

Abstract
Escherichia coli O157:H7 is a significant human pathogen that is continually responsible for sickness, and even death, on a worldwide scale. While the pathology of E. coli O157:H7 infection has been well studied, the effect of it’s multiple resulting cytotoxic mechanisms on host metabolic activity has not been well characterized. To develop a more thorough understanding of these effects, several bioluminescence assays were evaluated for their ability to track both toxicity and host metabolic activity levels in real-time. The use of continuously autobioluminescent human cells was determined to be the most favorable method for tracking these metrics, as its self-sufficient autobioluminescent phenotype was unaffected by the presence of the infecting bacteria and its signal could be measured without cellular destruction. Using this approach, it was determined that infection with as few as 10 CFU of E. coli O157:H7 could elicit cytotoxic effects. Regardless of the initial infective dose, an impact on metabolic expression was not observed until bacterial populations reached levels between 5 × 105 and 1 × 106 (R2 = 0.933), indicating that a critical bacterial infection level must be reached prior to the onset of cytotoxic effects. Supporting this hypothesis, it was found that cells displaying infection-mediated metabolic activity reductions could recover to wild type metabolic activity levels if the infecting bacteria were removed prior to cell death. These results indicate that rapid treatment of E. coli O157:H7 infection could serve to limit host metabolic impact and reduce overall host cell death.
Keywords: Toxicology, Cell culture, E. coli O157:H7, Bioluminescence, Optical imaging
Introduction
Escherichia coli O157:H7 is the primary pathogen responsible for inducing hemolytic uremic syndrome in the United States (Klein et al. 2002; Tarr et al. 1990), but is also a widespread ecotoxicological concern due to its ability to survive asymptomatically within a number of species such as cattle, horses, pigs, dogs, deer, and seagulls (Karch et al. 2005). As a result of its widespread prevalence, it commonly infects humans and leads to the onset of significant health problems or even death. Due to both its prevalence in the environment and the significant human health concerns it presents, its basic pathology has been well reviewed in the scientific literature (Law 2000). These analyses have revealed that the primary cytotoxic mediators of E. coli O157:H7 infection are its Shiga toxins, a collection of excreted proteins that recognize specific host cell surface receptors. Following host receptor recognition, the Shiga toxin is internalized via macroinocytosis and becomes cytotosolically available (Lukyanenko et al. 2011). Once present within the cell, it then functions as an N-glycosidase to inhibit protein synthesis via ribosomal inactivation, leading to cell death (Sandvig et al. 2010).
While much effort has been put forth to develop methods for detecting E. coli O157:H7 (Valderrama et al. 2015), comparatively less work as been performed to elucidate the metabolic activity level dynamics that occur throughout the infection process, which could provide data for improving treatment options by developing an improved understanding of when and how human cells recognize the presence and effects of Shiga toxin exposure. This is especially warranted given the discovery of a potential secondary route for cytotoxicity through the activation of host mRNA synthesis, which was demonstrated to overexpress endothelin-1 to deleterious levels following toxin exposure (Bitzan et al. 1998). This process, in contrast to ribosomal inhibition and its corresponding reduction in protein synthesis, presents an opposing metabolic burden, with transcriptional over-activation requiring increased metabolic effort and translational inhibition removing a significant sink of metabolic activity. Therefore, by tracking the metabolic activity dynamics and cytotoxic effects imposed throughout the infection process, it may be possible to determine which of these mechanism is most impactful towards limiting cellular activity or inducing cytotoxicity. If successfully applied, this approach would represent a novel tool for investigating the activation of metabolically based toxicological mechanisms within a targeted group of organisms in a mixed population, without necessitating investigator interaction. This approach would therefore provide a rapid, simplistic, and non-influential means for monitoring metabolic dynamics under natural conditions at an ecological scale.
However, due to the time scale at which protein synthesis inhibition and mRNA expression occur, as well as the uncertainty surrounding when the resulting metabolic or cytotoxic phenotypes will manifest, this approach necessitates the use of a real-time or near real-time detection system that is highly sensitive to changes in metabolic activity levels. Ideally, this system should be capable of determining cell death as well. These criteria suggest the use of a bioluminescent system as opposed to a fluorescent or chemical detection system. Bioluminescent systems, such as firefly luciferase or bacterial luciferase, rely on the presence of metabolic intermediates to produce a luminescent signal. Therefore, unlike fluorescent proteins, which may remain active following cellular death, these systems provide an increased correlation between metabolic activity and signal generation (Close et al. 2010). While microscopy-based fluorescent dye assays could potentially serve in this role as well, the time required for sample preparation and analysis precludes their use by necessitating the use of longer time points between observations or the inclusion of large numbers of additional samples, which has the potential to increase assay error, personnel requirements, and costs. While there are currently no bioluminescent assays commercially available that are specifically designed to co-monitor metabolic activity levels and cytotoxicity, three potential bioluminescent systems were evaluated to serve this role. Two were based on firefly luciferase, and one was based on bacterial luciferase, however, each should be similarly capable of tracking metabolic activity and reporting on cell death through an elimination of bioluminescent signal due to a complete cessation of cellular metabolism (Class et al. 2014).
Materials and methods
Strain maintenance
Autobioluminescent embryonic kidney (HEK293) cells (490 BioTech, TN, USA) were routinely maintained in Dulbecco’s Modified Eagle Medium (DMEM) (GE Healthcare Hyclone, UT, USA) supplemented with 10 % fetal bovine serum (GE Healthcare Hyclone), 1 mM sodium pyruvate (Thermo/Life Technologies, CA, USA), Penicillin and Streptomycin at 100 U/mL and 100 μg/mL, respectively (GE Healthcare Hyclone), and G418 (EMD Millipore, MA, USA) at 100 μg/mL. All cells were maintained at 37 °C and 5 % CO2 for routine growth. Antibiotic-free DMEM supplemented with 10 % fetal bovine serum and 1 mM sodium pyruvate was used in infection assays to prevent the interference of antibiotics with bacterial viability and activity. E. coli O157:H7 (ATCC #43895) was grown in Luria–Bertani (LB) medium at 37 °C with shaking at 200 rpm.
Comparison of cytotoxicity and metabolic activity detection assays
Actively growing autobioluminescent HEK293 cells were harvested, re-suspended in antibiotic-free DMEM assay medium, and seeded in 100 μl medium volumes in individual wells of black 96-well tissue culture plates (Corning) at 5 × 104 cells/well. The plates were then covered with a lid and housed in the incubator for 2–4 h until bacterial addition. E. coli O157:H7 was grown in LB medium to an optical density at 600 nm (OD600) of 0.54–0.56 (∼4 × 108 CFU/mL) and diluted to ∼1 × 108 CFU/mL (1:4 dilution) in antibiotic-free DMEM assay medium. Ten microliters of the 1 × 108 CFU/ml bacteria dilution was then added to 100 μl of cells to initiate the infection assay. Triplicate wells of cells treated with only assay medium were included in each plate as controls for bioluminescent signal normalization, and triplicate wells containing cell-free medium were included in each plate as negative controls to survey background light detection levels.
At 2 and 4 h post infection (hpi), bioluminescent output was determined using an IVIS Lumina imaging system (PerkinElmer, MA, USA) followed by multiplexed CellTiter-Glo and ROS-Glo assays (Promega, WI, USA) to determine cell viability and the level of reactive oxygen species, respectively. To determine bioluminescence, cells were removed from the incubator, equilibrated at room temperature for 5 min as previously described (Class et al. 2014), and imaged using an IVIS Lumina imaging system with a 5 min integration time. Immediately after imaging, the same cells were subjected to CellTiter-Glo and ROS-Glo assays according to the manufacturer’s instructions, and signal output was determined in a Synergy 2 plate reader (BioTek, VT, USA) using a 1 s/well luminescent integration time.
Real-time tracking of bacterial infection of autobioluminescent HEK293 cells
E. coli O157:H7 was grown in LB medium to an OD600 of 0.54–0.56 (∼4 × 108 CFU/mL) and diluted to various concentrations ranging from ∼1 × 108 CFU/mL (1:4 dilution) to ∼1 × 103 CFU/mL (1:400,000 dilution) in antibiotic-free DMEM assay medium. Ten microliters of each bacterial concentration was then added to 5 × 104 HEK293 cells to reach a 100 μl final volume and seeded in black 96-well tissue culture plates as described above. Each infection dose was performed in triplicate wells of each plate. Cells treated with only assay medium and wells containing cell-free medium were also included in triplicate wells of each plate as positive and negative controls, respectively.
Immediately prior to infection, cells were removed from the incubator, equilibrated at room temperature for 5 min, and imaged using an IVIS Lumina imaging system with a 5 min integration time. Cells were then infected as described above and imaged similarly every 1 h for 18 h. Between readings, cells were maintained in the incubator at 37 °C and 5 % CO2 to limit the effect of uncontrolled environmental factors on cellular health and metabolism.
Correlation between bacteria counts and cellular metabolic activities
To investigate the correlation between the number of bacteria present in the medium and the metabolic activity of infected cells, triplicate sets of 5 × 104 autobioluminescent HEK293 cells were infected with 1:4, 1:40, 1:400, 1:4000, 1:40,000, and 1:40,0000 dilutions of an E. coli O157:H7 culture at an OD600 between 0.54 and 0.56 as described above. The actual initial infection dose was determined to be 7.3 ± 0.7 (mean ± standard deviation) × 105 – 7.3 ± 0.7 CFU by serial plating on LB/agar plates. To determine the number of bacteria at a given time point following assay initiation, 10 μl of assay medium was sampled periodically from wells containing infected cells across a 12 h period post-infection, serially diluted, and plated on LB/agar plates to determine the total CFU count. Cells representative of each initial infection concentration were sampled once throughout the 12 h period, with wells infected with higher doses being sampled earlier than those infected with lower doses. Cells were imaged immediately after sampling to determine their corresponding bioluminescence and metabolic activities at the same time points. This experiment was repeated twice, with each initial infection concentration analyzed at two different time points (for example, cells initially infected with the 1:40 bacteria dilution were sampled at 3 and 4 hpi, whereas cells initially infected with the 1:400,000 dilution were analyzed at 11 and 12 hpi) to represent the full range of the bioluminescent intensity dynamics observed following the onset of cellular metabolic activity level diminishment.
Rescue and recovery of infected cells
Autobioluminescent HEK293 cells were seeded at 5 × 104 cells/well and infected with ∼1 × 106 CFU of E. coli O157:H7 as described above. Cells were incubated for 2 h prior to infection to allow for attachment to the culture plates. At 2, 4, or 6 hpi, bioluminescence was measured in an IVIS Lumina system as described above. Immediately following imaging, infected cells were rescued by removing the bacteria-containing medium, washing with sterile phosphate buffer saline (PBS) pre-warmed to 37°C, and refreshing with fresh pre-warmed DMEM medium supplemented with Penicillin and Streptomycin. To account for any possible perturbation in bioluminescence resulting from the rescue procedure itself, non-infected cells were subjected to the same process at 2, 4, or 6 hpi to serve as internal controls. Rescued cells were returned to the incubator for recovery and imaged repeatedly for up to 16 hpi. For all readings, the bioluminescent output of the rescued cells was normalized to that of the similarly processed non-infected cells.
Data and statistical analysis
Bioluminescent images were analyzed using Living Image 4.2 (PerkinElmer) to determine the signal intensity from individual wells. The bioluminescence of infected cells was reported as a percentage of that emitted from non-infected control cells. The standard deviation of the percentage value was propagated using mean and standard deviation values of both infected and non-infected groups accordingly. A 4-parameter sigmoidal formula was used to analyze the relationship between number of bacteria and relative bioluminescence in SigmaPlot 11.0 (SYSTAT Software, CA, USA).
Results
Selection of a dual cytotoxicity and metabolic activity detection assay
The CellTiter-Glo assay uses the presence of ATP to serve a limiting reagent for bioluminescent production. Therefore, in this assay, the amount of ATP liberated following cellular lysis serves as a proxy for the metabolic activity level within the surveyed cellular population. Application of this assay at 2 hpi demonstrated a reduction in bioluminescent output to 58.4 % (±13.7 %) of uninfected control cells, however, counterintuitively, by 4 hpi bioluminescent output had rebounded to 70.8 % (±16.2 %) (Fig. 1a). These results were in contrast to the use of autobioluminescent cells, which displayed a reduction in autobioluminescent output to 11.2 % (±4.2 %) by 2 hpi, and a further reduction in output to 2.5 % (±0.6 %) by 4 hpi (Fig. 1a). Comparisons of these assays using homogenous cellular cultures have demonstrated strong correlations between their results (Class et al. 2014; Xu et al. 2014), suggesting that the increased output levels of the CellTiter-Glo assay were due to the liberation of ATP from lysed bacterial cells in addition to the targeted human cellular populations. This hypothesis was supported by the observation that a stationary phase pure culture of E. coli O157:H7 was able to elicit a signal >1500-fold over medium background using the CellTiter-Glo assay reagents. Therefore, as an alternative approach to detecting the metabolic results of E. coli O175:H7 exposure, the ROS-Glo assay, which produces a luminescent signal in response to the presence of reactive oxygen species such as those generated upon bacterial exposure (Brunder et al. 1996), was evaluated, but was found to be incapable of distinguishing significant output levels between infected cells and uninfected controls (Fig. 1b). The use of autobioluminescent cells was therefore selected as the method for tracking cellular metabolic activity levels since its continuous autobioluminescent signal could be repeatedly assayed without lysing the cells and therefore exposing the culture to extraneous bacterial metabolic intermediate components, and because it has been previously demonstrated to strongly correlate with metabolic activity levels in human cells (Class et al. 2014; Xu et al. 2014).
Fig. 1.
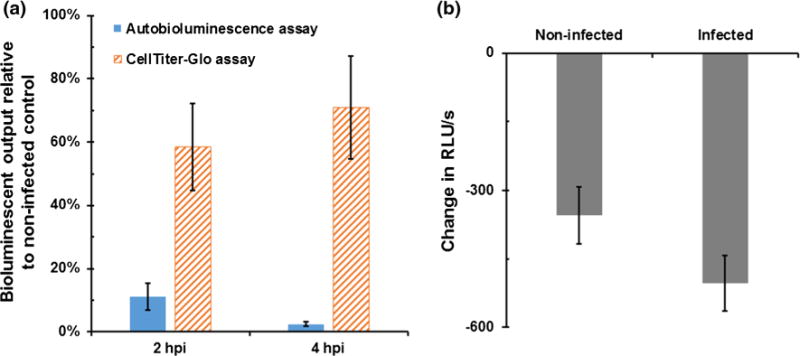
a Autobioluminescence from HEK293 cells demonstrated increasing reductions in metabolic activity levels at 2 and 4 hpi, while the liberated ATP from co-lysed E. coli O157:H7 artificially inflated CellTiter-Glo assay results as bacterial population sizes increased over time. b The alternative use of reactive oxygen species as a reporter for detecting the impacts of bacterial infection was similarly limited, as this assay could not differentiate between infected and non-infected control cells at 4 h post-infection. RLU relative light units
Correlation of cytotoxicity with initial infection dose
In order to determine the effect of initial infection doses on the dynamics of metabolic activity over the course of infection, equal numbers of cells were infected with a range of E. coli O157:H7 populations from ∼10 to 1 × 106 CFU and monitored hourly for their bioluminescent production (Fig. 2). It was observed that all infection levels resulted in diminishing metabolic activity levels as monitored through a reduction in autobioluminescent output. A correlation between the delay in the onset of metabolic diminishment and the infection dose was also observed. Cells infected with the highest infection dose displayed the earliest reduction in bioluminescence to 76.6 % (±10.1 %) at 2 hpi, and their relative metabolic activity continuously declined to ∼3.1 % (±0.5 %) by 6 hpi, at which point the signal was no longer significantly differentiable from background light detection. With the lower infection doses, the onset of cytotoxicity was delayed approximately 2 h for every order of magnitude decrease in initial infection dose, with the slowest onset observed at 12 hpi from cells infected with 10 CFU. Despite the observed delays, metabolic activity subsided rapidly within the 4 h following its onset, diminishing from an average of ∼ 88 to ∼5 % across all exposure levels.
Fig. 2.

Real-time tracking of the metabolic activity via bioluminescence production in HEK293 demonstrated that the initial infection dose of E. coli O157:H7 inversely correlated with the time required to observe reductions in metabolic activity levels. Bioluminescence reported as the mean (±standard deviation) percentage of signal relative to non-infected cells
Correlation of cytotoxic onset with E. coli O157:H7 population size
It was observed that the major difference between various infection doses appeared to be the length of incubation time required prior to the onset of cytotoxicity, with a higher infection dose correlating with an earlier initiation of metabolic shutdown. Considering the fast growth rate of O157:H7 under the experiment conditions (37 °C, aerobic), it was postulated that a minimum bacterial population threshold was therefore being achieved prior to the manifestation of impaired metabolism within the host cells. This hypothesis was supported by the observation that decreases in metabolic activity levels were not observed at any time point when exposure levels remained below 1 × 105 CFU (Fig. 3). Regardless of the initial infection dose, the bacterial population sizes observed concurrent with the diminishment of metabolic activity were determined to be within the range of ∼7.7 × 105 to 3.6 × 107 CFU, which corresponded to relative metabolic activity reductions to ∼65 to 4 % of uninfected controls. Mathematical analysis of all CFU count and corresponding relative bioluminescence data pairs produced a strong sigmoidal correlation between bacterial population sizes and cellular metabolism (R2 = 0.933) (Fig. 3) and predicted that a bacterial population size between ∼5 × 105 to 1 × 106 and ∼5 × 107 to 1 × 108 CFU was required to initiate the progression of cytotoxicity leading to cell death. These results were consistent with the 4 h window observed in Fig. 2, presuming a 20-minute doubling time for E. coli O157:H7 under the conditions employed.
Fig. 3.
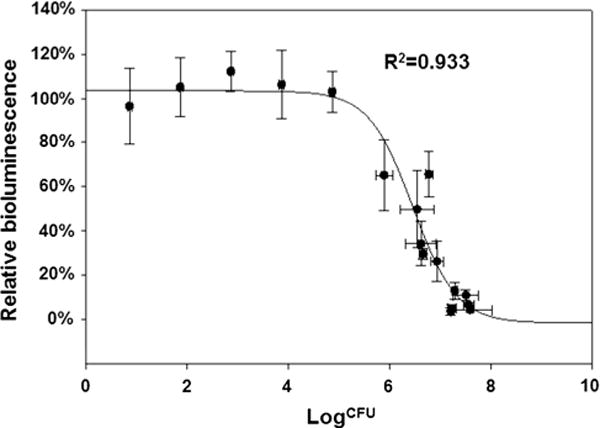
A strong correlation between bacterial population size and the onset of cytotoxicity indicated that a minimum of ∼5 × 105 to 1 × 106 CFU were required to induce changes in metabolic activity levels. Bioluminescence reported as the mean (±standard deviation) percentage of signal relative to non-infected cells
Metabolic recovery of E. coli O157:H7 exposed cells
Although 2 and 4 h exposures to E. coli O157:H7 reduced metabolic activity levels to 15.3 % (±2.4 %) and 2.6 % (±0.4 %) relative to uninfected cells, respectively, removal of the bacteria-containing medium and replenishment with fresh medium permitted a full recovery of metabolic activity to native levels within 2 h post-rescue (Student t test p = 0.39 and 0.76, respectively), with rescued cells maintaining their metabolic activity rates up to 12 h post-rescue (p > 0.05) (Fig. 4). However, cells rescued after 6 h of exposure manifest an inconsistent and partial recovery of metabolic activities, increasing from metabolic levels of ∼1.7 % (±1.6 %) relative to uninfected controls pre-rescue to 6.3 % (±6.3 %) by 2 h post-rescue. An extension of the recovery period up to 10 h post-rescue permitted a further increase in these cells’ relative metabolic activity to 19.4 % (±10.1 %), but they were unable to return to their pre-infection activity levels, likely indicating the occurrence of widespread cell death within the culture prior to rescue.
Fig. 4.
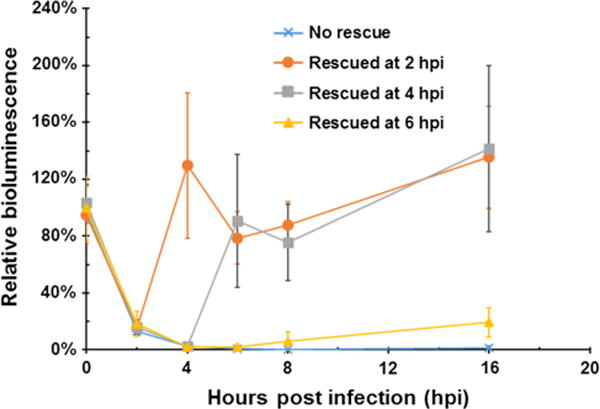
If infective pressure is removed prior to the onset of cellular death, HEK293 cells can reestablish native metabolic activity levels within 2 h of rescue. Bioluminescence reported as the mean (±standard deviation) percentage of signal relative to mock rescued non-infected cells
Discussion
In general, bioluminescent detection assays can be broken down into two categories. The first category relies on production of a luciferin protein from the subject cell, which is then exposed to an activating luciferin compound that is oxidized to produce the resulting luminescent signal. The second category externally applies both the luciferase protein and the luciferin, but relies on the liberation of a necessary co-factor (such as ATP) from the subject cell to serve as the limiting reagent for bioluminescent production. Due to the fact that protein synthesis inhibition is one of the hallmarks of E. coli O157:H7 toxicity, the former approach could not be employed because it was not possible develop a reporter system capable of producing the luciferase only after protein synthesis was inhibited. Similarly, due to the number of samples and frequency of screening that would be needed, it was not logistically feasible to develop a system that monitored for the reduction of luciferase protein expression. As such, the systems evaluated in this work were all based on the availability of metabolic intermediates. The two firefly luciferase systems were both found to be limited in that the destructive nature of their assay formats resulted in the liberation of metabolic intermediates from both the subject human cells and the infecting E. coli O157:H7 cells. This made it difficult to obtain reliable readings. The autobioluminescent bacterial luciferase system, on the other hand, did not require cellular destruction, and was therefore not subject to this limitation. While less well established than the firefly luciferase systems, there is nonetheless sufficient evidence to suggest that this system can be used to monitor for both metabolic activity levels and cytotoxic effects, and that its reported results correlate well with those of the firefly luciferase assays under monoculture conditions (Class et al. 2014; Xu et al. 2014).
Using this system, it was possible to simultaneously track the onset of metabolic activity dynamics in real-time across a variety of infective doses (Fig. 2). These results demonstrated that initial infections of as few as 10 bacteria were capable of leading to cytotoxic effects. This is consistent with previous reports suggesting that 50–80 cells are required to establish an infection in humans, and that even lower initial infection doses can lead to the enactment of deleterious cellular effects such as inflammation (Nataro and Kaper 1998). Although increases in cellular metabolic activity levels resulting from Shiga toxin-mediated transcriptional up-regulation were predicted (Bitzan et al. 1998), they were not observed using the employed system. It is possible that the increase in metabolic activity required to sustain these increases was not sufficient to increase autobioluminescent activity, but the possibility that these effects were not manifested until after ribosomal inactivation occurred cannot be ruled out. Indeed, if the latter hypothesis is correct, it is unlikely that the increase in metabolic activity required by transcriptional activation would be detectable in light of the significant decrease in translational activity resulting from protein synthesis inhibition, which can account for up to 87 % of metabolic activity in some organisms (Pace and Manahan 2007).
Interestingly, it was observed that the metabolic activity reduction patterns were similar for each infective dose, only with a corresponding delay in cytotoxic onset that correlated with decreasing infection rates. This observation led to the hypothesis that a minimum bacterial population threshold must be achieved before Shiga toxin production can reach a level capable of inducing cytotoxicity within the host cell population. To investigate this hypothesis, bacterial counts were performed concurrent with the onset of the reduction in metabolic activity levels. This analysis highlighted a strong correlation between bacterial population levels and host metabolic activity (Fig. 3). No reduction in metabolic activity was observed prior to reaching a total bacterial population size of approximately 5 × 105–1 × 106 CFU. However, once this population size was achieved, there was a rapid diminishment of metabolic activity. Exposure to bacterial populations greater than 1 × 106 CFU continued this trend, with full cellular death consistently observed prior to reaching bacterial population sizes of 5 × 107–1 × 108 CFU.
This rapid onset of cell death following reductions in metabolic activity levels indicates that there are few detoxification mechanism that can be employed by the host cell to avoid death following Shiga toxin exposure. However, the presence of both general and stressor-specific response pathways have been documented in human cells (Murray et al. 2004), providing a multitude of potential routes for detoxification. It was hypothesized that the exposed human cells could therefore leverage at least a subset of these mechanisms to survive if the infectious pressure was removed. To examine this hypothesis, infected cells were rescued at various time points post-E. coli O157:H7 exposure by removing all bacterial cells and non-internalized Shiga toxins. It was observed that the rescue survival window using this procedure was relatively short, with cells exposed for greater than 4 h unable to regain metabolic activity levels similar to uninfected control cells (Fig. 4). Interestingly, those cells that were rescued within the 4 h window demonstrated both a full and rapid recovery of metabolic activity. Cells rescued at both 2 and 4 hpi had retained full native metabolic activity levels within 2 h post-rescue, indicating that the stress response mechanisms responsible for overcoming Shiga toxin exposure are efficient, but also indicating that the continued inhibition of protein synthesis resulting from constitutive toxin exposure can prevent the enactment or effectiveness of these pathways. This suggests that the key detoxification methods employed by the host cells are proteomic in nature, rather than transcriptional or membrane modulation-based, and also indicates that a significant amount of Shiga toxin is required to manifest toxicity within the cell and that the protein may not remain active for prolonged time periods after entering into a human cell.
Conclusion
Autobioluminescent human cells allowed for real-time tracking of cytotoxicity and metabolic activity levels within a mixed population of human and bacterial cells because they do not rely on cellular destruction prior to signal generation. By tracking metabolic activity using these cells throughout the infection process, it was observed that cytotoxicity occurs rapidly following exposure to large populations of E. coli O157:H7. Human cells do appear to possess detoxification pathways for surviving an E. coli O157:H7 infection event, however, the detoxification process is likely dependent on protein synthesis activity, and can therefore only be employed if E. coli O157:H7 exposure levels can be maintained below the threshold required to reduce metabolic activity levels. Fortunately, the rapid recovery of metabolic activity following the removal of infection indicates that the cytotoxic Shiga toxin is relatively unstable within the human host cell, and demonstrates that cells can be rescued with no deleterious long term effects on metabolic activity levels if the infection is rapidly treated and removed.
Acknowledgments
Funding support for this work was provided to the University of Tennessee by the National Science Foundation Division of Chemical, Bioengineering, Environmental, and Transport Systems (CBET) under award number CBET-0853780.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare they have no conflict of interest.
References
- Bitzan MM, Wang Y, Lin J, et al. Verotoxin and ricin have novel effects on preproendothelin-1 expression but fail to modify nitric oxide synthase (ecNOS) expression and NO production in vascular endothelium. J Clin Invest. 1998;101:372–382. doi: 10.1172/JCI522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunder W, Schmidt H, Karch H. KatP, a novel catalase-peroxidase encoded by the large plasmid of enterohaemorrhagic Escherichia coli O157: H7. Microbiology. 1996;142:3305–3315. doi: 10.1099/13500872-142-11-3305. [DOI] [PubMed] [Google Scholar]
- Class B, Thorne N, Aguisanda F, et al. High-throughput viability assay using an autonomously bioluminescent cell line with a bacterial lux reporter. J Lab Autom. 2014 doi: 10.1177/2211068214560608. [DOI] [PubMed] [Google Scholar]
- Close D, Xu T, Sayler GS, et al. In vivo bioluminescent imaging (BLI): noninvasive visualization and interrogation of biological processes in living animals. Sensors. 2010;11:180–206. doi: 10.3390/s110100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch H, Tarr PI, Bielaszewska M. Enterohaemorrhagic Escherichia coli in human medicine. Int J Med Microbiol. 2005;295:405–418. doi: 10.1016/j.ijmm.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Klein EJ, Stapp JR, Clausen CR, et al. Shiga toxin-producing Escherichia coli in children with diarrhea: a prospective point-of-care study. J Pediatr. 2002;141:172–177. doi: 10.1067/mpd.2002.125908. [DOI] [PubMed] [Google Scholar]
- Law D. The history and evolution of Escherichia coli O157 and other Shiga toxin-producing E. coli. World J Microbiol Biotechnol. 2000;16:701–709. [Google Scholar]
- Lukyanenko V, Malyukova I, Hubbard A, et al. Enterohemorrhagic Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis and transcytosis across intestinal epithelial cells. Am J Physiol Cell Physiol. 2011;301:C1140–C1149. doi: 10.1152/ajpcell.00036.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JI, Whitfield ML, Trinklein ND, et al. Diverse and specific gene expression responses to stresses in cultured human cells. Mol Biol Cell. 2004;15:2361–2374. doi: 10.1091/mbc.E03-11-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace DA, Manahan DT. Cost of protein synthesis and energy allocation during development of antarctic sea urchin embryos and larvae. Biol Bull. 2007;212:115–129. doi: 10.2307/25066589. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Bergan J, Dyve AB, et al. Endocytosis and retrograde transport of Shiga toxin. Toxicon. 2010;56:1181–1185. doi: 10.1016/j.toxicon.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Tarr PI, Neill MA, Clausen CR, et al. Escherichia coli O157:H7 and the hemolytic uremic syndrome: importance of early cultures in establishing the etiology. J Infect Dis. 1990;162:553–556. doi: 10.1093/infdis/162.2.553. [DOI] [PubMed] [Google Scholar]
- Valderrama WB, Dudley EG, Doores S, et al. Commercially available rapid methods for detection of selected foodborne pathogens. Crit Rev Food Sci Nutr. 2015 doi: 10.1080/10408398.10402013.10775567. [DOI] [PubMed] [Google Scholar]
- Xu T, Ripp S, Sayler G, et al. Expression of a humanized viral 2A-mediated lux operon efficiently generates autonomous bioluminescence in human cells. PLoS One. 2014;9:e96347. doi: 10.1371/journal.pone.0096347. [DOI] [PMC free article] [PubMed] [Google Scholar]