

Pharmacological inhibition of PAI-1 augments proteolytic clearance of FGF23.
Abstract
Elevated levels of fibroblast growth factor 23 (FGF23), a bone-derived phosphaturic hormone, are associated with a number of pathologic conditions including chronic kidney disease, cardiac hypertrophy, and congestive heart failure. Currently, there are no specific treatments available to lower plasma FGF23 levels. We have recently reported that genetic plasminogen activator inhibitor–1 (PAI-1) deficiency provided a significant reduction in circulating FGF23 levels while simultaneously prolonging the life span of Klotho-deficient mice. We extend our investigations into the effect of PAI-1 on FGF23 homeostasis. Transgenic overexpression of PAI-1 resulted in threefold increase in FGF23 levels compared to wild-type littermates. Moreover, pharmacological modulation of PAI-1 activity with the small-molecule PAI-1 antagonist TM5441 significantly reduced FGF23 levels in PAI-1 transgenic and Klotho-deficient mice. In addition, TM5441 treatment or PAI-1 deficiency significantly accelerated the clearance of endogenous FGF23 and recombinant human FGF23 from circulation in mice with acute kidney injury. On the basis of these observations, we studied the effects of plasminogen activators (PAs), tissue-type PA (tPA) and urokinase-type PA (uPA), on FGF23. We demonstrate that both PAs directly cleave FGF23; however, it is not known whether the PA-generated FGF23 peptides retain or acquire functions that affect binding and/or signaling properties of intact FGF23. PAI-1 inhibits the PA-dependent cleavage of FGF23, and TM5441 inhibition of PAI-1 restores the proteolysis of FGF23. Furthermore, top-down proteomic analysis indicates that tPA cleaves FGF23 at multiple arginines including the proconvertase sensitive site R176. In summary, our results indicate that PAI-1 prevents the PA-driven proteolysis of FGF23 and PAI-1 inhibition provides a novel therapeutic approach to prevent the pathologic consequences of increased FGF23.
INTRODUCTION
The mammalian fibroblast growth factor (FGF) family contains 22 members (1), with a subfamily containing FGF19, FGF21, and FGF23 that display endocrine properties due, in part, to a lower affinity for heparin than the other FGF members (2, 3). Elevated serum levels of FGF23 are associated with a variety of human diseases and conditions including chronic kidney disease (CKD), left ventricular hypertrophy (LVH) and congestive heart failure (CHF), autosomal dominant hypophosphatemic rickets (ADHR), osteomalacia, vitamin D deficiency, fibrous dysplasia, and aging (4–10). FGF23 is synthesized in bone by osteocytes and osteoblasts (6), is secreted into the circulation, and functions in the kidney to regulate renal phosphate excretion (8, 11) and the enzymatic activation of vitamin D (9). FGF23 promotes phosphate excretion through the activation of the FGF receptor 1 (FGFR1) and the co-receptor klotho (12) complex. Although elevated levels of FGF23 have predictable effects on bone mineralization, they also predict future adverse events in patients with CKD and CHF (13). These clinical correlations suggest that FGF23 may have extrarenal effects that contribute to the progression or manifestations of cardiovascular disease (CVD).
Proteolytic cleavage influences the secretion of FGF23 from cells as well as the metabolic inactivation of FGF23 in the circulation. A proform of FGF23 is synthesized with a 24–amino acid secretory signal peptide that facilitates secretion into the Golgi apparatus, where it can be further processed by a protein convertase into the inactive N- and C-terminal fragments depending on the phosphorylation and glycosylation states of intact FGF23 (iFGF23) (2, 7). Mutations at or near residues R176 and R179 have been identified in ADHR and confer increased stability of iFGF23 (5, 11). Because an RXXR motif exists at this position in FGF23, it is thought that a subtilisin-like proprotein convertase (SPC), such as furin (5, 7, 14), mediates the proteolytic inactivation of FGF23, but the identity of other specific proteases that may be involved in this process has not been definitively demonstrated.
Klotho (kl/kl) mice, deficient in klotho protein, lack the ligand-dependent signaling through FGFRs and have increased levels of FGF23 (>1200-fold), vitamin D, and phosphate (15). Furthermore, kl/kl mice display an age-dependent increase in plasminogen activator inhibitor–1 (PAI-1) levels in blood (>45-fold increase in 8 weeks old) and tissues including kidney and aorta (16) and develop an accelerated aging phenotype with an average life span of ~70 days (15). We have recently reported that genetic deficiency of PAI-1 reduced the FGF23 levels by 98% and increased the average life span of kl/kl mice by fourfold (17). This observation indicates that PAI-1 plays a critical role in FGF23 homeostasis. PAI-1 is a serine protease inhibitor (SERPIN) and has a well-documented association with many human pathologies including CKD, myocardial infarction, and diabetes (18, 19). Although PAI-1 mainly inhibits tissue-type PA (tPA) and urokinase-type PA (uPA) irreversibly (20), it is known to inhibit other serine proteases as well. Thus, PAI-1 has the capacity to affect a number of physiological or pathological pathways. Both tPA and uPA convert plasminogen into the active protease plasmin by cleaving the activation peptide bond between R561-V562 residues. Once generated, plasmin is a highly active proteolytic enzyme and degrades a number of proteins including fibrin, procollagenases, fibronectin, thrombospondin, laminin, and von Willebrand factor (21, 22). Although the plasminogen activation system is historically viewed as the primary modifier of endogenous fibrinolysis, it also contributes to extracellular matrix remodeling, growth factor activation, and senescence (23, 24). A recent report describing the tPA-mediated proteolysis of IGFBP3 (insulin-like growth factor binding protein 3) (25) indicates that both tPA and uPA regulate the metabolism of peptides and hormones affecting many physiological and pathophysiological processes independent from their ability to activate plasminogen. Epidemiological data link elevated FGF23 levels with LVH (4, 10) and an increased risk of death in patients with CKD (26, 27). Therefore, therapeutic agents that reduce FGF23 levels in plasma may provide important clinical benefits. These observations led us to hypothesize that PAs and PAI-1 directly regulate FGF23 homeostasis. Here, we describe the effects of selective pharmacological inhibition of PAI-1 by TM5441 on FGF23 metabolism in vitro and in vivo.
RESULTS
PAI-1 inhibition affects FGF23 levels in kl/kl and Pai-1stab mice
We first investigated the role of PAI-1 in FGF23 homeostasis by administering an orally active PAI-1 antagonist, TM5441, mixed into the standard chow diet to Pai-1stab, transgenic mice overexpressing a functionally stable variant of human PAI-1 under the control of mouse endothelin-1 promoter (28, 29), and kl/kl mice. We tested various doses of TM5441 in Pai-1stab and kl/kl mice and found that doses as low as 10 mg/kg per day are effective in promoting significant and reproducible inhibition of plasma PAI-1 activity in these animals. Plasma levels of FGF23 were determined using specific enzyme-linked immunosorbent assay (ELISA) kits that detect both iFGF23 and cleaved FGF23 (cFGF23), which represents the sum of iFGF23 and cFGF23 in circulation. We observed that Pai-1stab mice have threefold elevated levels of cFGF23 (n = 14, P < 0.001) and TM5441 treatment reduced it by ~50% (n = 9, P < 0.01) and returned plasma levels of FGF23 that are not significantly different from those observed in wild-type (WT) mice (Fig. 1A). In contrast, iFGF23 levels in Pai-1stab mice (with and without TM5441 treatment) were similar to those observed in WT mice, indicating tight control over iFGF23 synthesis. We also measured cFGF23 and iFGF23 levels in TM5441-treated and PAI-1–deficient kl/kl (kl/klpai-1−/−) mice. Specifically, 4-week-old kl/kl mice (n = 8) received TM5441 (10 mg/kg per day) mixed in standard chow diet. Pharmacological inhibition and genetic deficiency of PAI-1 activity in kl/kl mice yielded 66% (P = 0.0003) and 98% (n = 10, P = 0.0001) reductions in cFGF23 levels, respectively, compared to those of untreated kl/kl mice (n = 14), but remained significantly elevated over WT control mice (Fig. 1B). Likewise, iFGF23 levels were reduced by 75% with TM5441 treatment (n = 8, P = 0.004) and by 96% with genetic PAI-1 deficiency (n = 8, P < 0.001) in kl/kl mice. These findings indicate improved clearance of FGF23 from circulation with either PAI-1 deficiency or pharmacologic inhibition and further confirm our initial observation in kl/kl mice that PAI-1 plays a critical role in FGF23 homeostasis (17).
Fig. 1. Effect of PAI-1 on circulating levels of cFGF23 and iFGF23 in Pai-1stab transgenic and Klotho mice.
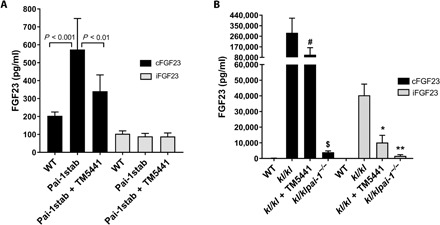
Plasma levels of cFGF23 (black filled bars) and iFGF23 (gray filled bars) were measured in WT, untreated, and TM5441-treated Pai-1stab mice (A) and in WT, kl/kl, TM5441-treated kl/kl, and kl/klpai-1−/− mice (B). As seen in (A), changes in cFGF23 levels due to PAI-1 overexpression and TM5441 treatment were both significant in Pai-1stab mice. Similarly, TM5441 treatment and PAI-1 deficiency significantly reduced cFGF23 and iFGF23 levels in kl/kl mice. #P = 0.0003, $P = 0.0001 versus kl/kl cFGF23; *P = 0.03, **P = 0.001 versus kl/kl iFGF23. Analysis of the measurements by one-way analysis of variance (ANOVA) yielded significant differences (P < 0.0001) for both Pai-1stab and kl/kl mice groups.
Modulating PAI-1 activity improves FGF23 homeostasis in acute kidney injury
Patients with acute kidney injury (AKI) have markedly increased circulating levels of FGF23 that are associated with increased risk of end-stage renal disease (ESRD) and mortality (30, 31). Next, we investigated whether PAI-1 contributes to the elevated levels of FGF23 in AKI. We induced AKI by intraperitoneal injection of folic acid (FA; 240 mg/kg) into mice, as described previously (32). FGF23 levels are significantly elevated as early as 1 hour after FA injection and reach 18-fold higher levels in 24 hours (33). Plasma samples were collected for PAI-1 and FGF23 measurements 24 hours after FA injection into animals. Plasma PAI-1 levels increased by 10-fold (n = 13, P = 0.0001) in mice with AKI (Fig. 2A). As expected, FGF23 levels increased by 30-fold in mice with AKI compared to those in vehicle-treated animals (Fig. 2B). The group of mice that received the chow diet containing TM5441 at 10 mg/kg per day for 7 days before the FA treatment displayed 20% reduction in iFGF23 levels (n = 15, P = 0.007). Similarly, PAI-1–deficient mice treated with FA (Fig. 2B) displayed a 45% reduction in iFGF23 compared to WT mice with AKI (n = 9, P = 0.005). At baseline, PAI-1−/− mice have lower levels of FGF23 than WT mice (P = not significant). Together, these results indicate that PAI-1 affects FGF23 homeostasis in mice with AKI.
Fig. 2. Effect of pharmacological inhibition or genetic deficiency of PAI-1 on the circulating levels of endogenous iFGF23 and clearance of rhFGF23 injected via the tail vein in mice with FA-induced AKI.
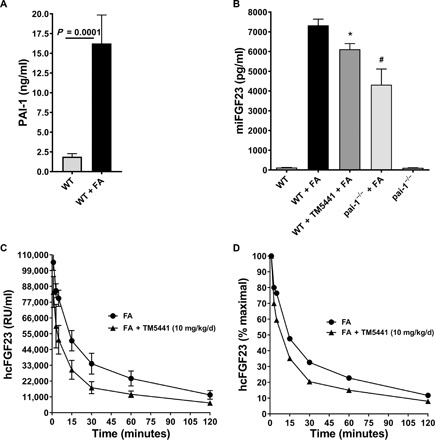
(A) AKI induces plasma PAI-1 levels 10-fold higher in FA-treated WT mice (n = 13) compared to vehicle-treated WT mice (n = 5). (B) Mouse iFGF23 (miFGF23) levels in WT (n = 7), WT with AKI (n = 21), TM5441-treated WT with AKI (n = 15), PAI-1−/− with AKI (n = 9), and pai-1−/− untreated (n = 6). *P = 0.007, #P = 0.005. (C) Effect of PAI-1 inhibition on clearance of rhFGF23 in WT AKI mice (n = 7) and TM5441 (10 mg/kg per day)–treated WT AKI mice (n = 7). The rhFGF23 was injected into the tail vein, and human cFGF23 (hcFGF23) levels were measured in plasma samples collected at time points 1, 3, 5, 15, 30, 60, and 120 min. (D) The cFGF23 levels at 1 min were considered as 100% to plot percent remaining values of rhFGF23 over the time course of the experiment.
To further investigate the effect of PAI-1 on clearance of FGF23 from circulation, we injected recombinant human FGF23 (rhFGF23) via the tail vein into FA-treated mice that received chow with and without TM5441 (10 mg/kg per day). PAI-1 inhibition preceded the FA treatment by 7 days and continued during the development of AKI. We collected plasma samples at 1, 3, 5, 15, 30, 60, and 120 min after rhFGF23 injection. Using an ELISA method that detects only the C terminus of human FGF23, we determined plasma levels of rhFGF23. As shown in Fig. 2 (C and D), rhFGF23 elimination from the plasma appears to be a biphasic process. Analysis of the FGF23 clearance data in Fig. 2C by two-phase decay nonlinear best-fit method using GraphPad Prism showed that the T1/2 (half-life) of FGF23 is shortened with PAI-1 inhibition from 5.4 to 1.2 min and from 45 to 11.6 min in fast and slow phases of clearance, respectively (P = 0.0001). This corresponds to a 4.5-fold acceleration of the fast phase and 3.9-fold acceleration of the slow phase. These results further confirm the effects of PAI-1 on FGF23 homeostasis in AKI.
Phosphate and parathyroid hormone levels in AKI and kl/kl mice
In the folate-induced AKI model, serum phosphate levels and plasma parathyroid hormone (PTH) levels increase markedly, whereas calcium levels remain unchanged and FGF23 levels increase independently of signaling by PTH, vitamin D3, and dietary phosphate (33). In kl/kl mice, we have previously reported that PAI-1 deficiency significantly reduces FGF23 and vitamin D3 levels but has negligible effects on phosphate and calcium levels (17). Here, we investigated whether the PAI-1 inhibition by TM5441 affects serum phosphate levels in the FA-induced AKI model and kl/kl mice. We observed that neither TM5441 treatment nor PAI-1 deficiency affects the high levels of phosphate in FA-treated and kl/kl mice (Fig. 3A). Furthermore, neither PAI-1 inhibition nor PAI-1 deficiency has any effect on the elevated PTH levels observed in the FA-induced AKI. In kl/kl mice, PTH levels were the same as those in WT mice (Fig. 3B). These findings indicate that FGF23 is incapable of regulating phosphate clearance in these mouse models, which likely reflects the deficiency of functional receptor heterodimer (FGFR and Klotho) in kl/kl mice and in mice with AKI.
Fig. 3. Effect of PAI-1 on circulating levels of phosphate and PTH in AKI and kl/kl mice.
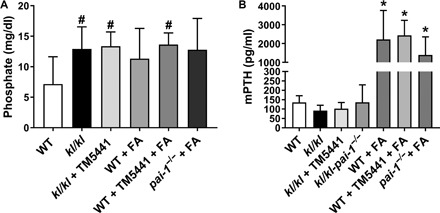
TM5441 treatment or PAI-1 deficiency did not affect the increased serum phosphate levels (#P < 0.05 versus WT) (A) and elevated plasma levels of PTH (*P < 0.01 versus WT) (B) in kl/kl and FA-treated WT and pai-1−/− mice. mPTH, mouse PTH.
In vitro proteolysis of FGF23 by PAs and plasmin
Our results from kl/kl and Pai-1stab mice suggest that one or more serine proteases inhibited by PAI-1, most likely tPA and/or uPA, are involved in the metabolism of FGF23. To test this hypothesis, we investigated the effects of tPA and uPA on FGF23 in vitro using recombinant human proteins. Under reaction conditions optimized for tPA or uPA activity, we incubated FGF23 with tPA or uPA at 37°C and resolved the resulting peptide fragments on silver-stained SDS-PAGE (polyacrylamide gel electrophoresis) gels. When incubated alone up to 6 hours at 37°C, we did not detect any spontaneous proteolysis or degradation of the recombinant proteins used in this study including tPA, uPA, PAI-1, and FGF23 (Fig. 4A). This indicates that any new peptide fragments detected on SDS-PAGE likely come from the proteolysis of FGF23 by tPA or uPA. We observed that incubation of FGF23 with tPA or uPA yielded new peptide fragments at approximately 17 and 13 kDa in size (Fig. 4A, lanes 3 and 4) that are not present in reactions containing only tPA or uPA (Fig. 4A, lanes 1 and 2). The appearance of these new bands indicates that both tPA and uPA cleave FGF23 and produce fragments consistent in size with the previously reported proteolytic fragments of FGF23 (7). The Arg179 residue of FGF23 is thought to be the most labile proteolytic site, and it is mutated to a Gln for production purposes of the recombinant iFGF23. Thus, this mutation (R179Q) eliminates a potential cleavage site for tPA and/or uPA. We used an anti–6×His-tag antibody for immunoblotting to confirm that the lower molecular fragments originated from FGF23 and to determine which fragment (if any) contained the C terminus of FGF23. The reactions containing FGF23 alone after 4 hours at 37°C displayed 6×His-tag signal at only 30 kDa (Fig. 4B, lane 1), whereas reactions containing both FGF23 and tPA showed a reduction in 6×His-tag signal at 30 kDa and the appearance of a new 6×His-tag signal at around 13 kDa (Fig. 4B, lane 2). This result confirmed that tPA cleaves the full-length FGF23 generating the lower–molecular weight peptide fragments seen in the silver-stained gels, of which the lower band at 13 kDa contained the C terminus of FGF23.
Fig. 4. Proteolysis of rhFGF23 by tPA, uPA, and plasmin in vitro.
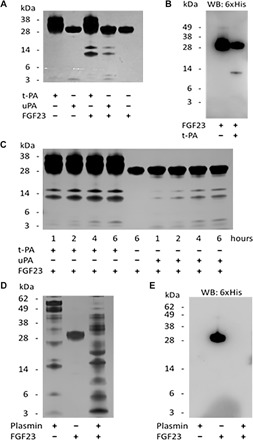
(A) Silver-stained SDS-PAGE gel of reactions containing purified recombinant human tPA, uPA, and FGF23. (B) Western blot (WB) using anti–6×His-tag antibody and tPA reactions from (A). (C) Silver-stained SDS-PAGE gel of in vitro time courses of FGF23 proteolysis by tPA and uPA. (D) Silver-stained gel showing degradation of FGF23 by plasmin. (E) Immunoblot of the gel in (D) with anti–6×His-tag antibody shows iFGF23 (lane 2) and a complete degradation of FGF23 by plasmin (lane 3).
The temporal pattern of proteolysis of FGF23 incubated in vitro at 37°C for 1, 2, 4, and 6 hours with tPA or uPA displayed a time-dependent increase in the intensity of the lower–molecular weight bands at approximately 17 and 13 kDa (Fig. 4C). Faint bands at 4 kDa were also visualized in a time-dependent manner in the protease-containing reactions as well, which were not present in the reactions containing the proteases alone. Smaller–molecular weight reaction products also appeared at earlier time points in reactions of FGF23 containing tPA than in reactions containing uPA, despite the fact that both reactions used molar equivalent amounts of each protease. The 17- and 13-kDa bands produced by tPA at every time point tested stained more intensely than those generated by uPA, indicating that tPA is likely to be more efficient at FGF23 proteolysis than uPA. This observation suggests that tPA may be the more relevant serine protease in FGF23 homeostasis in blood.
Next, we tested the effects of plasmin, the active enzyme of fibrinolysis, on FGF23 proteolysis. Under the identical reaction conditions used for tPA proteolysis for 4 hours at 37°C, self-incubation of plasmin yielded many peptide bands that indicate the autocatalytic activity of the enzyme (Fig. 4D, lane 1). Coincubation of FGF23 and plasmin produced new bands (Fig. 4D, lane 3) that were not present in the FGF23 alone (Fig. 4D, lane 2) or plasmin alone reactions. Because many peptides were generated in these reactions, we immunoblotted with the anti–6×His-tag antibody to identify the fragments derived from FGF23. As expected, no 6×His-tag signal was detected in the plasmin alone reaction (Fig. 4E, lane 1). In reactions of FGF23 alone, a single band was detected by the 6×His-tag antibody at 30 kDa, the molecular weight of full-length FGF23 (Fig. 4E, lane 2). Upon plasmin degradation of FGF23, the His-tag signal disappeared altogether (Fig. 4E, lane 3). These results suggest that plasmin can carry out proteolytic degradation of FGF23, and it may provide additional catabolism of FGF23 downstream from the PAs.
Identification of tPA cleavage sites of FGF23
To determine which peptide bonds of FGF23 are cleaved by tPA specifically, we analyzed tPA-generated peptide fragments with a liquid chromatography–mass spectrometry (LC-MS)–based top-down proteomics approach (Fig. 5). Reactions containing FGF23 incubated with or without tPA for 4 hours at 37°C were subjected to reversed-phase LC-MS to detect specific FGF23 fragments, and their amino acid identity was determined to locate the specific positions of proteolytic cleavage. The largest peptide fragment of FGF23 detected in the tPA-containing reactions had a mass of 17,267 Da (Fig. 5A), corresponding to amino acids 25 to 175 with an intact disulfide bond between cysteine residues 96 and 113 (Fig. 5B). No fragments of FGF23 were detected in the FGF23 alone reactions. Six other peptide fragments of FGF23 were detected in the tPA-catalyzed reactions as well, and a peptide map was generated summarizing these fragments (Fig. 5C), with numbers indicating which amino acids were present at each end. The table generated from this summary shows the specific tPA-dependent FGF23 cleavage sites (Fig. 5D). Amino acid numbers on the left side of the table, and standard amino acid single-letter code on the right side, indicate the positions within full-length human FGF23 where cleavage occurs, represented by the “^” symbol. The flanking four residues on each side of the cleavage site are also included for reference.
Fig. 5. Identification of tPA cleavage sites in FGF23 by LC-MS–based top-down proteomics.
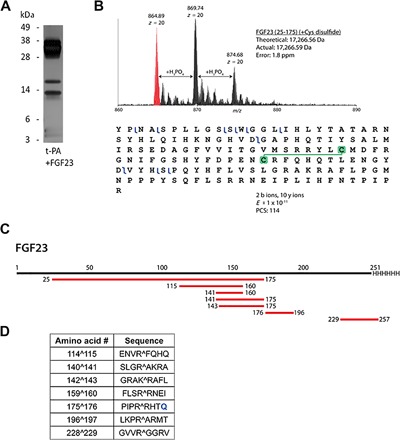
(A) Silver-stained SDS-PAGE gel image of the reaction subjected to proteomics. (B) Example data for proteolytic peptide identification of tPA-induced cleavage of FGF23. Mass spectrum of FGF23 (amino acids 25 to 175) showing the high mass accuracy of the match of MS data to theoretical mass [within 2 parts per million (ppm)]. Adducts spaced by ~98 Da are indicative of incomplete removal of large amounts of phosphate-containing buffer from the reaction mixture. A graphical fragment map of FGF23 (amino acids 25 to 175) is shown after collisionally induced dissociation of the 20+ charge state, demonstrating its confident identification and retention of its disulfide bond (49). PCS, proteoform characterization score. (C) Map of the peptides confidently identified (E ≤ 1 × 10−11) with proteomics from FGF23 in the sample. (D) Table summarizing the amino acid numbers and sequences of identified cut sites denoted by the “^” symbols. R179Q mutation is shown in blue font color.
TM5441 prevents PAI-1 inhibition of tPA-catalyzed FGF23 proteolysis
Because PAI-1 is the main physiological inhibitor of tPA and uPA, we next tested whether or not FGF23 proteolysis by the PAs in vitro could occur in the presence of PAI-1. This mimics physiological conditions where increased circulating levels of PAI-1 are present, such as those seen in CKD, systemic inflammation, or aging (18, 34, 35). Again using purified human recombinant proteins, reactions containing FGF23, tPA, and PAI-1 were performed for 4 hours at 37°C. Incubation of FGF23 with tPA expectedly produced lower–molecular weight bands of approximately 17 and 13 kDa, which were completely absent if PAI-1 was present in the reaction (Fig. 5, lanes 4 and 5). Finally, addition of the novel small-molecule PAI-1 antagonist TM5441 to the reaction restored the proteolytic activity of tPA on FGF23 in the presence of PAI-1, as evidenced by the presence of the 17- and 13-kDa bands (Fig. 5, lane 6).
DISCUSSION
The findings reported in this study expand our understanding of the regulation of the complex and highly dynamic homeostasis of the phosphaturic hormone FGF23. Previous investigations of FGF23 proteolysis have revealed that inactivating proteolysis can occur at an RXXR motif located at FGF23 residues R176 and R179, a process that is thought to be mediated by SPCs (7). Here, we demonstrate for the first time that extracellular proteases of the plasminogen activation system, tPA and uPA, display proteolytic activity against FGF23 in vitro and both produce reaction products that have identical mobility on SDS-PAGE, suggesting that tPA and uPA most likely cleave FGF23 at the same sites. Although both proteases recognize similar target sequences and cleave substrates at arginine residues, the optimum target consensus sequences for each differ substantially (36). In time course comparisons of FGF23 proteolysis by tPA and uPA, tPA appears to exhibit greater catalytic efficiency in FGF23 proteolysis than does uPA.
Previous reports have identified the susceptibility of FGF23 to proteolytic cleavage by furin, which is confined to the same RXXR motif. In contrast, we report here for the first time that tPA cleavage of FGF23 occurs at multiple RX motifs, although R176 seems to be the preferred cleavage site to both tPA and furin. Because of the R179Q mutation present in the rhFGF23 protein used in this study, we did not detect any peptides generated by cleavage at R179. Although the largest peptide product that we found in these experiments was a 17-kDa fragment produced from cleavage at the R176 site (Fig. 5A), MS analysis of the tPA-generated peptides of FGF23 revealed several other novel tPA peptide targets. Some of these novel sites lie in additional RXXR motifs present in FGF23, such as those seen at residues R140 and R228 (Fig. 6D). Most of these novel proteolytic cleavage sites are present in the C-terminal portion of FGF23. In contrast, we detected an intact disulfide bond in the N-terminal fragment, and it appears likely that this disulfide bond stabilizes the conformation of the N-terminal region that renders it resistant to proteolysis. One novel proteolytic site is located just distal to the disulfide bonding cysteine residue C113 (Fig. 6, B and D). Future investigations are warranted to examine the possible contribution of the disulfide bond to the metabolic stability of the N-terminal fragment and its potential effects on FGF23 signaling through the FGFR-klotho receptor complex.
Fig. 6. TM5441 restores tPA-mediated FGF23 proteolysis in the presence of PAI-1.
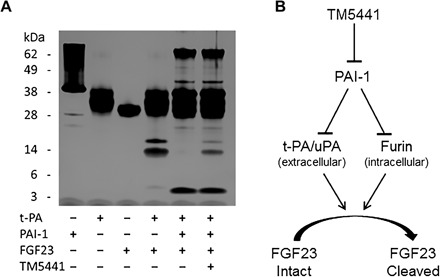
(A) Silver-stained SDS-PAGE gel of reactions containing purified recombinant human tPA, PAI-1, and FGF23 with and without the PAI-1 antagonist TM5441. (B) Model depicting that inhibition of PAI-1 leads to restoration of PA- and furin-mediated FGF23 proteolysis.
Many studies have verified that posttranslational modification determines whether FGF23 polypeptide is secreted as an intact biologically active molecule or subjected to intracellular proteolysis (37). FGF23 is O-glycosylated at the residue T178 by the enzyme ppGalNAc-T3 (polypeptide N-acetylgalactosaminyltransferase 3), and this modification blocks its proteolysis and is required for the release of iFGF23 (38). Furthermore, a secretory kinase, FAM20C, phosphorylates FGF23 on S180 within the furin recognition motif R176XXR179/S180, and this phosphorylation blocks the O-glycosylation of FGF23 and renders it susceptible to proteolysis by furin (14, 39). It seems that these dynamic posttranslational processes including phosphorylation, glycosylation, and proteolysis play a critical role in the intracellular regulation of FGF23 homeostasis. In light of these findings, it is important to note that both tPA and uPA can cleave glycosylated FGF23, because glycosylated rhFGF23 was used in this study. This contention is supported by the fact that PAI-1 inhibition decreased FGF23 levels in kl/kl and Pai-1stab transgenic mice, both of which likely have glycosylated FGF23, because the activity of the enzyme ppGalNAc-T3 has not been genetically altered in these animals.
Reductions in elevated plasma levels of FGF23 mediated by either TM5441 inhibition or genetic deficiency of PAI-1 in four different murine models including kl/kl, Pai-1stab, WT, and PAI-1−/− mice with AKI serve as an important proof of concept. These findings have therapeutic implications for conditions and diseases associated with high levels of FGF23, including LVH, CKD, AKI, and ADHR. TM5441 inhibition of PAI-1 activity resulted in reduced levels of the total FGF23 in plasma measured by the C-term FGF23 ELISA, an assay that detects both iFGF23 and cFGF23, through recognition of the C-terminal fragment generated by cleavage at residue R176 (peptide 176–251) or R179 (peptide 179–251) of iFGF23. Because we identified novel cleavage sites of FGF23 by tPA within these peptides, namely, R196 and R228 (Fig. 6D), epitopes used by the capture and detection antibodies of the ELISA kit are most likely altered by the tPA proteolysis. Thus, the C-term FGF23 ELISA assay used in this study likely does not recognize advanced PA-digested peptides of C-terminal fragment of FGF23 and underestimates the TM5441-induced decreases in total FGF23, which further supports the conclusion that the inhibition of PAI-1 activity reduces circulating FGF23 levels.
This study demonstrates that the small-molecule PAI-1 antagonist TM5441 permits FGF23 proteolysis in the presence of PAI-1. We have observed that PAI-1 deficiency and TM5441 treatment decreased plasma FGF23 levels by 98% (17) and 66% (this study) in kl/kl mice, respectively. Although the resultant FGF23 levels are significantly higher than those of WT mice, modulating PAI-1 activity by either TM5441 treatment or genetic deficiency caused important physiological changes that extended the life span of kl/kl mice by fourfold. In addition, we detected that PAI-1–deficient kl/kl mice exhibit normalized plasma levels of creatinine, senescence messaging secretory factors, and reduced vitamin D3 (17). Many of the diseases and conditions associated with elevated FGF23 levels also display elevated PAI-1 levels, such as CKD (19, 40–42). Thus, pharmacological inhibition of PAI-1 might be useful in this setting to reduce FGF23 levels and possibly to restore homeostasis to other targets of PA extracellular proteolysis, including IGFBP3 (25). It has also been previously demonstrated that PAI-1 can inhibit furin (43), another serine protease implicated in FGF23 proteolysis (14). However, because furin is thought to be mainly intracellular, it likely has no or minimal effect on extracellular processing of FGF23. Together, it seems likely that furin serves as the intracellular mechanism of regulating FGF23 processing, whereas the PAs play a critical role in the extracellular proteolysis of FGF23. PAI-1 has the potential to regulate both intracellular and extracellular homeostasis of FGF23 by inhibiting furin in the Golgi space and tPA and uPA in the extracellular space, respectively. Although PAI-1 inhibition accelerates FGF23 clearance and tPA cleaves FGF23 in multiple sites, including the furin cleavage site R176 that inactivates FGF23, it is not known whether the tPA-generated FGF23 peptides retain or acquire the capacity to bind to FGFR and act as a receptor agonist or as an antagonist to iFGF23. Although we speculate that the tPA-mediated proteolysis of FGF23 inactivates the protein, it is certainly possible that proteolysis may generate fragments with important functional properties.
Coding sequence mutations in FGF23 cause ADHR in humans. In this rare disorder, the furin cleavage site is mutated, leading to iFGF23 accumulation, unrelenting renal phosphate excretion, and bone disease. In this circumstance, the PA system is likely to be the only protease/protease inhibitor system that can play a role in FGF23 homeostasis. Thus, inhibition of PAI-1 activity in these patients may be desirable. More commonly, patients with CKD have elevated FGF23 levels, even though the genetic mutations causing the above disorders do not exist. This suggests that elevated FGF23 levels are likely caused by augmented FGF23 expression or impaired clearance of FGF23 due to possible alterations in the balance between the activities of proteases and protease inhibitors. Although pharmacological inhibition of PAI-1 will not directly affect FGF23 expression, this approach will likely increase elimination of FGF23 from the circulation.
Our findings indicate that either genetic deficiency or pharmacological inhibition of PAI-1 can reduce circulating FGF23 levels by improving its proteolytic clearance but does not improve mineral homeostasis in kl/kl mice and mice with AKI, both of which lack functional FGFR. In addition to its roles in phosphate excretion and reducing the vitamin D3 levels in blood, emerging evidence implicates FGF23 in LVH. The majority of patients with CKD and ESRD die from CVD that features LVH (4, 26, 44). FGF23 has been shown to induce LVH through FGFR but independent of klotho, the kidney-specific co-receptor of FGF23, via the PLC-γ (phospholipase C–γ)–calcineurin–NFAT (nuclear factor of activated T cell) pathway (4). Because the common treatment to prevent CVD is generally less effective in patients with CKD and ESRD, the potential value of PAI-1 inhibitors in cardiovascular mortality in these patient groups merits further investigation.
Phosphaturic mesenchymal tumors are rare, difficult to locate, and often linked to tumor-induced osteomalacia (TIO). Excessively high levels of FGF23, secreted by mesenchymal tumors, have been established as the causative factor for TIO, in which patients present with hypophosphatemia due to increased renal phosphate wasting, low vitamin D3 levels, and reduced bone density (45–47). Although a number of different therapeutic approaches are currently being explored to treat this rare disorder, our observations indicate that pharmacological inhibition of PAI-1 may provide some therapeutic benefit in patients with TIO due to elevated FGF23.
In the FA-induced AKI model, FGF23 levels rise sharply due to increased FGF23 synthesis primarily in bone and possibly in other tissues as well. We present evidence that increased PAI-1 levels contribute to increased plasma FGF23 levels in mice with AKI. We observed that genetic PAI-1 deficiency reduces FGF23 levels in kl/kl mice and mice with AKI more than PAI inhibition. This finding suggests that PAI-1 itself may influence a receptor-dependent feedback loop that actually governs FGF23 production. This hypothesis is supported by our findings that PAI-1 inhibition was more effective in lowering plasma FGF23 levels in Pai-1stab transgenic mice, which likely have impaired clearance of FGF23 but not pathologically induced FGF23 synthesis. Together, the present study indicates that inhibition of PAI-1 activity significantly augments the clearance of endogenous or exogenously delivered FGF23 in mice. These findings suggest that selective PAI-1 inhibition may provide a multifaceted therapeutic benefit in humans with elevated FGF23 levels.
MATERIALS AND METHODS
Study design
The purpose of this study was to explore the effects of serine proteases, tPA and uPA, and the SERPIN PAI-1 of the plasminogen activation system on the clearance of FGF23, an endocrine hormone that primarily affects kidneys to regulate phosphate homeostasis. On the basis of our previous observation that PAI-1 deficiency reduced the extremely high levels of FGF23 in kl/kl mice, we hypothesized that PAs and PAI-1 directly regulate FGF23 homeostasis and this will result in altering FGF23 signaling. We tested this hypothesis using several in vivo models that have elevated levels of FGF23 and PAI-1 including kl/kl mice, PAI-1 transgenic mice, mice with AKI, and mice that were tail vein–injected with rhFGF23. We modulated PAI-1 activity either by administering a small-molecule PAI-1 antagonist, TM5441, or by genetic deficiency of PAI-1. Using recombinant human proteins, we further tested in vitro the proteolysis of FGF23 by tPA and uPA in the presence and absence of PAI-1 and additionally identified the peptide bonds of FGF23 cleaved by tPA. To avoid variations due to circadian rhythm, plasma samples were collected at the same time of the day. The end points of the study were measuring plasma levels of cFGF23, iFGF23, and PAI-1. We used a sample size varying from 6 to 14 mice in study groups to achieve a 90% power and a significance at P < 0.05 using the PS software to calculate power and sample size available from http://biostat.mc.vanderbilt.edu/wiki/Main/PowerSampleSize.
Mice
All mice used in this study were housed in the Northwestern University Feinberg School of Medicine, and all the procedures were approved by the Institutional Animal Care and Use Committee of Northwestern University. kl/kl mice were obtained from M. Kuro-o and were of mixed background. Breeder pairs of all other mice strains were obtained from The Jackson Laboratory and of the C57BL/6J background. Study groups were 10- to 12-week-old mice except for kl/kl mice. Because of their short life span, kl/kl mice were 4 weeks old when admitted into the study.
Recombinant proteins and quantitation of circulating factors
Purified recombinant human two-chain tPA (catalog no. HTPA-TC), uPA (catalog no. UPA-LMW), and active PAI-1 (catalog no. CPAI) were obtained from Molecular Innovations. Purified recombinant human FGF23 (rhFGF23) protein containing R179Q mutation (native FGF23 is not available commercially) was obtained from R&D Systems (catalog no. 2604-FG-025). Plasma levels of FGF23 were determined using the following ELISA kits from Immutopics Inc.: Mouse/Rat FGF23 (C-Term) ELISA Kit (catalog no. 60-6300), Mouse/Rat FGF23 (Intact) ELISA Kit (catalog no. 60-6800), and Human FGF23 (C-Term) ELISA Kit (catalog no. 60-6100). The Mouse PTH 1-84 ELISA Kit was purchased from Immutopics (catalog no. 60-2305). Total PAI-1 antigen levels in mouse plasma samples were measured using the ELISA kit from Molecular Innovations (catalog no. MPAIKT-TOT). Serum phosphate levels were measured using a kit from Stanbio Laboratories (catalog no. 0830-125).
Pharmacological inhibition of PAI-1
The PAI-1 antagonist TM5441 was obtained from T. Miyata. A 2.5 mM stock solution was prepared in dimethyl sulfoxide (DMSO) and used for the in vitro studies. Rodent diet containing the inhibitor was prepared by mixing a homogeneous suspension of TM5441 in 0.5% carboxymethyl cellulose into powder standard rodent diet (Harlan Teklad LM-485 number 7012), dried at room temperature, and stored at 4°C until use. Our preliminary tests have shown that the effective inhibitor doses were 10 mg/kg per day for kl/kl mice and 100 mg/kg per day for Pai-1stab mice. The reason for the higher dose of TM5441 required for Pai-1stab is likely to be the levels of PAI-1 stab in plasma and tissues as well as the transgenic expression of functionally stabilized human PAI-1 in Pai-1stab mice.
FGF23 clearance under normal physiological and AKI conditions
We analyzed the elimination of FGF23 from circulation and induced AKI, as described previously (33). Briefly, we reconstituted rhFGF23 in sterile saline solution and injected into the mice via the tail vein at a dose of 40 μg/kg 24 hours after they were treated with either vehicle or FA (240 mg/kg). Plasma samples were collected at 1, 3, 5, 15, 30, 60, and 120 min after injection of rhFGF23. Plasma samples were diluted 100-fold, and iFGF23 and cFGF23 levels were measured by ELISA method in duplicate of each sample. Results were analyzed by using nonlinear curve fitting statistical analysis function of GraphPad Prism.
In vitro protease reactions and Western blotting
rhFGF23 (1 to 1.5 μg) was incubated at 37°C for the indicated time points with either recombinant human tPA (1.1 μg) or uPA (1.1 μg). As needed, tPA or uPA was inhibited with 1.7 μg of recombinant active human PAI-1. Reactions were carried out in 25-μl final volume containing 50 mM tris-HCl (pH 8.0), 150 mM NaCl, 0.2 mM CHAPS, 0.1% PEG-8000 (polyethylene glycol, molecular weight 8000), and 1% DMSO. To terminate the reaction, samples were incubated at 95°C for 5 min in the presence of 1× NuPAGE LDS sample buffer and 1× NuPAGE sample reducing agent. Gels were silver-stained using the Pierce Silver Stain Kit from Thermo Fisher Scientific according to the manufacturer’s protocol. The anti–6×His-tag antibody was obtained from Abcam, and the FGF23 antibody was obtained from Santa Cruz Biotechnology. Secondary horseradish peroxidase (HRP)–conjugated goat anti-rabbit antibodies were obtained from Molecular Innovations. NuPAGE 4-12% Bis-Tris Gels, PVDF iBlot Gel Transfer Stacks, 4× NuPAGE LDS sample buffer, and 10× NuPAGE sample reducing agent were from Life Technologies. Luminata Forte Western HRP substrate detection reagent was from EMD Millipore.
Determination of FGF23 fragments and sample preparation
Proteins [2.2 μg of tPA and 2 μg of FGF23 alone or in combination, incubated for 4 hours at 37°C in a 50-μl final volume of tris-buffered saline (pH 8.0)] were precipitated using the methanol-chloroform-water method (48). After precipitation, the sample was resuspended in 35 μl of 95% water, 5% acetonitrile + 0.1% formic acid. For MS, samples (5 μl) were injected onto a trap column [150 μm inside diameter (ID) × 3 cm] coupled with a nanobore analytical column (75 μm ID × 15 cm). The trap and analytical columns were packed with polymeric reversed-phase (PLRP-S, Phenomenex) medium (5 μm, 1000 Å pore size). Samples were separated using a linear gradient of solvent A and solvent B (5% water, 95% acetonitrile, and 0.1% formic acid) over 60 min. MS data were obtained on a 12T Velos FT Ultra (Thermo Fisher Scientific) mass spectrometer fitted with a custom nanospray ionization source. Intact MS data were obtained at a resolving power of 170,000 [mass charge ratio (m/z) 400]. The top 2 m/z species were isolated within the Velos ion trap and fragmented using CID. The data were initially processed with the cRAWler algorithm to create PUF files for searching with ProSightPC. The data were searched against a custom database containing only tissue plasminogen activator (UniProt: P00750) and fibroblast growth factor 23 (UniProt: Q9GZV9, modified to include C-terminal 6×His). The search strategy used was a biomarker search (looking for any subsequences of either of the proteins) with a 15-ppm intact mass tolerance and 15-ppm fragment mass tolerance, allowing the potential for disulfide bonds.
Statistical analysis
Nonlinear curve fit analysis was used to evaluate biphasic clearance of FGF23 from the circulation. We report the data group means ± SD. Results were considered statistically significant if P < 0.05. T test and ANOVA were used.
Acknowledgments
We thank S. Misener (Center for Translational Imaging, Department of Radiology) for technical assistance with the tail vein injections and C. DeHart and P. Gottlieb (Proteomics Center of Excellence, Northwestern University Feinberg School of Medicine, Chicago, IL) for technical assistance with MS assays. Funding: This work was supported by NIH/National Heart, Lung, and Blood Institute grants 2 R01 HL051387-18A1, 1P01HL108795-01, and T32 (National Institute of Diabetes and Digestive and Kidney Diseases KD007169). Author contributions: M.E. and A.T.P. conducted the experiments, analyzed the data, and wrote the manuscript. P.M.T. performed the proteomics analysis. P.F. analyzed the data. T.M. contributed reagents. D.E.V. analyzed the data and wrote the manuscript. Competing interests: M.E., A.T.P., T.M., and D.E.V. are authors on a provisional patent application filed by Northwestern University (U.S. Provisional Application number 62/082,969, filed 21 November 2014). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Itoh N., Ornitz D. M., Evolution of the Fgf and Fgfr gene families. Trends Genet. 20, 563–569 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Goetz R., Mohammadi M., Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 14, 166–180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goetz R., Beenken A., Ibrahimi O. A., Kalinina J., Olsen S. K., Eliseenkova A. V., Xu C., Neubert T. A., Zhang F., Linhardt R. J., Yu X., White K. E., Inagaki T., Kliewer S. A., Yamamoto M., Kurosu H., Ogawa Y., Kuro-o M., Lanske B., Razzaque M. S., Mohammadi M., Molecular insights into the Klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 27, 3417–3428 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faul C., Amaral A. P., Oskouei B., Hu M.-C., Sloan A., Isakova T., Gutiérrez O. M., Aguillon-Prada R., Lincoln J., Hare J. M., Mundel P., Morales A., Scialla J., Fischer M., Soliman E. Z., Chen J., Go A. S., Rosas S. E., Nessel L., Townsend R. R., Feldman H. I., St. John Sutton M., Ojo A., Gadegbeku C., Di Marco G. S., Reuter S., Kentrup D., Tiemann K., Brand M., Hill J. A., Moe O. W., Kuro-o M., Kusek J. W., Keane M. G., Wolf M., FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 121, 4393–4408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White K. E., Carn G., Lorenz-Depiereux B., Benet-Pages A., Strom T. M., Econs M. J., Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 60, 2079–2086 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Riminucci M., Collins M. T., Fedarko N. S., Cherman N., Corsi A., White K. E., Waguespack S., Gupta A., Hannon T., Econs M. J., Bianco P., Gehron Robey P., FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J. Clin. Invest. 112, 683–692 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benet-Pagès A., Lorenz-Depiereux B., Zischka H., White K. E., Econs M. J., Strom T. M., FGF23 is processed by proprotein convertases but not by PHEX. Bone 35, 455–462 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Nakatani T., Sarraj B., Ohnishi M., Densmore M. J., Taguchi T., Goetz R., Mohammadi M., Lanske B., Razzaque M. S., In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J. 23, 433–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haussler M. R., Whitfield G. K., Kaneko I., Forster R., Saini R., Hsieh J.-C., Haussler C. A., Jurutka P. W., The role of vitamin D in the FGF23, klotho, and phosphate bone-kidney endocrine axis. Rev. Endocr. Metab. Disord. 13, 57–69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu H. J., Wu M.-S., Fibroblast growth factor 23: A possible cause of left ventricular hypertrophy in hemodialysis patients. Am. J. Med. Sci. 337, 116–122 (2009). [DOI] [PubMed] [Google Scholar]
- 11.ADHR Consortium , Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 26, 345–348 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Urakawa I., Yamazaki Y., Shimada T., Iijima K., Hasegawa H., Okawa K., Fujita T., Fukumoto S., Yamashita T., Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Wohlfahrt P., Melenovsky V., Kotrc M., Benes J., Jabor A., Franekova J., Lemaire S., Kautzner J., Jarolim P., Association of fibroblast growth factor-23 levels and angiotensin-converting enzyme inhibition in chronic systolic heart failure. JACC Heart Fail. 3, 829–839 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Tagliabracci V. S., Engel J. L., Wiley S. E., Xiao J., Gonzalez D. J., Nidumanda Appaiah H., Koller A., Nizet V., White K. E., Dixon J. E., Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. U.S.A. 111, 5520–5525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuro-o M., Matsumura Y., Aizawa H., Kawaguchi H., Suga T., Utsugi T., Ohyama Y., Kurabayashi M., Kaname T., Kume E., Iwasaki H., Iida A., Shiraki-Iida T., Nishikawa S., Nagai R., Nabeshima Y.-i., Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Takeshita K., Yamamoto K., Ito M., Kondo T., Matsushita T., Hirai M., Kojima T., Nishimura M., Nabeshima Y., Loskutoff D. J., Saito H., Murohara T., Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “Klotho” mouse. Semin. Thromb. Hemost. 28, 545–554 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Eren M., Boe A. E., Murphy S. B., Place A. T., Nagpal V., Morales-Nebreda L., Urich D., Quaggin S. E., Budinger G. R. S., Mutlu G. M., Miyata T., Vaughan D. E., PAI-1–regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc. Natl. Acad. Sci. U.S.A. 111, 7090–7095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park M. Y., Herrmann S. M., Saad A., Eirin A., Tang H., Lerman A., Textor S. C., Lerman L. O., Biomarkers of kidney injury and Klotho in patients with atherosclerotic renovascular disease. Clin. J. Am. Soc. Nephrol. 10, 443–451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy A. A., Fogo A. B., Plasminogen activator inhibitor-1 in chronic kidney disease: Evidence and mechanisms of action. J. Am. Soc. Nephrol. 17, 2999–3012 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Vaughan D. E., Angiotensin, fibrinolysis, and vascular homeostasis. Am. J. Cardiol. 87, 18C–24C (2001). [DOI] [PubMed] [Google Scholar]
- 21.Gebbink M. F. B. G., Tissue-type plasminogen activator-mediated plasminogen activation and contact activation, implications in and beyond haemostasis. J. Thromb. Haemost. 9 (suppl. 1), 174–181 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Tersteeg C., de Maat S., De Meyer S. F., Smeets M. W. J., Barendrecht A. D., Roest M., Pasterkamp G., Fijnheer R., Vanhoorelbeke K., de Groot P. G., Maas C., Plasmin cleavage of von Willebrand factor as an emergency bypass for ADAMTS13 deficiency in thrombotic microangiopathy. Circulation 129, 1320–1331 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Lijnen H. R., Elements of the fibrinolytic system. Ann. N. Y. Acad. Sci. 936, 226–236 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Kortlever R. M., Higgins P. J., Bernards R., Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 8, 877–884 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elzi D. J., Lai Y., Song M., Hakala K., Weintraub S. T., Shiio Y., Plasminogen activator inhibitor 1-insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc. Natl. Acad. Sci. U.S.A. 109, 12052–12057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isakova T., Xie H., Yang W., Xie D., Anderson A. H., Scialla J., Wahl P., Gutiérrez O. M., Steigerwalt S., He J., Schwartz S., Lo J., Ojo A., Sondheimer J., Hsu C.-y., Lash J., Leonard M., Kusek J. W., Feldman H. I., Wolf M. Chronic Renal Insufficiency Cohort (CRIC) Study Group , Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305, 2432–2439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf M., Molnar M. Z., Amaral A. P., Czira M. E., Rudas A., Ujszaszi A., Kiss I., Rosivall L., Kosa J., Lakatos P., Kovesdy C. P., Mucsi I., Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J. Am. Soc. Nephrol. 22, 956–966 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eren M., Painter C. A., Atkinson J. B., Declerck P. J., Vaughan D. E., Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation 106, 491–496 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Eren M., Gleaves L. A., Atkinson J. B., King L. E., Declerck P. J., Vaughan D. E., Reactive site-dependent phenotypic alterations in plasminogen activator inhibitor-1 transgenic mice. J. Thromb. Haemost. 5, 1500–1508 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Leaf D. E., Wolf M., Waikar S. S., Chase H., Christov M., Cremers S., Stern L., FGF-23 levels in patients with AKI and risk of adverse outcomes. Clin. J. Am. Soc. Nephrol. 7, 1217–1223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang M., Hsu R., Hsu C.-y., Kordesch K., Nicasio E., Cortez A., McAlpine I., Brady S., Zhuo H., Kangelaris K. N., Stein J., Calfee C. S., Liu K. D., FGF-23 and PTH levels in patients with acute kidney injury: A cross-sectional case series study. Ann. Intensive Care 1, 21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long D. A., Price K. L., Ioffe E., Gannon C. M., Gnudi L., White K. E., Yancopoulos G. D., Rudge J. S., Woolf A. S., Angiopoietin-1 therapy enhances fibrosis and inflammation following folic acid-induced acute renal injury. Kidney Int. 74, 300–309 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Christov M., Waikar S. S., Pereira R. C., Havasi A., Leaf D. E., Goltzman D., Pajevic P. D., Wolf M., Jüppner H., Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. 84, 776–785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu F.-C., Kritchevsky S. B., Liu Y., Kanaya A., Newman A. B., Perry S. E., Visser M., Pahor M., Harris T. B., Nicklas B. J.; Health ABC Study , Association between inflammatory components and physical function in the health, aging, and body composition study: A principal component analysis approach. J. Gerontol. A Biol. Sci. Med. Sci. 64, 581–589 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baune B. T., Smith E., Reppermund S., Air T., Samaras K., Lux O., Brodaty H., Sachdev P., Trollor J. N., Inflammatory biomarkers predict depressive, but not anxiety symptoms during aging: The prospective Sydney memory and aging study. Psychoneuroendocrinology 37, 1521–1530 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Coombs G. S., Bergstrom R. C., Madison E. L., Corey D. R., Directing sequence-specific proteolysis to new targets. The influence of loop size and target sequence on selective proteolysis by tissue-type plasminogen activator and urokinase-type plasminogen activator. J. Biol. Chem. 273, 4323–4328 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Blau J. E., Collins M. T., The PTH-vitamin D-FGF23 axis. Rev. Endocr. Metab. Disord. 16, 165–174 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Kato K., Jeanneau C., Tarp M. A., Benet-Pagès A., Lorenz-Depiereux B., Bennett E. P., Mandel U., Strom T. M., Clausen H., Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 281, 18370–18377 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Lindberg I., Pang H. W., Stains J. P., Clark D., Yang A. J., Bonewald L., Li K. Z., FGF23 is endogenously phosphorylated in bone cells. J. Bone Miner. Res. 30, 449–454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertoni E., Rosati A., Larti A., Merciai C., Zanazzi M., Rosso G., Gallo M., Marcucci R., Salvadori M., Chronic kidney disease is still present after renal transplantation with excellent function. Transplant. Proc. 38, 1024–1025 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Makulska I., Szczepańska M., Drożdż D., Polak-Jonkisz D., Zwolińska D., Skin autofluorescence as a novel marker of vascular damage in children and adolescents with chronic kidney disease. Pediatr. Nephrol. 30, 811–819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norata G. D., Baragetti I., Raselli S., Stucchi A., Garlaschelli K., Vettoretti S., Piloni G., Buccianti G., Catapano A. L., Plasma adiponectin levels in chronic kidney disease patients: Relation with molecular inflammatory profile and metabolic status. Nutr. Metab. Cardiovasc. Dis. 20, 56–63 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Bernot D., Stalin J., Stocker P., Bonardo B., Scroyen I., Alessi M.-C., Peiretti F., Plasminogen activator inhibitor 1 is an intracellular inhibitor of furin proprotein convertase. J. Cell Sci. 124, 1224–1230 (2011). [DOI] [PubMed] [Google Scholar]
- 44.de Simone G., Gottdiener J. S., Chinali M., Maurer M. S., Left ventricular mass predicts heart failure not related to previous myocardial infarction: The Cardiovascular Health Study. Eur. Heart J. 29, 741–747 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Chong W. H., Andreopoulou P., Chen C. C., Reynolds J., Guthrie L., Kelly M., Gafni R. I., Bhattacharyya N., Boyce A. M., El-Maouche D., Crespo D. O., Sherry R., Chang R., Wodajo F. M., Kletter G. B., Dwyer A., Collins M. T., Tumor localization and biochemical response to cure in tumor-induced osteomalacia. J. Bone Miner. Res. 28, 1386–1398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nawrot-Wawrzyniak K., Varga F., Nader A., Roschger P., Sieghart S., Zwettler E., Roetzer K. M., Lang S., Weinkamer R., Klaushofer K., Fratzl-Zelman N., Effects of tumor-induced osteomalacia on the bone mineralization process. Calcif. Tissue Int. 84, 313–323 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Shiba E., Matsuyama A., Shibuya R., Yabuki K., Harada H., Nakamoto M., Kasai T., Hisaoka M., Immunohistochemical and molecular detection of the expression of FGF23 in phosphaturic mesenchymal tumors including the non-phosphaturic variant. Diagn. Pathol. 11, 26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wessel D., Flügge U. I., A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 (1984). [DOI] [PubMed] [Google Scholar]
- 49.Dang X., Scotcher J., Wu S., Chu R. K., Tolić N., Ntai I., Thomas P. M., Fellers R. T., Early B. P., Zheng Y., Durbin K. R., Leduc R. D., Wolff J. J., Thompson C. J., Pan J., Han J., Shaw J. B., Salisbury J. P., Easterling M., Borchers C. H., Brodbelt J. S., Agar J. N., Paša-Tolić L., Kelleher N. L., Young N. L., The first pilot project of the consortium for top-down proteomics: A status report. Proteomics 14, 1130–1140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]