90% yield (Scheme 3). After removal of all benzoyl protecting groups by the Zemplén condition, several methods were tested to reduce the azido group while keep the alkene group intact. Methods using PPh3,36 PMe3,37 or Lindlar catalyst38 led to poor yields (30–50%). The combination of 1,3-propanedithiol and triethylamine39 were found to be the most efficient for selective reduction of the azido group to produce LacβSph (5) in an excellent yield (94%). The total yield for the glycosylation was 85% in three steps and the total yield for chemical synthesis of LacβSph (5) was 52% in nine steps.
Scheme 3.

Synthesis of lactosyl sphingosine (LacβSph, 5). Reagents and conditions: (a) BF3·OEt2, CH2Cl2, -18 °C, 3 h, 90%; (b) NaOMe, MeOH, r.t., 14 h; (c) 1,3-propanedithiol, Et3N, pyridine-water (1:1 v/v), 50 °C, 36 h, 94% in two steps.
Lactosyl sphingosine (LacβSph, 5) was readily soluble in aqueous solution for up to 30 mM, allowing it be used efficiently as a starting glycosyltransferase acceptor for enzymatic extension using one-pot multienzyme (OPME) reactions23, 40-45 for the synthesis of more complex structurally diverse glycosphingosines. The sphingosine (lipid) component of the acceptor and the product can be used as an anchor to allow facile purification of the glycosphingosines by reverse phase column chromatography such as simple C18 cartridge-based purification.
For the synthesis of the target α-Gal pentasaccharyl ceramide Galα3nLc4βCer (1), α-Gal pentasaccharyl sphingosine Galα3nLc4βSph (2) was synthesized by enzymatic extension from LacβSph (5) using a sequential OPME strategy involving three OPME reactions (Scheme 4). Formation of trisaccharyl sphingosine Lc3βSph (GlcNAcβ3Galβ4GlcβSph, 4) from LacβSph (5) was achieved by a one-pot four-enzyme N-acetylglucosamine (GlcNAc) activation and transfer system containing Bifidobacterium longum (strain ATCC55813) N-acetylhexosamine-1-kinase (BLNahK),46 Pasteurella multocida N-acetylglucosamine uridylyltransferase (PmGlmU),47 Pasteurella multocida inorganic pyrophosphatase (PmPpA),48 and Neisseria meningitidis β1–3-N-acetylglucosaminyltransferase (NmLgtA)49 in Tris-HCl buffer (100 mM, pH 8.0) at 37 °C for 52 hours. The BLNahK, PmGlmU, and PmPpA allowed in situ formation of uridine 5′-diphosphate-N-acetylglucosamine (UDP-GlcNAc), the sugar nucleotide donor substrate, efficiently and directly from monosaccharide GlcNAc for NmLgtA-catalyze formation of Lc3βSph (4). Upon the completion of the enzymatic reaction as monitored by thin-layer chromatography (TLC) and high-resolution mass spectrometry (HRMS), the reaction mixture was diluted with the same volume of ethanol. The solution was incubated at 4 °C for 30 minutes and centrifuged to remove precipitates. The supernatant was concentrated and the residue was dissolved in 2–3 mL of water at 40–45 °C. The solution was directly loaded to a pre-conditioned C18 cartridge. The cartridge was then washed with 0.01% TFA in water (10 mL) using a syringe. The unreacted sugar, adenosine 5′-triphosphate (ATP), uridine 5′-triphosphate (UTP), adenosine 5′-diphosphate (ADP), uridine 5′-diphosphate (UDP), UDP-sugar, and salts were completely removed in this step. The product Lc3βSph (4) was eluted by 37% acetonitrile in 0.01% TFA/H2O and unreacted starting material was eluted with 50% acetonitrile in 0.01% TFA/H2O. The purification process took less than 30 min in contrast to several hours using standard silica gel chromatography. A yield of 83% was achieved after purification.
Scheme 4.

High-yield synthesis of α-Gal pentasaccharyl ceramide Galα3nLc4βCer (1) by enzymatic extension of lactosyl sphingosine (LacβSph, 5) using sequential one-pot multienzyme (OPME) reactions with C18-cartridge purification for the formation of α-Gal pentasaccharyl sphingosine Galα3nLc4βSph (2) followed by a simple acylation reaction.
The obtained Lc3βSph (4) was used for synthesizing nLc4βSph (3) using an improved OPME galactose (Gal) activation and transfer system50 containing Streptococcus pneumoniae TIGR4 galactokinase (SpGalK),51 Bifidobacterium longum UDP-sugar pyrophosphorylase (BLUSP),50 PmPpA, and Neisseria meningitidis β1–4-galactosyltransferase (NmLgtB)48, 49 in Tris-HCl buffer (100 mM, pH 8.0) at 37 °C for 30 hours. The SpGalK, BLUSP, and PmPpA allowed in situ formation of uridine 5′-diphosphate-galactose (UDP-Gal), the donor substrate of NmLgtB, from monosaccharide galactose (Gal) for the formation of LNnTβSph (3). A similar C18-cartridge purification procedure was carried out as described above for Lc3βSph (4) except that 35% acetonitrile in 0.01% TFA/H2O was used as an eluant to purify nLc4βSph (3). The acceptor was completely consumed. After purification, a yield of 92% was obtained.
The last OPME reaction was carried out to convert the obtained nLc4βSph (3) to Galα3nLc4βSph (2) using a galactose activation and transfer system containing SpGalK, BLUSP, PmPpA, and a recombinant bovine α1–3-galactosyltransferase (Bα1–3GalT)23, 52 in Tris-HCl buffer (100 mM, pH 7.5) at 37 °C for 48 hours. The donor substrate UDP-Gal was generated from galactose in situ as described in the previous step for stereo-selective production of the desired Galα3nLc4βSph (2). The enzymatic introduction of the terminal α1–3-linked galactoside by the Bα1–3GalT was especially advantageous as the 1,2-cis-glycosylation is more challenging to achieve by chemical glycosylation strategies. The product (88% yield) was purified by C18-cartridge with elution using 32% acetonitrile in 0.1% TFA in H2O and the unreacted acceptor was eluted with 35% or a higher concentration (50%) of acetonitrile in 0.1% TFA/H2O.
It is worth to mention that 1.5 equivalents of nucleoside triphosphates (ATP and UTP) were used in each OPME reaction to optimize the glycosylation yields. With decreased costs of these compounds, in situ recycling of ATP and UTP was not necessary for preparative or gram-scale reactions.
The target Galα3nLc4βCer (1) was readily obtained in 85% yield by N-acylation of Galα3nLc4βSph (2) with palmitic acid in the presence of EDC·HCl, HOBt and Et3N.18 Overall, the chemoenzymatic route provided the target Galα3nLc4βCer (1) with an overall 30% yield in 13 steps. Although Galα3nLc4βCer (1) had low solubility in methanol as noticed previously,15 its solubility in CD3OD was sufficient to allow detailed nuclear magnetic resonance characterization of the product.
In conclusion, using α-Gal pentasaccharyl ceramide synthesis as an example, we have demonstrated that efficient sequential one-pot multienzyme (OPME) chemoenzymatic systems can be combined with facile C18-purification processes for high-yield production of glycosphingosines and glycosylceramides. The strategy can be extended to the synthesis of other complex glycosphingolipids. The method and the established protocols will allow non-specialists for synthesizing, purifying, and study desired glycosphingolipids of interest in their own labs with a general research lab setting.
Supplementary Material
Acknowledgments
This work was supported by NIH grant U01GM120419 and NSF grant CHE-1300449. Bruker Avance-800 NMR spectrometer was funded by NSF grant DBIO-722538. Y.L., H.Y., and X.C. are co-founders of Glycohub, Inc., a company focused on the development of carbohydrate-based reagents, diagnostics, and therapeutics. Glycohub, Inc. played no role in the design, execution, interpretation, or publication of this study.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details for chemical and enzymatic synthesis, NMR and HRMS data, and NMR spectra. See DOI: 10.1039/x0xx00000x
References
- 1.Lowther J, Naismith JH, Dunn TM, Campopiano DJ. Biochem Soc Trans. 2012;40:547–554. doi: 10.1042/BST20110769. [DOI] [PubMed] [Google Scholar]
- 2.Schulze H, Sandhoff K. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004804. pii: a004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X, Kiechle FL. Ann Clin Lab Sci. 2004;34:3–13. [PubMed] [Google Scholar]
- 4.Cummings RD. Mol Biosyst. 2009;5:1087–1104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 5.Schnaar RL. J Mol Biol. 2016;428:3325–3336. doi: 10.1016/j.jmb.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mather AR, Siskind LJ. Adv Exp Med Biol. 2011;721:121–138. doi: 10.1007/978-1-4614-0650-1_8. [DOI] [PubMed] [Google Scholar]
- 7.Merrill AH., Jr J Biol Chem. 2002;277:25843–25846. doi: 10.1074/jbc.R200009200. [DOI] [PubMed] [Google Scholar]
- 8.Rearick JI, Sadler JE, Paulson JC, Hill RL. J Biol Chem. 1979;254:4444–4451. [PubMed] [Google Scholar]
- 9.Basu SC. Glycobiology. 1991;1:469–475. doi: 10.1093/glycob/1.5.469. [DOI] [PubMed] [Google Scholar]
- 10.Guilbert B, Flitsch SL. J Chem Soc Perkin Trans 1. 1994:1181–1186. [Google Scholar]
- 11.Vaughan MD, Johnson K, DeFrees S, Tang X, Warren RA, Withers SG. J Am Chem Soc. 2006;128:6300–6301. doi: 10.1021/ja058469n. [DOI] [PubMed] [Google Scholar]
- 12.Masson EA, Sibille E, Martine L, Chaux-Picquet F, Bretillon L, Berdeaux O. J Lipid Res. 2015;56:1821–1835. doi: 10.1194/jlr.D060764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Svennerholm L, Fredman P. Biochim Biophys Acta. 1980;617:97–109. doi: 10.1016/0005-2760(80)90227-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Huang X, Zhang LH, Ye XS. Org Lett. 2004;6:4415–4417. doi: 10.1021/ol0483246. [DOI] [PubMed] [Google Scholar]
- 15.Gege C, Kinzy W, Schmidt RR. Carbohydr Res. 2000;328:459–466. doi: 10.1016/s0008-6215(00)00145-2. [DOI] [PubMed] [Google Scholar]
- 16.Bommer R, Schmidt RR. Liebigs Annalen der Chemie. 1989;1989:1107–1111. [Google Scholar]
- 17.Chen W, Xia C, Wang J, Thapa P, Li Y, Talukdar A, Nadas J, Zhang W, Zhou D, Wang PG. J Org Chem. 2007;72:9914–9923. doi: 10.1021/jo701539k. [DOI] [PubMed] [Google Scholar]
- 18.Schutte OM, Ries A, Orth A, Patalag LJ, Romer W, Steinem C, Werz DB. Chem Sci. 2014;5:3104–3114. [Google Scholar]
- 19.Nakashima S, Ando H, Saito R, Tamai H, Ishida H, Kiso M. Chem Asian J. 2012;7:1041–1051. doi: 10.1002/asia.201100928. [DOI] [PubMed] [Google Scholar]
- 20.Fujikawa K, Nakashima S, Konishi M, Fuse T, Komura N, Ando T, Ando H, Yuki N, Ishida H, Kiso M. Chem Eur J. 2011;17:5641–5651. doi: 10.1002/chem.201003357. [DOI] [PubMed] [Google Scholar]
- 21.Yao Q, Song J, Xia C, Zhang W, Wang PG. Org Lett. 2006;8:911–914. doi: 10.1021/ol053070p. [DOI] [PubMed] [Google Scholar]
- 22.Rich JR, Cunningham AM, Gilbert M, Withers SG. Chem Commun. 2011;47:10806–10808. doi: 10.1039/c1cc13885e. [DOI] [PubMed] [Google Scholar]
- 23.Hwang J, Yu H, Malekan H, Sugiarto G, Li Y, Qu J, Nguyen V, Wu D, Chen X. Chem Commun. 2014;50:3159–3162. doi: 10.1039/c4cc00070f. [DOI] [PubMed] [Google Scholar]
- 24.Eto T, Ichikawa Y, Nishimura K, Ando S, Yamakawa T. J Biochem. 1968;64:205–213. doi: 10.1093/oxfordjournals.jbchem.a128881. [DOI] [PubMed] [Google Scholar]
- 25.Samuelsson BE, Rydberg L, Breimer ME, Backer A, Gustavsson M, Holgersson J, Karlsson E, Uyterwaal AC, Cairns T, Welsh K. Immunol Rev. 1994;141:151–168. doi: 10.1111/j.1600-065x.1994.tb00876.x. [DOI] [PubMed] [Google Scholar]
- 26.Hallberg EC, Strokan V, Cairns TD, Breimer ME, Samuelsson BE. Xenotransplantation. 1998;5:246–256. doi: 10.1111/j.1399-3089.1998.tb00035.x. [DOI] [PubMed] [Google Scholar]
- 27.Fryer JP, Leventhal JR, Matas AJ. Transpl Immunol. 1995;3:21–31. doi: 10.1016/0966-3274(95)80002-6. [DOI] [PubMed] [Google Scholar]
- 28.Sandrin MS, McKenzie IF. Curr Opin Immunol. 1999;11:527–531. doi: 10.1016/s0952-7915(99)00011-4. [DOI] [PubMed] [Google Scholar]
- 29.Macher BA, Galili U. Biochim Biophys Acta. 2008;1780:75–88. doi: 10.1016/j.bbagen.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Benedetto R, Zanetti L, Varese M, Rajabi M, Di Brisco R, Panza L. Org Lett. 2014;16:952–955. doi: 10.1021/ol403688t. [DOI] [PubMed] [Google Scholar]
- 31.Rai AN, Basu A. J Org Chem. 2005;70:8228–8230. doi: 10.1021/jo051069y. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Toyokuni T, Ruan F, Hakomori Si. Glycoconj J. 2001;18:557–563. doi: 10.1023/a:1019682102486. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, Lee S, Lee T, Ko H, Kim D. J Org Chem. 2006;71:8661–8664. doi: 10.1021/jo0614543. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Ding N, Xiao H, Li Y. J Carbohydr Chem. 2006;25:471–489. [Google Scholar]
- 35.Zimmermann P, Sommer R, Bär T, Schmidt RR. J Carbohydr Chem. 1988;7:435–452. [Google Scholar]
- 36.Long DE, Karmakar P, Wall KA, Sucheck SJ. Bioorg Med Chem. 2014;22:5279–5289. doi: 10.1016/j.bmc.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyffeler PT, Liang CH, Koeller KM, Wong CH. J Am Chem Soc. 2002;124:10773–10778. doi: 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- 38.Reddy PG, Pratap TV, Kumar GDK, Mohanty Subhendu K, Baskaran S. Eur J Org Chem. 2002;2002:3740–3743. [Google Scholar]
- 39.Bayley H, Standring DN, Knowles JR. Tetrahedron Lett. 1978;19:3633–3634. [Google Scholar]
- 40.Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X. J Am Chem Soc. 2009;131:18467–18477. doi: 10.1021/ja907750r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang S, Yu H, Chen X. Sci China Chem. 2011;54:117–128. doi: 10.1007/s11426-010-4175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Li Y, Yu H, Sugiarto G, Thon V, Hwang J, Ding L, Hie L, Chen X. Angew Chem Int Ed Engl. 2013;52:11852–11856. doi: 10.1002/anie.201305667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H, Lau K, Thon V, Autran CA, Jantscher-Krenn E, Xue M, Li Y, Sugiarto G, Qu J, Mu S, Ding L, Bode L, Chen X. Angew Chem Int Ed Engl. 2014;53:6687–6691. doi: 10.1002/anie.201403588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng X, Yao W, Cheng J, Zhang X, Jin L, Yu H, Chen X, Wang F, Cao H. J Am Chem Soc. 2014;136:5205–5208. doi: 10.1021/ja5000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santra A, Yu H, Tasnima N, Muthana MM, Li Y, Zeng J, Kenyon NJ, Louie AY, Chen X. Chem Sci. 2016;7:2827–2831. doi: 10.1039/c5sc04104j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Yu H, Chen Y, Lau K, Cai L, Cao H, Tiwari VK, Qu J, Thon V, Wang PG, Chen X. Molecules. 2011;16:6396–6407. doi: 10.3390/molecules16086396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X. Chem Commun. 2011;47:10815–10817. doi: 10.1039/c1cc14034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X. Chem Commun. 2010;46:6066–6068. doi: 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Xue M, Sheng X, Yu H, Zeng J, Thon V, Chen Y, Muthana MM, Wang PG, Chen X. Bioorg Med Chem. 2016;24:1696–1705. doi: 10.1016/j.bmc.2016.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X, Fang J, Zhang J, Liu Z, Shao J, Kowal P, Andreana P, Wang PG. J Am Chem Soc. 2001;123:2081–2082. doi: 10.1021/ja005738v. [DOI] [PubMed] [Google Scholar]
- 51.Chen M, Chen LL, Zou Y, Xue M, Liang M, Jin L, Guan WY, Shen J, Wang W, Wang L, Liu J, Wang PG. Carbohydr Res. 2011;346:2421–2425. doi: 10.1016/j.carres.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 52.Fang J, Li J, Chen X, Zhang Y, Wang J, Guo Z, Zhang W, Yu L, Brew K, Wang PG. J Am Chem Soc. 1998;120:6635–6638. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.