

Abstract
Physiologically based pharmacokinetic (PBPK) modeling is a powerful tool used to characterize maturational changes in drug disposition to inform dosing across childhood; however, its use is limited in pediatric drug development. Access to pediatric pharmacokinetic data is a barrier to widespread application of this model, which impedes its development and optimization. To support the development of a pediatric PBPK model, we sought to leverage opportunistically-collected plasma concentrations of the commonly used antibiotic, clindamycin. The pediatric PBPK model was optimized following development of an adult PBPK model that adequately described literature data. We evaluated the predictability of the pediatric population PBPK model across 4 age groups, and found that 63–93% of the observed data where captured within the 90% prediction interval of the model. We then used the pediatric PBPK model to optimize intravenous clindamycin dosing for a future prospective validation trial. The optimal dosing proposed by this model are 9 mg/kg/dose in children ≤5 months of age, 12 mg/kg/dose in children >5 months to 6 years of age, and 10 mg/kg/dose in children 6-18 years of age, all administered every 8 hours. The simulated exposures achieved with the dosing regimen proposed were comparable to adult plasma and tissue exposures for treatment of community acquired methicillin resistant Staphylococcus aureus infections. Our model demonstrated the feasibility of using opportunistic pediatric data to develop pediatric PBPK models, extending the reach of this powerful modeling tool and potentially transforming the pediatric drug development field.
1. INTRODUCTION
The 2012 U.S. Food and Drug Administration (FDA) recommendation in support of modeling and simulation in pediatric drug development included an endorsement of physiologically based pharmacokinetic (PBPK) modeling (1). PBPK models are particularly useful for modeling drug disposition in infants and children because they incorporate growth changes and maturation in processes that are known to alter drug disposition. By integrating both the known physiological changes that alter drug disposition in children, with known drug specific parameters, these models can accurately predict changes in drug disposition as a function of age. Because PBPK models leverage prior knowledge of drug disposition and physiology, their development is less reliant on drug concentration data than their population pharmacokinetic (PK) counterparts (2). As such, non-traditional clinical trials collecting minimal number of drug concentrations may be sufficient to successfully develop PBPK models.
A successful, non-traditional alternative to PK trials in children is the use of opportunistic PK data collection (3). Opportunistic PK studies capitalize on standard of care procedures by timing collection of samples to occur optimally at the time of routine laboratory blood draws. Because participants are already receiving the drug of interest, and additional samples do not need to be collected for research purposes only, this study design minimizes the risk to the study participant and often has higher informed consent rates. Data from opportunistic PK trials have previously served as the basis for empirical compartmental population PK model development (4, 5). Opportunistic PK data may also be useful for the development and validation of pediatric PBPK models. We hypothesized that combining opportunistic and literature data would allow us to develop a well parameterized pediatric PBPK model for the lincosamide antibiotic, clindamycin.
We chose clindamycin to evaluate this approach due to its frequent and increasing use to treat severe infections with methicillin resistant Staphylococcus aureus (MRSA) in children despite remaining relatively understudied in this population (6). Also, the availability of adult literature data and pediatric opportunistic data collected from infants to adolescents provided the necessary in vivo data needed to evaluate model performance. Last, clindamycin has pharmacologic properties that make it ideally suited for PBPK modeling: as a CYP3A4 substrate, its clearance will be affected by developmental changes; and target tissue concentrations for MRSA therapy in the lung, bones, and skin have been established as pharmacodynamic endpoints that can be simulated by the PBPK model (7, 8).
2. METHODS
2.1 Description of PBPK Model Development Workflow
We followed the FDA guidance on PBPK model development and workflow in children to build our pediatric PBPK model (Figure 1) (9-11). We first developed an adult PBPK model and extracted clindamycin IV concentration versus time data from 3 adult PK studies (12-14). To assess the predictive accuracy of the adult population PBPK model, we compared the distribution of observed clindamycin plasma concentration data to the simulated plasma concentration data for a virtual population with demographic characteristics representative of all 3 studies (14). The finalized adult model served as the basis for developing the pediatric PBPK model. In the pediatric model, we maintained the physicochemical and drug-specific absorption, distribution, metabolism and elimination (ADME) parameters of clindamycin and replaced the anthropometric and physiological information with pediatric values using pre-established age-dependent algorithms in PK-Sim® (Version 5.5, Bayer Technology Services, Leverkusen, Germany). To assess the predictive accuracy of the pediatric model, we performed simulations with virtual populations ranging from term neonates to 18 years of age.
Figure 1.
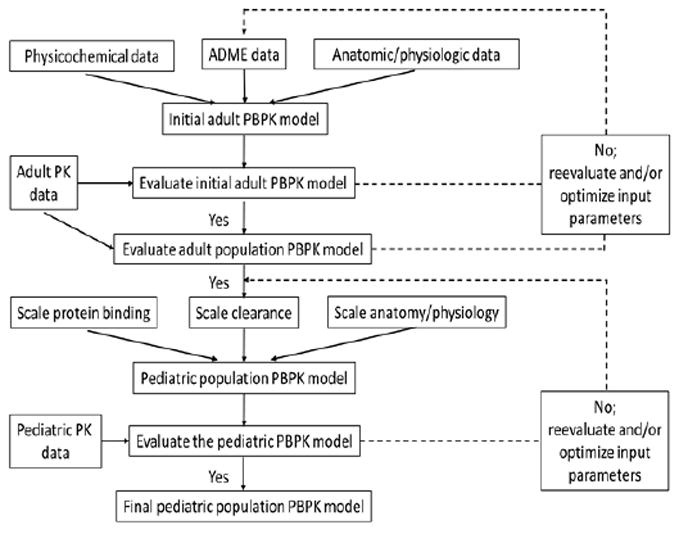
Model building workflow for pediatric PBPK model.
2.2 Clinical Data and Software Used
We identified relevant adult clindamycin concentration versus time data reported in the literature using a systematic search of Pubmed® with the following search terms: clindamycin, pharmacokinetics, and pharmacokinetic. We excluded data obtained using microbiologic assay techniques that may not have reliably distinguished clindamycin from clindamycin metabolite concentrations. To provide the concentration time data for the adult PBPK model development, we selected 3 adult clindamycin PK studies that were most appropriate based on route of administration, study participant demographics and clinical characteristics, and bioanalytical method used (Supplementary Table 1) (13-15). We used the software Plot Digitizer® (version 2.6.6) to extract the concentration versus time data.
To develop the pediatric PBPK model, we leveraged individual clindamycin plasma concentration versus time data collected after IV administration of clindamycin phosphate in an opportunistic PK study (POPS, ClinicalTrials.gov #NCT01431326) (5). This is a multi-center, prospective, PK, and safety study of understudied drugs administered to children (<21 years of age) per standard of care. In this study, we collected PK samples either in an opportunistic fashion at the time of clinical laboratory collections, or if the parent or patient consented, following a specific collection for study purposes (end of infusion, 2-5 hours after infusion, within 1 hour prior to next dose, and 16-24 hours after the final dose). Because this was a standard-of-care study, dosing and PK sample collection times varied between participants. Clindamycin concentrations were measured using a validated bioanalytical assay previously described (5). In the analyses described herein, we excluded clindamycin plasma concentration versus time data collected from pre-term infants, obese participants, and those on extracorporeal membrane oxygenation support due to the significant physiologic alterations expected to be present in these populations.
We used the software PK-Sim® (Version 5.5, Bayer Technology Services, Leverkusen, Germany) and MoBi® (Version 3.5, Bayer Technology Services, Leverkusen, Germany) for model development and simulation. We used Stata® (Version 14.1, StataCorp LP, College Station, TX) for data manipulation and visualization.
2.3 Adult PBPK Model Development
We used the standard whole-body 15-organ PBPK model implemented in PK-Sim® (16, 17). Organs were kinetically equivalent to well-stirred compartments. Clindamycin phosphate is extensively metabolized to clindamycin, with only 0.35% of unchanged clindamycin phosphate excreted in urine after IV administration (14). The conversion to clindamycin has been shown to be complete within approximately 2 hours, which was incorporated into the model using a plasma alkaline phosphatase conversion rate. The physicochemical parameters and ADME data for clindamycin were extracted from the literature (Table 1) (18). We predicted tissue or plasma partition coefficients (Kp) using the in silico tissue composition approach proposed by Rodgers and Rowland (19-21). While clindamycin is 78-94% bound to plasma α1-acid glycoprotein, the fraction of plasma protein bound clindamycin phosphate and the preferred protein to which it is bound are not known (21-24). Therefore, we assumed the same degree of protein binding to α1-acid glycoprotein for clindamycin phosphate and clindamycin.
Table 1.
Physico-chemical, ADME, and Anatomic/Physiologic Data for clindamycin phosphate and clindamycina
| Clindamycin phosphate | Clindamycin | |
|---|---|---|
| Physico-chemicalb | ||
| LogP | 0.95 | 2.16 |
| pKa | Base 6.78 | Base 7.55 |
| MW, g/mol | 504.963 | 424.98 |
| Solubility at pH7, mg/L | 3220 | 30.6 |
| ADME | ||
| fup | 0.22 | 0.06 (22-24, 27) |
| Binding proteinc | α1-acid glycoprotein | α1-acid glycoprotein (22-24, 27) |
| CLint(plasma-ALP) | 0.8 L/min | |
| CLint(hep-CYP3A4) | - | 2.21 ul/min/pmol P450 |
| CLint(hep-CYP3A5) | - | 0.28 ul/min/pmol P450 |
| Tubular secretion | - | 0.1038 L/min |
| Renal filtration | 0.044 | 1 |
LogP, logarithm of the octanol-water partition coefficient (lipophilicity); pka, negative logarithm of the acid dissociation constant; MW, molecular weight; ADME, absorption distribution metabolism elimination; fup, plasma fraction unbound; CL int(plasma-ALP), intrinsic clearance of plasma alkaline phosphatase; CL int(hep-CYP3A4), intrinsic clearance of hepatic isozyme CYP3A4; CL int(hep-CYP3A5), intrinsic clearance of hepatic isozyme CYP3A5.
All physicochemical data came from DrugBank (18).
We assumed the same degree of protein binding to α1-acid glycoprotein for clindamycin phosphate and clindamycin.
For both clindamycin phosphate and clindamycin, we approximated renal clearance using 2 methods: 1) based on total plasma clearance and percent of the dose recovered in urine as unchanged drug and 2) based on glomerular filtration rate and unbound fraction of the drug in plasma. Renal clearance estimated using methods 1 and 2 were then compared to determine whether an active secretion or reabsorption process was needed in the model. The hepatic clearance of clindamycin in the adult PBPK model was calculated as a sum of the clearance mediated by CYP3A4/A5. The relative contribution of these 2 clearance pathways was determined based on results from an in vitro metabolism study of clindamycin (8). We calculated the total hepatic clearance of clindamycin phosphate and clindamycin as the sum of scaled values of 3 individual clearance pathways: CLint(plasma-ALP), intrinsic clearance of plasma alkaline phosphatase; CLint(hep-CYP3A4), intrinsic clearance of hepatic isozyme CYP3A4; CLint(hep-CYP3A5), intrinsic clearance of hepatic isozyme CYP3A5. We compared the observed concentration versus time data from the selected adult PK studies with the concentrations simulated using the initial adult PBPK model. To evaluate model performance, we simulated concentration versus time data in an adult using average demographics (age and body weight) representative of all 3 adult PK studies. We evaluated the central tendency of model predictions using a visual check of any substantial discrepancy between simulated and observed mean concentration versus time data. We optimized the model parameters, including the intrinsic clearance via CYP3A4/A5, tubular secretion, and unbound fraction of clindamycin and intrinsic clearance via alkaline phosphatase, renal absorption, and unbound fraction of clindamycin phosphate using the MoBi® Toolbox for MATLAB® (Bayer Technology Services GmbH, Leverkusen, Germany/The Mathworks Inc., Natick, MA).
To account for inter-individual PK variability, we developed an adult population PBPK model by incorporating virtual populations into its framework, while maintaining the physico-chemical and ADME parameters from the finalized adult PBPK model. We created a virtual population of adults (N=100) using the demographic (sex, age, and weight) distribution reported in the adult PK studies of clindamycin selected from the literature. In addition, we included in the model inter-individual variability (expressed as the percentage coefficient of variation) for enzymes and transporters (CYP3A4, 81%; CYP3A5, 185%; plasma alkaline phosphatase, 31%; and transporter for tubular secretion, 25%) extracted from the literature (25, 26). From the virtual population, we evaluated the adult population PBPK model by comparing the distribution of observed clindamycin plasma concentration versus time data (mean and standard deviation [SD]) to the simulated data (geometric mean and SD). We considered a model final if the mean ±SD of the simulated data captured >80% of the observed data.
2.4 Pediatric PBPK Model Development
2.4.1 Anatomical and physiological parameterization
We used the pre-established age-dependent algorithms in PK-Sim® to generate anatomical and physiological parameters including bodyweight, height, organ weights, blood flows, cardiac output, total body water, lipid and protein concentrations (16, 17).
2.4.2 Scaling of unbound fraction
We estimated the unbound fraction of clindamycin in children using the unbound fraction of clindamycin in adults and the default α1 acid glycoprotein ontogeny function in PK-Sim® (22-24, 27). Prior to selecting the default ontogeny function, we compared it to ontogeny functions previously described in the literature by plotting empirical Bayesian estimates of clearance derived from a published population PK model by age over the clearance predicted by the PBPK model using various ontogeny functions (Supplementary Figure 1) (5). The default function was retained after visual inspection revealed that its use resulted in predicted clearance most in agreement with the population model.
2.4.3 Scaling of renal clearance
We used the default age-dependent value for glomerular filtration rate in PK-Sim® in the pediatric model. Because the specific transporter for tubular secretion of clindamycin is unknown, we used the age-dependence of tubular secretion published by Hayton in the pediatric model (28). Since this function was based on the excretion of aminohippuric acid, we are obligated to assume that the ontogeny of tubular secretion of clindamycin is reflected by the ontogeny of aminohipurric acid tubular secretion.
2.4.4 Scaling of hepatic clearance
We calculated total hepatic clearance of clindamycin phosphate and clindamycin as the sum of scaled values of its individual clearance pathways using a physiologically-based approach. The process of physiologic hepatic clearance scaling is based on the following assumptions (29):
Pathways of clearance in children are the same as those observed in adults.
Well-stirred model conditions hold.
Enzyme metabolism follows first-order kinetics.
For clindamycin, we used the default setting for hepatic CYP3A4/A5 ontogeny in PK-Sim® (30). Prior to selecting the default ontogeny function, we again compared it to ontogeny functions previously described in the literature by plotting empirical Bayesian estimates of clearance derived from a published population PK model by age over the clearance predicted by the PBPK model using various ontogeny functions (Supplementary Figure 1) (5). Upon visual inspection, use of the default PK Sim® ontogeny functions for hepatic CYP3A4/ and CYP3A5 resulted in predicted clearance most in agreement with the population model, and the default functions were retained in the PBPK model.
These functions assume that enzyme activity per gram of tissue of CYP3A4 is on average 12% of the adult value at term gestation, increases to 80% by the age of 1.3 years, and reaches adult activity by 5 years of age, while CYP3A5 reaches adult enzyme activity levels at term. We calculated scaled pediatric intrinsic clearance estimates for CYP3A4/A5 participant from adult values using the following formulas:
Where CLint,CYP3A4(child)/g liver is the scaled intrinsic clearance for CYP3A4 per gram of liver; CLint,CYP3A5(child)/g liver is the scaled intrinsic clearance for CYP3A5 per gram of liver; CLint,CYP3A4(adult)/g liver is the intrinsic clearance due to CYP3A4 per gram of liver in adults; CLint,CYP3A5(adult)/g liver is the intrinsic clearance due to CYP3A5 per gram of liver in adults; OSFCYP3A4 is the ontogeny scaling factor for CYP3A4 corresponding to the age of the child; OSFCYP3A5 is the ontogeny scaling factor for CYP3A5 corresponding to the age of the child.
To scale the conversion of clindamycin phosphate to clindamycin mediated by plasma alkaline phosphatase, we derived the ontogeny of the enzyme using mean normal plasma from Pediatric Normal Laboratory Values (31). We scaled intrinsic clearance of clindamycin phosphate similarly to that of CYP3A4/A5.
2.4.5 Pediatric dose optimization
Using the population module in PK-Sim®, we created 5 virtual pediatric populations (N=100) of full-term gestation stratified by postnatal age: 0-5 months, 1-2 years, 2-5 years, 6-11 years, and 12-8 years. Race and gender distributions in the virtual populations were based on those observed in the pediatric PK trial. We maintained the adult inter-patient variability (percentage coefficient of variation) values associated with CYP3A4/A5, plasma alkaline phosphatase, and the transporter that mediates tubular secretion of clindamycin in the simulation of virtual pediatric populations.
We used the final developed pediatric PBPK model to evaluate optimal dosing for children of different developmental ages. We selected 5 age groups of 100 children each: 0-5 months, >5 months-1 year, >1-6 years, >6-12 years, and >12-18 years. We assigned race and gender in the simulated children using the approximate distribution of race (85% white;15% black) and gender (50% male) in the general U.S. pediatric population. We optimized pediatric dosing regimen to match (within 20%) median adult (70-kg body weight) clindamycin exposure. As recommended in clinical practice guidelines for community-acquired MRSA infections, the clindamycin adult dose selected to generate reference clindamycin exposure was 600 mg every 8 hours (32). We calculated the area under the concentration versus time curve from 0 to infinity after a single dose (AUC0-∞) to represent the area under the concentration versus time curve from 0 to tau at steady state (AUC0-8,ss) using a non-compartmental analysis. We also calculated the percent of participants with simulated clindamycin concentrations in target organs and tissues (bone, lung, and skin) >MRSA MIC (0.5 mg/L) for at least 50% of the dosing interval following the optimized pediatric dosing regimen (7).
3.0 RESULTS
3.1 Adult PBPK model evaluation
The observed plasma concentrations of clindamycin and fractions of clindamycin and clindamycin phosphate excreted unchanged in urine were adequately described by the adult PBPK model for all 3 dosing regimens. This is demonstrated by the plots overlaying simulated data from the PBPK model with plasma concentrations of clindamycin and the fraction of the drug excreted unchanged in urine for clindamycin and clindamycin phosphate after a single IV dose of 600 mg clindamycin phosphate (12, 15) and after multiple IV doses of 600 mg or 1200 mg clindamycin phosphate (14) (Figure 2). The observed clindamycin phosphate plasma concentrations were slightly overestimated by the adult PBPK model (Figure 2).
Figure 2.
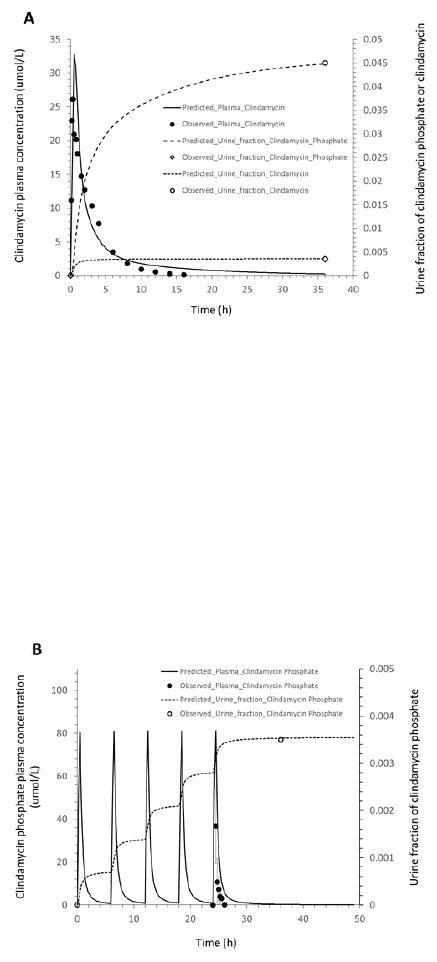
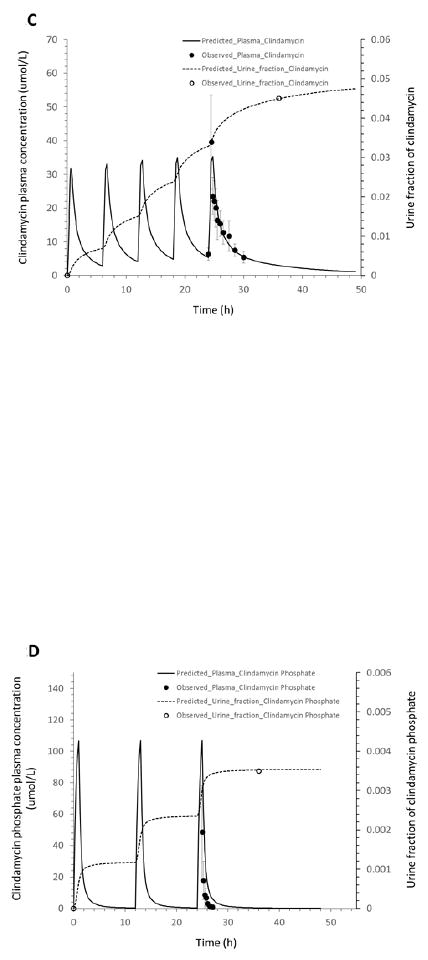

Mean observed (dots) and simulated (lines) plasma concentrations and fractions of the drug excreted unchanged in urine in healthy adults: (A): clindamycin concentration in plasma, and clindamycin and clindamycin phosphate fractions in urine after a 30-min intravenous infusion of 600 mg clindamycin phosphate; (B): clindamycin phosphate concentration in plasma and fraction in urine after IV administration of 600 mg clindamycin phosphate every 6 hours; (C): clindamycin concentration in plasma and fraction in urine after IV administration of 600 mg clindamycin phosphate every 6 hours; (D): clindamycin phosphate concentration in plasma and fraction in urine after IV administration of 1200 mg clindamycin phosphate every 12 hours; (E): clindamycin concentration in plasma and fraction in urine after IV administration of 1200 mg clindamycin phosphate every 12 hours.
Mean (± SD) weight, age and gender were comparable between our virtual adult population and the patient population that provided the observed clindamycin concentration-time data (Supplementary Table 1). The distribution (mean ± SD) of plasma concentrations of clindamycin and clindamycin phosphate after multiple IV doses of 600 mg clindamycin phosphate from this study was plotted and overlaid with the distribution (geometric mean ± geometric SD) of simulated data from the adult population PBPK model. The mean and SD of observed plasma concentrations for clindamycin were acceptably predicted by the adult population PBPK model (Figure 3).
Figure 3.
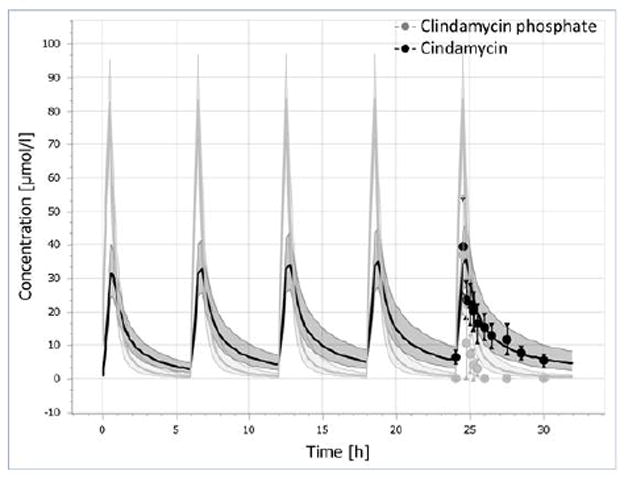
Observed (dots) and simulated (lines) plasma concentration-time profiles of clindamycin phosphate and clindamycin following IV administration of 600 mg clindamycin phosphate every 6 hours in healthy adults. Solid lines represent geometric mean of the simulated data. The shaded area represents geometric mean ± geometric SD for the simulated data. Symbols represents mean and SD for the observed data.
3.2 Pediatric PBPK model
To evaluate the predictive accuracy of the pediatric PBPK model, we used a total of 68 plasma concentrations from 48 POPS study participants. Participants received a median (range) of 5 (1-32) doses of clindamycin phosphate via IV administration at a median (range) dose of 9.97 (4.6-13.8) mg/kg. All drug concentrations were above the limit of quantification with a median (range) number of samples collected per participant of 1.4 (1-6). We summarized the demographic characteristics of children with PK data by age groups in Table 2. No concomitant medications known to alter clindamycin exposure were reported for these participants.
Table 2.
Demographic characteristics of children with PK data.
| Variable | Median (Range) or N (%) | |||
|---|---|---|---|---|
| 0-1 year | 2-5 years | 6-11 years | 12-18 years | |
| N | 9 | 11 | 10 | 18 |
| Gestational agea (weeks) | 39 (37.3 – 40) | - | - | - |
| Postnatal age (years) | 0.1 (0.03 – 0.3) | 3.3 (2.0 – 5.9) | 10.2 (7.5 – 11.5) | 17.0 (12.7 – 19.0) |
| Postmenstrual ageb (weeks) | 42 (41 – 53) | 212 (145 – 349) | 570 (430 – 639) | 928 (702 – 1030) |
| Body weight (kg) | 4.2 (2.7 – 6.0) | 17.2 (10.3 – 24.6) | 31.7 (23.6 – 47.7) | 67.9 (28.6 – 87) |
| Female (%) | 3 (33) | 5 (45) | 6 (60) | 7 (39) |
| Race | ||||
| White | 9 (100) | 9 (82) | 5 (50) | 13 (72) |
| Black or African American | 0 | 1 (9) | 3 (30) | 4 (22) |
| Asian | 0 | 0 | 1 (10) | 0 |
| Other Race | 0 | 1 (9) | 0 | 1 (6) |
Gestational age was only collected for participants with PNA < 120 days.
Postmenstrual age was calculated as PNA + 40 weeks when GA was missing.
Because pediatric participants in the POPS study had different dosing regimens, we generated a 90% prediction interval of clindamycin plasma concentrations for each individual dosing regimen. The number of observations and participants in each age group, and number of observations outside the 90% prediction interval are summarized in Table 3. These results showed that the developed PBPK model characterized 63–93% of the opportunistic clindamycin PK data across age groups.
Table 3.
Number of concentration data out of 90% prediction interval of the pediatric population PBPK model.
| 0-1 year | 2-5 years | 6-11 years | 12-18 years | |
|---|---|---|---|---|
| Total number of participants | 9 | 11 | 10 | 18 |
| Total number of data points | 14 | 16 | 11 | 27 |
| Number (%) of data points outside of the 90% prediction interval | 1 (7) | 6 (37.5) | 2 (18) | 4 (22) |
Using model simulations, we identified the following optimal dosing: 9 mg/kg/dose in children ≤5 months of age, 12 mg/kg/dose in children >5 months to 6 years of age, and 10 mg/kg/dose in children 6-18 years of age, all administered every 8 hours. The resulting AUC0-∞ in each age cohort was comparable with simulated adult exposure (70 kg) (Figure 4). The PBPK model also predicted therapeutic clindamycin concentrations in bone, lung and skin at the proposed dosing. Clindamycin concentrations in targeted organs and tissues were >MRSA MIC (0.5 mg/L) for at least 50% of the dosing interval (7) in ≥88% of patients across all age groups, and in ≥98% of children >6 years of age. Lastly, the proposed dosing regimen was consistent with previously published dosing recommendations derived using a population PK modeling approach, and is within the doses recommended by the Infectious Diseases Society of America for the treatment of community acquired MRSA (5, 33).
Figure 4.
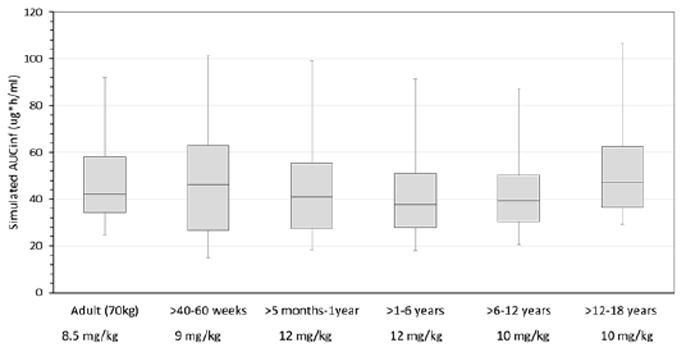
Simulated total drug exposure as concentration versus time curve from zero to infinity after a single dose (AUCinf) with age-based clindamycin dosage regimens.
4. DISCUSSION
To our knowledge, we are the first to report the successful use of opportunistic pediatric PK data for PBPK model development. Our model adequately characterized the PK of clindamycin in children and supported an age/body weight-based dosing regimen. This dosing regimen is now being applied in an ongoing pediatric PK trial to externally validate the developed model.
PBPK models are an attractive tool in pediatric drug development because of their inherent ability to facilitate extrapolation across different life stages (34). PBPK model development leverages existing knowledge of drug disposition and physiology, and may be less reliant on drug concentration data (35). Conducting the clinical trials necessary to garner population-specific PK data in children is challenging (2). Recently, an opportunistic study design, where participants receive the therapeutic of interest as part of standard-of-care and investigators collect PK samples at the time of routine laboratory draws, has been successfully applied to pediatric PK trials (3-5). The typically sparse PK data from opportunistic trials has previously served as the basis for successful empirical compartmental PK model development using population PK approaches for various drugs including clindamycin (5, 36, 37). We hypothesized that the spare PK data collected during an opportunistic PK trial would be well suited for the development of a PBPK model, which relies more heavily prior knowledge than clinical data (35).
Using the probe drug clindamycin, a known CYP3A4/A5 substrate, we found that our final model adequately characterized the opportunistic PK data. As is the case for all models, the applicability of model predictions is driven by the validity of its underlying assumptions. Key assumptions of our model included the ontogeny functions of α1 acid glycoprotein and CYP3A4/A5. For both, we used the standard PK-Sim® functions which have been previously validated. We further confirmed the adequacy of ontogeny functions for our data, by plotting individual empirical Bayesian estimates of clearance obtained for children of different ages from a previously published population PK model of clindamycin against our PBPK model predicted clearance (CL) using various CYP3A4 and α1 acid glycoprotein maturation functions. In general, the standard PK-Sim® functions characterized the ontogeny of CL as well as any of the other functions, and overall fit the individual estimates from the population PK model well. This suggests that our ontogeny function assumptions were reasonable and that our model predicted CL compares favorably to previously published clindamycin CL estimates derived from a population PK approach (5). Other model assumptions were made due to the lack of complete clinical pharmacology data for clindamycin. Because the specific transporter for tubular secretion of clindamycin is unknown, we were forced to use the age-dependence of tubular secretion published by Hayton and based on the excretion of aminohippuric acid in the pediatric model (28). While this assumes that the ontogeny of tubular secretion of clindamycin is reflected by the ontogeny of aminohipurric acid tubular secretion, we were reassured by the prior use of this ontogeny function in published pediatric PBPK models and its recommended use in situations where drug specific transporter ontogeny is unknown (9, 38).
Our pediatric PBPK model predicted doses fell within the range of MRSA treatment guidelines as published by the Infectious Disease Society of America (32). The body weight normalized doses were higher than in adults, suggesting a higher clearance per kg body weight in children. Given clindamycin’s low hepatic extraction ratio, lower plasma protein concentration resulting in reduced plasma protein binding and a higher liver weight: body weight ratio relative to adults could have contributed to the increased weight normalized clearance observed in children (39). Importantly, the PBPK model derived doses were consistent with dosing recommendations derived from a pediatric population PK modeling approach, and the resulting AUC0-∞ in each age cohort were comparable to adult exposures (5). Leveraging the PBPK model’s physiologic compartment structure, we were able to predict concentrations in target tissues including bone, skin, and lungs, and found them to be >MRSA MIC (0.5 mg/L) for at least 50% of the dosing interval in ≥88% of participants (7). This finding further supports the recommended dosing and demonstrates the strengths of the PBPK modeling approach in predicting drug exposure in target tissues.
Despite the satisfactory performance of our model, our study is not without limitations. In a subset of children, the model predictability was lower than expected. This discrepancy may be due to physiologic characteristics of children in the opportunistic PK trial hospitalized at the time of clindamycin administration, not matching the relatively uniform, healthy children used for model simulations in PK-Sim®. The discrepancy was also most pronounced in the 2-5 year old age stratum where only 16 opportunistic data points were available, and 1 appeared to be an outlier value. In addition to the sparsity of opportunistic data, our PBPK model was further limited by the assumptions made during its development, including the overarching assumption of physiologic scaling that pediatric clearance pathways are the same as adults, and regarding the ontogeny functions of α1 acid glycoprotein, renal clearance, and CYP3A4/A5. The prediction of concentration in target tissues is a major advantage of the PBPK model structure, and our identified dosing regimen resulted in adequate drug penetration into skin, bone, and lung tissue. It is important to remember, however, that the reliability of tissue concentration estimates derived from evaluation of plasma concentration time profiles can only be confirmed with the help of experimental tissue concentration data. We are currently conducting an open label PK trial (ClinicalTrials.gov #NCT02475876) to prospectively validate our model predicted dosing using external data. Results of this trial will be used to optimize the PBPK model, consistent with the proposed iterative approach to model development (40).
5. CONCLUSION
Opportunistic PK data were successfully used to evaluate the predictive accuracy of a clindamycin pediatric PBPK model. The model predicted clindamycin exposures reasonably well, and recommended doses similar to those predicted by a population PK approach. Given the greater access to opportunistic PK data, proof of its successful incorporation into the model development workflow may revolutionize the application of pediatric PBPK models. Once appropriately validated, this approach offers an alternative to the use of opportunistic data for population PK model development. By first developing an adult model using existing drug data, and scaling this model to children, prior information can be leveraged to mechanistically characterize changes in drug disposition with age. Once an opportunistic PK study is performed, PK data collected can be used to evaluate the pediatric population PBPK model. If the pediatric PBPK model is deemed robust, simulations can be performed to identify optimal pediatric dosing. This approach could potentially diminish the need for more complex PK trials with richer sampling and more extensively leverages available adult data. Future PBPK model development efforts should leverage opportunistic pediatric data whenever available to improve model predictability and increase the reach of this powerful modeling tool in the pediatric population.
Supplementary Material
Summary of clindamycin adult PK data sources.
Empirical Bayesian estimates of clearance in children derived from a population PK model and predicted clearance over time from PBPK models with various ontogeny functions of CYP3A4 and α1 acid glycoprotein.
Key points.
Opportunistic PK data were successfully used to evaluate the predictive accuracy of a clindamycin pediatric PBPK model.
The model predicted clindamycin exposures reasonably well, and recommended doses similar to those predicted by a population PK approach.
Given the greater access to opportunistic pediatric PK data, this method holds great promise to increase the development of PBPK models in children.
Acknowledgments
The assay measuring clindamycin concentrations was performed at OpAns Laboratory (Durham, NC, USA).
Funding source
This work was funded by the National Institutes of Health (1R01-HD076676-01A1; M.C.W.).
C.P.H. receives salary support for research from the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1TR001117) and the U.S. government for his work in pediatric and neonatal clinical pharmacology (Government Contract HHSN267200700051C, PI: Benjamin under the Best Pharmaceuticals for Children Act).
A.N.E. receives support for research from the National Institutes of Health (1R01-HD076676-01A1; PI: Cohen-Wolkowiez).
K.W. receives support from the Pediatric Critical Care and Trauma Scientist Development Program (5K12HD047349) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (1K23HD075891, 2K24HD058735).
M.C.W. receives support for research from the National Institutes of Health (1R01-HD076676-01A1), the National Institute of Allergy and Infectious Disease (HHSN272201500006I and HHSN272201300017I), the National Institute of Child Health and Human Development (HHSN275201000003I), the Biomedical Advanced Research and Development Authority (HHSO100201300009C), and industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp).
D.G. receives support for research from NICHD (K23HD083465), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and from industry (Cempra, Inc. and Jacobus Pharmaceutical Company, Inc.) for drug development in adults and children. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Clinical PK data used in this publication were collected during the POP01 clinical trial (ClinicalTrials.gov #NCT01431326).
Ethical approval: Clinical PK data used in this publication was collected during the POP01 clinical trial (ClinicalTrials.gov #NCT01431326). The POP01 study protocol was reviewed and approved by the institutional review board of each participating institution.
Informed consent: Informed consent and assent when applicable was obtained from all participants enrolled in the POP01 clinical trial who contributed clinical PK data used in this study.
References
- 1.Huang SM, Rowland M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin Pharmacol Ther. 2012;91(3):542–9. doi: 10.1038/clpt.2011.320. [DOI] [PubMed] [Google Scholar]
- 2.Laughon MM, Benjamin DK, Jr, Capparelli EV, Kearns GL, Berezny K, Paul IM, et al. Innovative clinical trial design for pediatric therapeutics. Expert review of clinical pharmacology. 2011;4(5):643–52. doi: 10.1586/ecp.11.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laughon MM, Benjamin DK., Jr Mechanisms to provide safe and effective drugs for children. Pediatrics. 2014;134(2):e562–3. doi: 10.1542/peds.2014-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen-Wolkowiez M, Watt KM, Zhou C, Bloom BT, Poindexter B, Castro L, et al. Developmental pharmacokinetics of piperacillin and tazobactam using plasma and dried blood spots from infants. Antimicrob Agents Chemother. 2014;58(5):2856–65. doi: 10.1128/AAC.02139-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez D, Melloni C, Yogev R, Poindexter BB, Mendley SR, Delmore P, et al. Use of opportunistic clinical data and a population pharmacokinetic model to support dosing of clindamycin for premature infants to adolescents. Clin Pharmacol Ther. 2014;96(4):429–37. doi: 10.1038/clpt.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herigon JC, Hersh AL, Gerber JS, Zaoutis TE, Newland JG. Antibiotic management of Staphylococcus aureus infections in US children’s hospitals, 1999-2008. Pediatrics. 2010;125(6):e1294–300. doi: 10.1542/peds.2009-2867. [DOI] [PubMed] [Google Scholar]
- 7.Reeves DS, Holt HA, Phillips I, King A, Miles RS, Paton R, et al. Activity of clindamycin against Staphylococcus aureus and Staphylococcus epidermidis from four UK centres. J Antimicrob Chemother. 1991;27(4):469–74. doi: 10.1093/jac/27.4.469. [DOI] [PubMed] [Google Scholar]
- 8.Wynalda MA, Hutzler JM, Koets MD, Podoll T, Wienkers LC. In vitro metabolism of clindamycin in human liver and intestinal microsomes. Drug Metab Dispos. 2003;31(7):878–87. doi: 10.1124/dmd.31.7.878. [DOI] [PubMed] [Google Scholar]
- 9.Maharaj AR, Barrett JS, Edginton AN. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 2013;15(2):455–64. doi: 10.1208/s12248-013-9451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leong R, Vieira ML, Zhao P, Mulugeta Y, Lee CS, Huang SM, et al. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther. 2012;91(5):926–31. doi: 10.1038/clpt.2012.19. [DOI] [PubMed] [Google Scholar]
- 11.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45(9):931–56. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 12.Gatti G, Flaherty J, Bubp J, White J, Borin M, Gambertoglio J. Comparative study of bioavailabilities and pharmacokinetics of clindamycin in healthy volunteers and patients with AIDS. Antimicrob Agents Chemother. 1993;37(5):1137–43. doi: 10.1128/aac.37.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaherty JF, Rodondi LC, Guglielmo BJ, Fleishaker JC, Townsend RJ, Gambertoglio JG. Comparative pharmacokinetics and serum inhibitory activity of clindamycin in different dosing regimens. Antimicrob Agents Chemother. 1988;32(12):1825–9. doi: 10.1128/aac.32.12.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plaisance KI, Drusano GL, Forrest A, Townsend RJ, Standiford HC. Pharmacokinetic evaluation of two dosage regimens of clindamycin phosphate. Antimicrob Agents Chemother. 1989;33(5):618–20. doi: 10.1128/aac.33.5.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatti G, Malena M, Casazza R, Borin M, Bassetti M, Cruciani M. Penetration of clindamycin and its metabolite N-demethylclindamycin into cerebrospinal fluid following intravenous infusion of clindamycin phosphate in patients with AIDS. Antimicrob Agents Chemother. 1998;42(11):3014–7. doi: 10.1128/aac.42.11.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willmann S, Hohn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401–31. doi: 10.1007/s10928-007-9053-5. [DOI] [PubMed] [Google Scholar]
- 17.Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clinical pharmacokinetics. 2006;45(10):1013–34. doi: 10.2165/00003088-200645100-00005. [DOI] [PubMed] [Google Scholar]
- 18. [05/06/2016];DrugBank Version 4.5 Drug&Drug Target Database. Available online at www.drugbank.ca.
- 19.Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238–57. doi: 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- 20.Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94(6):1259–76. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 21.Rodgers T, Leahy D, Rowland M. Tissue distribution of basic drugs: accounting for enantiomeric, compound and regional differences amongst beta-blocking drugs in rat. J Pharm Sci. 2005;94(6):1237–48. doi: 10.1002/jps.20323. [DOI] [PubMed] [Google Scholar]
- 22.Gordon RC, Regamey C, Kirby WM. Serum protein binding of erythromycin, lincomycin, and clindamycin. J Pharm Sci. 1973;62(7):1074–7. doi: 10.1002/jps.2600620704. [DOI] [PubMed] [Google Scholar]
- 23.Kiosz D, Simon C, Malerczyk V. The plasma-protein-binding of Clindamycin Cephazolin and Cephradin in neonates and adults (author’s transl) Klin Padiatr. 1975;187(1):71–80. [PubMed] [Google Scholar]
- 24.Kays MB, White RL, Gatti G, Gambertoglio JG. Ex vivo protein binding of clindamycin in sera with normal and elevated alpha 1-acid glycoprotein concentrations. Pharmacotherapy. 1992;12(1):50–5. [PubMed] [Google Scholar]
- 25.Achour B, Barber J, Rostami-Hodjegan A. Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: a meta-analysis. Drug Metab Dispos. 2014;42(8):1349–56. doi: 10.1124/dmd.114.058834. [DOI] [PubMed] [Google Scholar]
- 26.Dent CE, Harper CM. Plasma-alkaline-phosphatase in normal adults and in patients with primary hyperparathyroidism. Lancet. 1962;1(7229):559–63. doi: 10.1016/s0140-6736(62)91544-1. [DOI] [PubMed] [Google Scholar]
- 27.Flaherty JF, Jr, Gatti G, White J, Bubp J, Borin M, Gambertoglio JG. Protein binding of clindamycin in sera of patients with AIDS. Antimicrob Agents Chemother. 1996;40(5):1134–8. doi: 10.1128/aac.40.5.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayton WL. Maturation and growth of renal function: dosing renally cleared drugs in children. AAPS pharmSci. 2000;2(1):E3. doi: 10.1208/ps020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edginton AN, Schmitt W, Voith B, Willmann S. A mechanistic approach for the scaling of clearance in children. Clin Pharmacokinet. 2006;45(7):683–704. doi: 10.2165/00003088-200645070-00004. [DOI] [PubMed] [Google Scholar]
- 30.Willmann S, Lippert J, Sevestre M, Solodenko J, Fois F, Schmitt W. PK-Sim®: a physiologically based pharmacokinetic ‘whole-body’ model. BIOSILICO. 2003;1(4):121–4. [Google Scholar]
- 31.Connelly MA, Brown JT, Kearns GL, Anderson RA, St Peter SD, Neville KA. Pupillometry: a non-invasive technique for pain assessment in paediatric patients. Arch Dis Child. 2014;99(12):1125–31. doi: 10.1136/archdischild-2014-306286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2011;52(3):285–92. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 33.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18–55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 34.Barrett JS, Della Casa Alberighi O, Laer S, Meibohm B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin Pharmacol Ther. 2012;92(1):40–9. doi: 10.1038/clpt.2012.64. [DOI] [PubMed] [Google Scholar]
- 35.Edginton AN, Joshi G. Have physiologically-based pharmacokinetic models delivered? Expert Opin Drug Metab Toxicol. 2011;7(8):929–34. doi: 10.1517/17425255.2011.585968. [DOI] [PubMed] [Google Scholar]
- 36.Cohen-Wolkowiez M, Ouellet D, Smith PB, James LP, Ross A, Sullivan JE, et al. Population pharmacokinetics of metronidazole evaluated using scavenged samples from preterm infants. Antimicrob Agents Chemother. 2012;56(4):1828–37. doi: 10.1128/AAC.06071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen-Wolkowiez M, Benjamin DK, Jr, Ross A, James LP, Sullivan JE, Walsh MC, et al. Population pharmacokinetics of piperacillin using scavenged samples from preterm infants. Ther Drug Monit. 2012;34(3):312–9. doi: 10.1097/FTD.0b013e3182587665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maharaj AR, Edginton AN. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacometrics Syst Pharmacol. 2014;3:e150. doi: 10.1038/psp.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology--drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 40.Edginton AN, Theil FP, Schmitt W, Willmann S. Whole body physiologically-based pharmacokinetic models: their use in clinical drug development. Expert Opin Drug Metab Toxicol. 2008;4(9):1143–52. doi: 10.1517/17425255.4.9.1143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of clindamycin adult PK data sources.
Empirical Bayesian estimates of clearance in children derived from a population PK model and predicted clearance over time from PBPK models with various ontogeny functions of CYP3A4 and α1 acid glycoprotein.