

Abstract
PLA2G6-associated neurodegeneration (PLAN) and hereditary spastic paraplegia (HSP) are two groups of heterogeneous neurodegenerative diseases. In this study, we report PLA2G6 gene mutations in three families from Turkey, Morocco, and Romania. Two affected Turkish siblings presenting HSP adds the disease to PLAN phenotypes. They were homozygous for the PLA2G6 missense c.2239C>T, p.Arg747Trp variant and the ages of onset were 9 and 21. Parkinsonism, dystonia or cognitive decline were not the clinical elements in these patients contrary to the cases that has been previously reported with the same variant, however, iron accumulation was evident in their cranial MRI. The Moroccan patient was homozygous for a novel missense c.1786C>T, p.Leu596Phe variant and the Romanian patient had two novel mutations; c.1898C>T, p.Ala633Val and c.1765_1768del, p.Ser589ThrfsTer76. Both of these patients conformed better to childhood onset PLAN with the age of onset at four and seven years, respectively. Interestingly, all identified mutations were affecting the highly conserved patatin-like phospholipase domain of the PLA2G6 protein.
Graphical abstract
PLA2G6-associated neurodegeneration (PLAN) has been considered as the second most common cause of neurodegeneration with brain iron accumulation (NBIA). Still, it is a rare disease with an estimated prevalence of approximately 1/106 [1].
A classification was suggested for PLAN based on age of onset as; infantile, childhood, and adult onset PLAN [2]. Infantile PLAN refers to classic infantile neuroaxonal dystrophy (INAD) where the age of onset ranges between six months and three years. Initial clinical findings include developmental regression, hypotonia, progressive psychomotor delay, and progressive spastic tetraparesis. Disease progresses rapidly with addition of strabismus, nystagmus, and optic atrophy. Severe spasticity, progressive cognitive decline, and visual impairment follow as disease enters the final stage. Patients often die in the first decade of their life [1]. Phenotypic heterogeneity is higher in childhood PLAN that is traditionally known as atypical neuroaxonal dystrophy (atypical NAD). Age of onset is in a later stage and varies from one to six years and disease progression is not remarkable during early childhood unlike INAD. Patients might have gait instability, ataxia, speech delay or autistic features in the initial phases of the disease. Adult PLAN is known as PLA2G6-related dystonia-parkinsonism and can be accompanied by features including pyramidal tract signs, marked cognitive decline, and eye movement abnormalities. Age of onset is between late adolescence and early adulthood [2].
Hereditary spastic paraplegia (HSP) is another group of heterogeneous neurodegenerative disorders that is associated with lower limb spasticity and progressive weakness. ‘Complicated’ HSP is associated with additional neurological or systemic abnormalities including mental retardation or cognitive decline, cerebellar ataxia, and peripheral neuropathy, as well as thin corpus callosum on MRI. So far, HSP phenotype has not yet been associated with underlying PLA2G6 mutations.
The causative gene for PLAN, PLA2G6, is located on chromosome 22q13.1 and codes for Group VIA, cytosolic, calcium-independent phospholipase A2 (iPLA2β) that belongs to phospholipase A2 (PLA2) protein family. The enzymes in this family play a role in catalyzing the hydrolysis of the sn-2 fatty acyl bond of phospholipids to release free fatty acids and lysophospholipids [3]. Transcript variant 1 codes for diverse functional domains including N-terminal ankyrin repeat regions, a serine lipase consensus sequence (GXSXG), a nucleotide-binding domain, and a highly conserved patatin-like phospholipase domain that harbors the calmodulin-binding region (amino acids: 747–759) [4]. It has been proposed that, iPLA2 deficiency may alter membrane permeability, fluidity, or ion homeostasis [5, 6].
Next generation sequencing has allowed to re-define the phenotypic spectrum of many neurodegenerative diseases and has led to an increasing realization of the genetic overlaps among different clinical phenotypes. In one such report, FA2H gene that was known to be a causative gene of Hereditary Spastic Paraplegia (SPG35) and a progressive familial leukodystrophy, was identified to be responsible for NBIA in two families. Investigation of these patients revealed absence of common features of PLAN but similarity to that of neuroaxonal dystrophies extending the clinical spectrum of FA2H-associated neurodegeneration (FAHN) [7].
Here, we report three additional families with PLA2G6 mutations that were diagnosed in different centers in Turkey, Spain, and Germany. The studies were approved by the ethics committees of the respective universities or centers. Written consent was obtained from all individuals and/or from their parents.
Whole exome sequencing (WES) was performed for Cases 1 and 3 to identify the causative mutation. For exome capture, SureSelect Human All Exon 50Mb kit (Agilent, Santa Clara, CA, USA) and for sequencing Hiseq 2000 instruments (Illumina, San Diego, CA, USA) were used. 72,603,929 sequence reads of 100 bp length was produced for Case 1, 98.8% of which could be aligned to the target sequence. Mean sequencing depth was 57 and 69% of the target sequence was covered for at least 20 times. For Case 3, average read depth was 30 and the average variant quality was 603. BWA and GATK software were used for sequence alignment and variant calling, and the browser interface of the Genomes Management Application GEM.app was used for analysis of the data of Cases 1 and 3 [8]. Use of stringent filtering criteria (Genotyping quality >75, MAF <0.05, GERP score >4, PhastCons Score >0.9, phyloP Score >1.5) revealed homozygous missense variant in PLA2G6 in Case 1 (c.2239C>T, p.Arg747Trp) (PS1, PM1, PM2, PP1, and PP3) that has been previously reported [9, 10]. Sanger sequencing (Macrogen, Korea) revealed that his affected brother, Case 2, was homozygous and the consanguineous parents were heterozygous for the variant (Figure 1a). Moroccan patient Case 3 was homozygous for the novel c.1786C>T, p.Leu596Phe PLA2G6 variant (PM1, PM2, and PP3) and his mother was heterozygous for the same variant (Figure 1b).
Fig 1.
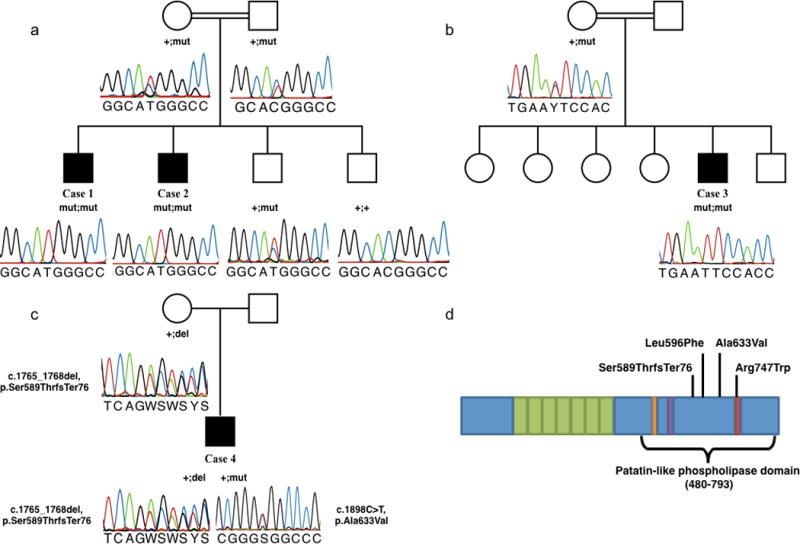
Variations identified in PLA2G6 gene. c.2239C>T, p.R747W variant in Cases 1 and 2 (a), c.1786C>T, p.L596F variation in Case 3 (b) and c.1765_1768delTCTG, p.Ser589Thrfs*76 and c.1898C>T, p.A633V variations in Case 4 (c). Black boxes indicate affected family members that are homozygous for the variation. Electropherograms are shown below. Domain structure of the PLA2G6 gene indicating the position of the mutations (modified from Engel et al. 2010) (d)
A high coverage HaloPlex gene panel kit (Agilent, Santa Clara, CA, USA) was used to analyze about 120 known genes associated with ataxia phenotypes on Illumina MiSeq instrument for Case 4 with Romanian origin as described previously [11]. Paired-end reads of approximately 150bp length were mapped against the hg19 standard reference genome (SAMtools) and two novel mutations were identified in PLA2G6. The missense mutation c.1898C>T, p.Ala633Val (PM2, PM3, and PP3) was absent in common databases (EVS6500, ExAC, 1000Genomes). The 4bp deletion at position c.1765_1768del leads to a shift of the reading frame and consecutive stop (p.Ser589ThrfsTer76) (PM2, PM3, and PM4). To determine phasing of the variant in the absence of parent DNA we amplified a 893bp fragment containing both variants, added sequencing adapters by processing amplicons with a Nextera DNA Library Preparation Kit (Illumina, Inc.) and sequenced the library on a MiSeq instrument. Analysis of individual reads clearly revealed trans-phase of the two mutations thus, compound heterozygosity in the patient (Figure 1c). Positions of the mutations on the domain structure of the protein is given in Figure 1d.
Transcript NP_003551.2 was used for the nomenclature of all the identified mutations in this study.
Homozygosity mapping using PLINK program from WES data of Cases 1 and 3 excluded linkage to known HSP and NBIA genes.
Clinical findings of the patients are summarized in Table 1 and available in PhenomeCentral database (https://phenomecentral.org/). The Turkish siblings, Cases 1 and 2, had predominant pyramidal signs throughout the disease course with the prominent symptom of spastic paraparesis. The age of onset was nine and 21, respectively. Parkinsonism, dystonia or cognitive decline were not the elements of the clinical picture, however, brain iron accumulation was evident in their MRI (Figure 2a, a′, a″). Besides, the clinical course was not identical in these siblings in terms of age of onset, rate of disease progression, and disease severity. Comparatively, the spectrum of symptoms mainly included early onset pyramidal signs and mild cerebellar findings in Case 1 whereas clinical picture encompassed pyramidal signs, that started later and progressed faster, severe ataxia, an implied rigidity, and cerebellar findings, that were present at initial presentation in Case 2. Case 3 with Moroccan origin had initial presentation with pyramidal signs at the age of four. He had cognitive involvement and axonal polyneuropathy with slow progression. MRI obtained at age seven showed cerebellar atrophy without brain iron accumulation (Figure 2b, b′). Finally, the Romanian patient, Case 4, had an onset of symptoms at the age of seven. He had dysarthria, dysphagia, and cognitive involvement added on to mild pyramidal and cerebellar signs. MRI presented cerebellar atrophy but no brain iron accumulation as for Case 3 (Figure 2c, c′, c″).
Table 1.
Clinical features of the cases presented in this study in comparison to two previously reported cases with the same PLA2G6 variant as in Cases 1 and 2. NA: Not available; (+): present, (−) absent.
| Turkish Patient 1 (Case 1) | Turkish Patient 2 (Affected brother- Case 2) | Moroccan Patient (Case 3) | Romanian Patient (Case 4) | Family 2 (Ref.9) | Index Case (Ref. 10) | |
|---|---|---|---|---|---|---|
| Genetic finding | c.2239C>T, p.R747W (Homozygous) | c.1786C>T, p.L596F (Homozygous) | c.1898C>T, p.A633V/c.1765_1768delTCTG, p.S589Tfs*76 (Compound heterozygous) | c.2239C>T, p.R747W (Homozygous) | c.2239C>T, p.R747W (Homozygous) | |
| Major phenotype | HSP | Atypical NAD | Atypical NAD | Dystonia Parkinsonism | Early-onset Parkinsonism | |
| Age of symptom onset (yrs) | 9 | 21 | 4 | 7 | 18 | 27 |
| Initial symptom | Spastic gait | Scissoring gait | Clumsiness, spasticity (hypertonia) | Frequent falls | Foot drag (subacute) | Foot shuffling and reduced arm swinging |
| Pyramidal signs | ||||||
| Spasticity | + | + | + | − | + | NA |
| Brisk reflexes | + | + | + | − | + | − |
| Achilles clonus | + | + | + | − | + | NA |
| Babinski sign | + | + | + | + | NA | NA |
| Cerebellar signs | ||||||
| Ataxia | + | + | − | + | − | NA |
| Intention tremor | + | − | − | − | − | NA |
| Dysdiadokinesia | − | − | + | + | − | NA |
| Abnormal heel-knee-shin test | + | − | NA | + | − | NA |
| Extrapyramidal signs | ||||||
| Tremor | − | − | − | − | − | + |
| Rigidity | − | +/− | − | − | + (cogwheeling) | NA |
| Bradykinesia | − | − | + | − | + | + |
| Dystonia | − | − | − | + (right foot) | + | NA |
| Dopaminergic response | NA | + | − | NA | + | + |
| Cognitive impairment | − | − | IQ 60 at age 15 | + (IQ 50 at age 16) | + | + |
| Psychiatric features | − | + | − | + (attention deficit disorder) | + | + |
| Eye movement abnormalities | − | − | − | + | + | − |
| Dysarthria | + | + | + | + | − | + |
| Autonomic involvement | − | − | − | − | + | NA |
| MRI findings | Brain iron accumulation | Brain iron accumulation | Cerebellar atrophy | Cerebellar atrophy | Mild cerebellar atrophy | Marked atrophy of frontal and temporal cortex |
Fig 2.
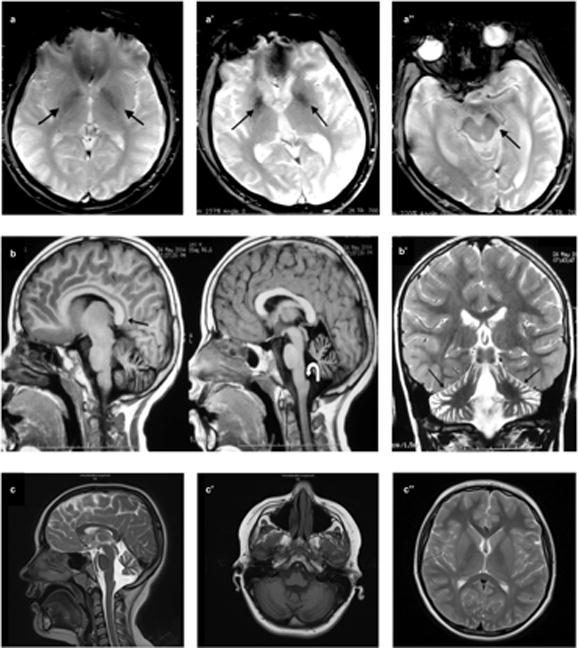
MRI findings. Axial gradient-recalled echo image shows bilateral hypointensity in both globi pallidi (arrows) consistent with iron accumulation in Case 1 at age 27. (a). Axial gradient-recalled echo sequence of Case 2 at age 32 depicts hypointensity in both globi pallidi (a′) and substantia nigra (a″) (arrows) consistent with iron accumulation. Sagittal T1-weighted images demonstrate callosal elongation (black arrow) and claval hypertrophy (curved white arrow) (b), marked atrophy of cerebellum on T2-weighted image (b′) of Case 3 at the age of seven. T2-weighted sagittal scan shows vermian atrophy and claval hypertrophy (arrow) (c), T1-weighted axial image demonstrates cerebellar atrophy (c′), and there was no evidence of iron deposition in basal ganglia on T2-weighted axial scan (c″) in Case 4 at age 10.
Taken together, in this study we report three different PLAN families and four mutations that are all affecting the highly conserved patatin-like phospholipase domain of the PLA2G6 protein. Although missense mutations are known to appear frequently in the 3′ end of the gene in atypical and adult-onset PLAN as in our cases, a genotype-phenotype correlation among our patients is still not possible since each manifest unique phenotypes that cover a spectrum ranging from complicated HSP to INAD [2]. In accordance, the mutation identified in the two Turkish siblings has been reported previously in a Pakistanian family with dystonia-parkinsonism [9] and in another Turkish patient with left sided parkinsonism at age 27 that responded to levodopa therapy [10]. These reports suggest further clinical heterogeneity for PLAN in independent cases and within the same family sharing exact same PLA2G6 mutation and implicating the need for further analysis to understand how incomplete penetrance, modifier genes, epigenetic factors, or environmental factors are involved in disease pathogenesis.
Turkish family stood out among the PLAN cases since spastic paraparesis was the prominent symptom that had started much earlier in the index case compared to the others. Besides, they also differed with the absence of progressive extrapyramidal degeneration, and preserved cognitive function. Clinical, imaging, and genetic investigations demonstrated that they had Hereditary Spastic Paraplegia with brain iron accumulation and PLA2G6 mutation; a combination that has not been reported before to our knowledge [1, 2, 9, 10, 13].
The Moroccan and the Romanian cases can be considered as childhood onset PLAN not only according to their early ages of onset but also with initial presentation of gait abnormalities and slower progression.
This is not the first study reporting a common causative gene for HSP and NBIA since C19ORF12 (SPG43) gene can be responsible for both diseases [12]. Our finding supports the previous hypothesis that the spectrum of clinical features associated with mutations in PLA2G6 is broader than what was suggested [13]. In the light of our new findings, we suggest screening of PLA2G6 in HSP patients with brain iron accumulation in MRI. The dust on the common pathways underlying neurodegeneration might be removed with more reports revealing involvement of common proteins in the pathogenesis of these diseases.
Supplementary Material
Supplementary Fig S1 Conservation of the serine residue at position 589 and leucine residue at position 596 (a), alanine residue at position 633 (b), and arginine residue at position 747 (c) across species.
Acknowledgments
We are grateful to the affected persons and their families for their valuable cooperation. We would like to thank Bogazici University Research Fund Grant Number 6047 to E.B., the National Institute of Health (NIH) grants 5R01NS072248 to S.Z., 1R01NS075764 to S.Z., 5R01NS054132 to S.Z., and 2U54NS065712 to S.Z. This work has also been supported, in part, by a E-RARE JTC grant “PREPARE” (BMBF, 01GM1607 to M.S. and as an associated unfunded member to S.Z.).
Footnotes
Disclosure statement: The authors have no conflict of interests to declare.
References
- 1.Paisan-Ruiz C, Li A, Schneider SA, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33:814–823. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurian MA, McNeill A, Lin JP, Maher ER. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA) Dev Med Child Neurol. 2011;53(5):394–404. doi: 10.1111/j.1469-8749.2011.03955.x. [DOI] [PubMed] [Google Scholar]
- 3.Ma Z, Turk J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 4.Engel LA, Jing Z, O’Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shinzawa K, Sumi H, Ikawa M, et al. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci. 2008;28:2212–20. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J Biol Chem. 1997;272:29317–21. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 7.Kruer MC, Paisan-Ruiz C, Boddaert N, et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA) Ann Neurol. 2010;68:611–8. doi: 10.1002/ana.22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez MA, Lebrigio RF, Van Booven D, et al. GEnomes Management Application (GEM.app): A New Software Tool for Large-Scale Collaborative Genome Analysis Hum Mutat. 2013;34:842–846. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paisan-Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurology. 2009;65(1):19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giri A, Guven G, Hanagasi H, et al. PLA2G6 Mutations Related to Distinct Phenotypes: A New Case with Early-onset Parkinsonism. Tremor Other Hyperkinet Mov. 2016;6:363. doi: 10.7916/D81G0M12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Synofzik M, Smets K, Mallaret M, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: findings from a large-scale multi-center screening. Brain. 2016;139:1378–93. doi: 10.1093/brain/aww079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartig MB, Iuso A, Haack T, et al. Absence of an orphan mitochondrial protein, C19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet. 2011;89:543–550. doi: 10.1016/j.ajhg.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregory A, Westaway SK, Holm IE, et al. Neurodegeneration associated with genetic defects in phospholipase A2. Neurology. 2008;71:1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig S1 Conservation of the serine residue at position 589 and leucine residue at position 596 (a), alanine residue at position 633 (b), and arginine residue at position 747 (c) across species.