

Abstract
Inflammatory bowel diseases (IBD), including Crohn’s disease and ulcerative colitis, are complex diseases that result from the chronic dysregulated immune response in the gastrointestinal tract. The exact etiology is not fully understood, but it is accepted that it occurs when an inappropriate aggressive inflammatory response in a genetically susceptible host due to inciting environmental factors occurs. To investigate the pathogenesis and etiology of human IBD, various animal models of IBD have been developed that provided indispensable insights into the histopathological and morphological changes as well as factors associated with the pathogenesis of IBD and evaluation of therapeutic options in the last few decades. The most widely used experimental model employs dextran sodium sulfate (DSS) to induce epithelial damage. The DSS colitis model in IBD research has advantages over other various chemically induced experimental models due to its rapidity, simplicity, reproducibility and controllability. In this manuscript, we review the newer publicized advances of research in murine colitis models that focus upon the disruption of the barrier function of the intestine, effects of mucin on the development of colitis, alterations found in microbial balance and resultant changes in the metabolome specifically in the DSS colitis murine model and its relation to the pathogenesis of IBD.
Keywords: Dextran sodium sulfate, Experimental colitis, Inflammatory bowel disease, Pathogenesis, Intestinal barrier
Core tip: In the last few decades the proliferation of research in experimental colitis models of inflammatory bowel diseases (IBD) has had profound effects in our understanding of human IBD pathophysiology as well as to exploit potential therapeutic avenues outside of immunologic therapy. The dextran sodium sulfate colitis model, through its rapidity, simplicity, reproducibility and controllability has been instrumental in our understanding of intestinal barrier function through the dysregulation of mucin, interaction with the intestinal microbiome and metabolome.
INTRODUCTION
Inflammatory bowel disease (IBD) is a group of inflammatory conditions of the colon and small intestine caused by a dysregulated immune response. Crohn’s disease (CD) and ulcerative colitis (UC) are the principal types of IBD. Both usually involve severe diarrhea, pain, fatigue and weight loss. IBD can be debilitating and sometimes leads to life-threatening complications. A convenient and time honored approach to study the pathogenesis and complexity of human IBD has been the development of a variety of animal models. These animal models have provided meaningful and indispensable insights into the histopathological and morphological changes in the intestinal tract related to the pathogenesis of human IBD[1]. While no single model has proven to sufficiently represent the complexity of the clinical and histopathological characteristics of human disease, the collective data obtained from these various animal models have provided a more detailed understanding of the underlying principles of human IBD pathogenesis[2,3]. These models have become an indispensable tool to elucidate the histopathological, immunological and morphological changes in the intestinal tract and potential therapeutic targets. These various models can be grouped into categories broadly defined as spontaneous colitis, chemically inducible colitis, genetically modified and adoptive transfer models[1,4-7].
The most widely used mouse model of colitis employs dextran sodium sulfate (DSS), a chemical colitogen with anticoagulant properties, to induce epithelial damage. The DSS colitis model lends itself to IBD research due to its rapidity, simplicity, reproducibility and controllability. Acute, chronic and relapsing models of intestinal inflammation can be achieved by modifying the concentration of DSS and the frequency of administration[8]. There are excellent and exhaustive reviews focused on the immunological aspects of experimental animal models in inflammatory bowel disease and we would recommend the reader to refer to these articles for the specifics in immunology[2,9]. In this review, we aim to provide an updated and concise review of the less publicized aspects of research in murine colitis models that focus upon the barrier function of the intestine in one specific chemically induced experimental colitis model, the DSS colitis murine model.
HUMAN IBD PATHOPHYSIOLOGY: MECHANISTIC INSIGHTS
IBD is a disorder of chronic intestinal inflammation without an exact etiology. The leading hypothesis on IBD pathogenesis states that it is an inappropriate and overly aggressive inflammatory response to enteric microbes in a genetically susceptible host with environmental factors precipitating the onset or reactivation of disease[10,11]. Epidemiologic data suggest these environmental factors include antibiotic use, microbial exposure and possibly dietary components[12-15]. Host genetics, luminal microbiome and its associated antigens and immune response, all have been implicated in playing important roles in IBD pathogenesis (Figure 1).
Figure 1.
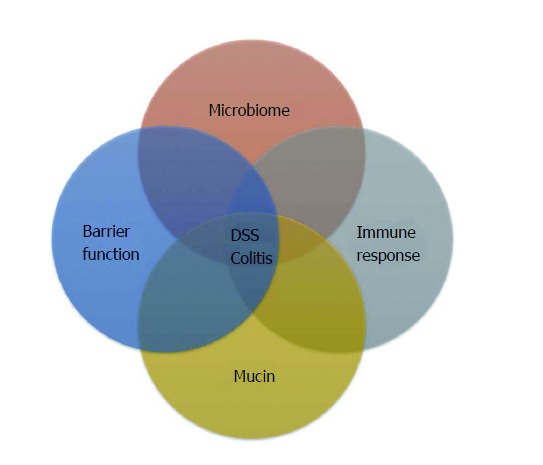
Factors that play important roles in the pathogenesis of inflammatory bowel diseases. DSS: Dextran sodium sulfate.
Genetics
Advances in IBD genetics have indicated modifications in genes regulating mucosal barrier integrity and function, innate immune response and microbial homeostasis[11]. Thus far, four genes have been associated with increased susceptibility to CD and one gene mutation has been associated with ulcerative colitis[11]. The most widely known and studied gene implicated in IBD is the gene for CARD15, formerly known as NOD2, which is responsible for luminal bacterial recognition. The leucine-rich repeat region of CARD15 binds muramyl dipeptide (MDP), which is the active moiety of peptidoglycan. This binding of bacterial peptidoglycan to MDP activates NFκB and mitogen-activated protein kinase signaling pathways, causing the production of various cytokines including TNF and IL-1β[10,16,17]. In normal circumstances, the pro-inflammatory cytokine secretion by intestinal APCs is minimal, yet bacterial killing occurs. This indicates that the intestinal immune system can defend against luminal microbiota without appreciable tissue injury[10]. The importance of NFκB activation in the clearance of invasive bacteria is underscored by the observations that CARD15 gene mutation fails to clear Salmonella from epithelial cells leading to increased bacterial interaction with the host’s luminal defense mechanisms. Indeed, heterozygosity for a NOD2 polymorphism confers a 1.75 to 4-fold increased risk whereas homozygosity confers even a much higher risk (11 to 27-fold) for development of IBD[10,18].
Microbiome and dysbiosis
A complex network of interactions exists between the host intestinal epithelial cells (IEC), host immune cells and the abundant intestinal microbiota. An altered balance of commensal pathogenic microbiota could lead to a pro-inflammatory milieu that exacerbates intestinal inflammation[11]. The dysbiosis theory suggests that intestinal microbiota of IBD patients change in diversity, composition and localization compared to that of healthy controls[19-24]. Fecal microbiota studies on IBD patients have revealed a decreased frequency in Bacteroidetes and Firmicutes and an increase of Proteobacteria and Actinobacteria phyla[21,23-26]. Advances in metagenomics sequencing of microbial RNA have confirmed decreases in bacterial composition and diversity in IBD patients compared to normal population[15]. Likewise, defects in the intestinal epithelial barrier integrity suggests increased uptake of luminal antigens that further leads to persistent immune activation[11]. Evidence suggests that reduced mucin production, through goblet cell depletion and epithelial cell tight junction dysfunction, by facilitating the access of gut luminal antigens to the intestinal mucosa, are also involved in the pathophysiology of IBD[15]. Additionally, Faecalibacterium prausnitzii, a member of the Firmicutes phylum and one of the most abundant species in the healthy human colon, is underrepresented in IBD patients and altered in terms of disease activity specifically in CD patients[19,20,24,27-30].
It is still uncertain as to whether the dysbiosis is the primary cause of inflammation or merely a secondary phenomenon of IBD. The uncertainty stems from the fact that intestinal microbiota analysis in IBD patients is performed after the development of the disease rather than prior to development[22]. However, the pathogenic role of the microbiome in IBD pathogenesis and therapy is indicated by studies showing an improvement in IBD by antibiotic treatment[31,32]. Indeed, treatment of UC patients with antibiotics improves mucosal inflammation[15]. Further, remission of inflammation and mucosal healing was observed in CD patient’s that underwent diversion of feces with subsequent disease reactivation after infusion of feces[33]. Likewise, defects in the intestinal epithelial barrier integrity suggests increased uptake of luminal antigens that further leads to persistent immune activation[11]. Evidence suggests that reduced mucin production, through goblet cell depletion and epithelial cell tight junction dysfunction, by facilitating the access of gut luminal antigens to the intestinal mucosa, are also involved in the pathophysiology of IBD[15].Collectively, these observations attest to the role of microbial dysbiosis in the induction of IBD[34].
Immune response
The innate and adaptive immune system activation and dysfunction as well as a loss of tolerance to enteric commensal bacteria contribute to the abnormal inflammatory response in the intestinal tract in patients with IBD[11,35]. Characteristic histological findings in IBD is an influx of innate immune cells (neutrophils, macrophages, dendritic cells and NK cells) as well as adaptive immune cells (B cells and T cells) into the lamina propria. With activation of immune cells, there is an elevation in the TNF-α, IL-1β, IFN-γ and cytokines levels. Recent advances in genome-wide associated studies and immunological studies suggest aberration in the mucosal innate response, innate microbial sensing, autophagy and unfolded protein response are potential pathogenic pathways in IBD[35].
Microbial antigen sensing by the innate immune system is mediated by pattern recognition receptors (PRR), such as Toll-like receptors (TLR) that recognize pathogen associated molecular patterns. The stimulation of PRRs results in a signaling cascade in which NFκB activation produces pro-inflammatory mediators, thereby ensuring an effective and appropriate innate response to presented antigens. Specific TLR that have been implicated in CD include nucleotide-binding oligomerization domain (NOD)-like receptors, specifically NOD2, which encodes an intracellular sensor and is stimulated by specific components of bacterial peptidoglycan that results in NFκB activation as mentioned in the earlier section on genetics[35].
Adhesion molecules, such as intercellular cell adhesion molecule 1 (ICAM1) are necessary for the circulating cells to adhere to the activated endothelium, which leads to extravasation of mononuclear and polymorphonuclear cells into the inflammatory tissue[11]. In addition, adhesion molecules mediate migration of the extravasated immune cells through the stroma to the source of maximal chemokine production, as well as through the epithelium to the lumen, where they produce crypt abscesses[11]. Historically, the NFκB pathway was thought to elicit a pro-inflammatory response, but selective gene-deletion studies have shown that this pathway produces both beneficial and deleterious effects[11]. NFκB activation stimulates expression of numerous molecules that are involved in the inflammatory response such as IL-1β, TNF, IL-6, IL-8, ICAM1 and other adhesion molecules[11]. NFκB also stimulates the expression of protective molecules, CARD15, cyclo-oxygenase 2, B defensins, TNF-induced protein 3 that inhibit the inflammatory response[11].
IL-23 is a key cytokine in orchestrating the crosstalk between innate and adaptive immunity and has a central role in driving early responses to microbes[35]. Interestingly, IL23R polymorphisms have been associated with both CD and UC, suggesting that the IL-23 axis might represent a shared inflammatory pathway in chronic intestinal inflammation[35]. Recent studies have shown that, besides its activity on Th17 cells, IL-23 can also act on cells of the innate immune system. Unconventional, innate-like T cell populations, which are particularly represented at mucosal sites, have been found to respond to IL-23 stimulation and secrete Th17-related cytokines[35].
The adaptive immune system is highly specific and confers long lasting immunity and is adaptable with its antigen specificity and maturation[35]. The inhibitory cytokines IL-10 and TGF-β in Peyer’s patches, mesenteric lymph nodes and lamina propria are involved in T-cell tolerance in the intestine[10]. Immunologically Th0 cells are activated and differentiate into Th1, Th2 or Th17 cells based upon the clearance of specific pathogens, but a dysregulated T cell response with abnormal development of activated T cells subsets may lead to the onset of inflammation by an excessive release of cytokines and chemokines with have multiple pathogenic effects[35]. Moreover, there is a genetic association between the inhibitory cytokine IL-10 and UC, in which IL-10 participates in the down-regulation of intestinal inflammation[10]. CD had been thought to be a Th1 mediated disease, secreting copious amounts of IFNγ, TNF-α, and IL-12; whereas UC was thought to be a Th2 mediated response, with signature cytokine secretion from IL-4, IL-5 and IL-13[15]. However, recent data has suggested this paradigm is not quite so straight-forward[15]. Furthermore, data suggests that Th17 cell production of IL-17 and IL-23 play important roles in the pathogenesis of IBD, with DCs isolated from CD patients producing more IL-23 than UC patients[15].
Th17 cells are a subset of helper T-cells that are induced by IL-6 and TGF-β and expanded by IL-23[15]. Signaling is mediated through the engagement of heterodimeric IL-23 with its heterodimeric receptor that activates the JAK-STAT signaling pathway, which then regulates the transcription of several genes[10]. IL-23 secreted by macrophages and dendritic cells may contribute to Th17 proliferation, survival or both[10].
PATHOPHYSIOLOGY OF DSS-INDUCED COLITIS: AN INVALUABLE ANIMAL MODEL
Numerous animal experiment in the last 25 years have employed the DSS colitis model as a chemical induction of intestinal inflammation model that morphologically and symptomatically resembles epithelial damage seen in human ulcerative colitis[1,36,37]. The DSS model, initially reported by Okayasu et al[38], and has been used to investigate the role of leukocytes in the development of colitis in animal models and is now one of the most extensively employed experimental models abetted by its simplicity and reproducibility[38,39].
DSS is a water soluble, negatively charged sulfated polysaccharide with a highly variable molecular weight ranging from 5 to 1400 kDa. Murine colitis results from administration of 40-50 kDa DSS added to drinking water. In the DSS model, the sulfated polysaccharide does not directly induce intestinal inflammation, but rather acts as a direct chemical toxin to colonic epithelium resulting in epithelial cell injury. The proposed and most accepted mechanism by which DSS induces intestinal inflammation results in the disruption of the intestinal epithelial monolayer lining, leading to the entry of luminal bacteria and associated antigens into the mucosa and allowing the dissemination of proinflammatory intestinal contents into underlying tissue[2,4,6,38,40-42].
The effectiveness of the DSS-induced colitis depends on several factors, including DSS concentration (usually 1%-5%), duration and frequency of administration (acute or chronic), molecular weight of the manufactured DSS, strain of animals (C3H/HeJ, C57BL/6 and BALB/c mice strains are more susceptible), and microbial environment of animals (i.e. germ-free versus specific pathogen-free). Furthermore, depending upon these various factors, the animals may develop acute colitis, chronic colitis or even colitis induced dysplastic lesions (Table 1).
Table 1.
Factors that influence effectiveness of dextran sodium sulfate to induce colitis
| Factors | Variables | Description |
| DSS | Molecular weight | 40-50 kDa for tissue penetration (larger molecule does not penetrate colonic tissue well and smaller molecule has poor distribution) |
| Dosage concentration | Ranges from 1.5%-3% used most frequently (1% with mild symptoms and delayed onset) | |
| Duration of therapy | Acute: 5-10 d administration | |
| Manufacturer/batch | Chronic: 4-5 repeating cycles of DSS and sterile water | |
| Various manufacturers with differing potency | ||
| Host | Genetically susceptible strain of animal | Certain strains are more susceptible to DSS colitis than other strains. |
| Susceptible strains: C3H/HeJ, C57BL/6, BALB/C | ||
| Environment | Housing Conditions | Group vs individual unit, frequency of cage changes alters coprophagy by host |
| Microbial State | Germ-free vs specific pathogen free vs wild type |
DSS: Dextran sodium sulfate.
In various protocols, 40-50 kDa DSS is added to sterilized drinking water at various concentrations to produce the desired inflammatory effect. At this molecular weight the tissue distribution reveals that DSS penetrates the mucosal membrane of the intestine[4]. Acute colitis is induced by administering DSS to the chosen strain of mice, often C57BL/6 or BALB/c male mice, for a period of 6-10 d (average 7 d). Alternatively, chronic colitis is induced by administering 4-5 repeated cycles of DSS, each cycle involves administration of various DSS concentrations for 1 wk followed by sterile water for 7-14 d[1,38,43,44]. The severity of colitis can be augmented based upon the duration of DSS administration as well as the concentration of DSS[1]. During DSS administration, mice can exhibit pronounced weight loss (about 5%-10% reduction by day 5), altered stool consistency leading to diarrhea and hematochezia[41]. A significant physiological indicator of animal stress and imminent demise occurs if weight loss is greater than 20% of initial weight[41]. Following the desired protocol, the mice are then sacrificed and tissue samples are collected for histological analysis and other assays[8,41].
Observations of the chronological changes induced by DSS administration have revealed that the signs of disease appear as early as 1 d of treatment primarily identified by changes in the expression of tight junction proteins such as occludin, Zonula occludens-1 (ZO-1) and various claudins[4,8,40]. The altered expression of tight junction complexes and increased epithelial apoptosis has been identified in human IBD and is thought to contribute to leaks in the epithelial barrier[4]. These modest initial effects are followed by increasingly worsening symptoms, including increased intestinal permeability, severe bleeding and mortality[8]. The typical histological changes induced by acute DSS include mucin and goblet cell depletion, epithelial erosion, ulceration and infiltration of granulocytes into the lamina propria and submucosa resulting in immune responses[2,8]. In chronic DSS protocols, additional histological changes such as crypt architecture disarray and widening of the gap between the base of the crypt and muscularis with deep mucosal lymphocytosis tend to appear a few weeks after DSS induction[8]. Furthermore, transepithelial migration of neutrophils resulting in cryptitis and crypt abscess, a common histologic finding in human IBD, is reproduced in mice subjected to the chronic DSS administration[8]. With prolonged administration of DSS in rodents, squamous metaplasia of rectal mucosa, adenomatous changes and adenocarcinoma can also be seen histologically[45].
It has been identified that the molecular weight of DSS is critically important to the induction, severity and of DSS induced colitis[4]. Carcinogenic activity in the colon is achieved by DSS of 50 kDa while larger and smaller molecular weights (520 kDa and 10 kDa, respectively) failed to induce activity due to inadequate tissue penetration[4]. Additionally, the repeated pulses of DSS administration utilized in chronic phases of DSS colitis results in dysplasia that frequently resembles the clinical course in human UC[31].
The knowledge of the unique or exact mechanisms that result in carcinogenesis underlying colitis-associated cancer (CAC) in humans is lacking. The transition from inflammation, to dysplasia and hence cancer is not fully elucidated, but it has been proposed that a host of multiple factors are integral in the role of CAC including; immune response, activation of oncogenes, inhibition of tumor suppressors, as well as alterations in intestinal microbiota[46]. A variety of murine models of CAC have been developed. The most widely used and best studied is a chemically induced colitis-associated model that incorporates a combination of a potent carcinogen, azoxymethane (AOM) and DSS[47]. Long term administration or repeated cycles of DSS has been shown to induced chronic colitis and subsequent dysplasia in rodents[48]. In this murine model, as in human CAC, the degree of inflammation correlates with dysplasia and is associated with nuclear translocation and mutational activation of β-catenin which results in increased activity of the Wnt signaling pathway[46,48]. The change in Wnt signaling results in enhanced inflammatory immune response with release of pro-inflammatory cytokines (IL-6 and TNF-α), which results in elevated levels of c-myc, a known oncogene and activator of cell cycle progression[46]. Additionally, many important inflammatory components are increased and activated during CAC (NFκB, Janus Kinase, cyclooxygenase-2 and inducible nitric oxide synthase) leading to further infiltration of lymphocytes, plasma cells and macrophage to sites of chronic inflammation[46]. The limitation of the models is that Kras or p53 mutations, that are typical in human CAC, are not present in murine AOM/DSS-induced CAC models[46].
DSS colitis is associated with increased production of various cytokines and chemokines. Following the induction of DSS colitis various tissue derived cytokines have been shown to be upregulated as early as the first day of DSS-induction[4]. The different inflammatory mediators assessed include TNF-α, the hallmark of DSS induced colitis, IL-6, IL-10, IL-17, IL-1β, TGF-β, mucin, TLR2/4 gene expression, MPO activity[1,4]. Differences in inflammatory profiles was expressed between acute and chronic DSS phases[4]. It was shown that in acute inflammation in DSS converts to a predominant Th-2 mediated response in the chronic state with noted decreased levels of TNF-α, IL-17 and elevated levels of IL-4,-6,-10 and IFNγ[4,49]. IL-6, IL-1β tissue levels correlate with IBD activity and IFNγ secretion has been linked to IL-17 section which tends to be expressed during chronic inflammation[31]. It has been shown that the cytokine profiles in DSS colitis phases correlates with barrier function, histological and clinical parameters lending the model as an integral tool in the study of cytokine role in induction and recovery from inflammation[4]. The bulk of research on IBD pathophysiology till date has focused on the immunological response and less attention has been paid to defects in barrier function that can potentially increase bacterial contact with the epithelium[50]. In animal studies assessing epithelial damage, DSS has been considered a toxicity model as induction of intestinal inflammation is not direct. Rather the chemical exerts an epithelial injury resulting in intestinal epithelial barrier disruption causing an influx of luminal bacteria and associated antigens into the mucosa and submucosa and thus triggering an inflammatory reaction[51]. An alternative mechanistic action for the induction of inflammation in the DSS model is through dysbiosis of murine gut microbiota leading to immunoregulation defects, mucin and goblet cell depletion and barrier dysfunction. Although the exact mechanism through which DSS induces colonic mucosal inflammation is not completely understood, recent results indicate that sulfate groups of the DSS molecules destabilize the mucus layers and make it more permeable to bacteria[52]. Hence, the DSS model is not simply a toxicity model, but also a barrier dysfunction model that encompasses mucus loss and the eventual bacterial penetration frequently found during intestinal trauma[53].
DSS DISRUPTS INTESTINAL BARRIER FUNCTION
The function of the intestinal epithelium is to simultaneously provide a barrier between the host and external environment while facilitating selective permeability that limits migration of harmful molecules but allows appropriate absorption of nutrients, ions and water[19]. This dynamic relationship of selective permeability is dependent upon specialized structures composed of tight junction complexes[40]. Tight junctions (TJ) are protein complexes consisting of Occludin, ZO, Claudins and junctional adhesion molecules (JAM) that are located at the apical ends of the lateral membranes of IEC and form a physiologically active barrier that can alter permeability based on the cellular environment[40,54-56] .
Dysfunctions of intestinal barrier lead to increased intestinal permeability that have been associated with the pathogenesis of IBD[2]. Barrier dysfunction in IBD has been identified as shifts in tight junction protein expression, and function with poorly adherent mucosa in the inflamed intestinal mucosa[2,40,43,54,57,58]. This change in composition and function corresponds to an increase in intestinal permeability with entry of commensal bacteria and decrease in transepithelial resistance[2,40,57]. The question remains whether the alteration in the intestinal barrier function is a primary necessity for the inflammatory response or a secondary development from the inflammatory response[40].
Several studies have shown that the appearance of intestinal inflammation is not the initial, inciting event, but rather the TJ complex changes and the subsequent increase in colonic permeability precede the development of intestinal inflammation[38,40,42,43,57,59]. In mice DSS colitis leads to a decrease in TJs expression that is followed by an increase in permeability and clinical manifestations of colonic inflammation[19]. Specifically, the protein pattern of TJ undergo rapid changes, as evident in the increased expression of claudin-2 and depletion of various claudins and zona occludens-1[19,40,54]. Therefore, the breach in the mucosa barrier is seen as a secondary event to the increase in colonic mucosal permeability resulting in the influx of inflammatory cells into the intestinal mucosa[40,57].
As noted earlier, the TJ are comprised of protein complexes that include occludin, ZO, claudins and junctional adhesion molecules. Importantly, claudin proteins are intrinsically involved in the formation of the IEC barrier function[60]. Claudins comprise a multigene family of 27 members and are expressed in a pattern that is both organ and segment-specific. A majority of these claudins (claudins 1, 3, 4, 5, 7, 8) confer barrier properties and are often found in tight epithelia of distal intestine. Claudins interact in a tissue-specific manner to form a charge-selective and size-selective barrier and predominantly contribute to epithelial barrier function and regulate paracellular permeability in intact epithelium[61]. Several mouse studies have assessed the expression of various claudins in conjunction to DSS administration. So far, the results have been varied. One study found enhanced expression of the claudin-1 protein, whereas other studies found decreased expression of claudin-1[54,60]. These contradictory findings could be related to species differences as claudin-1 was increased in rats while decreased in mice when exposed to DSS. Further studies conducted by independent laboratories reported similar findings demonstrating an up-regulation of the pore-forming claudin-2 and decreased expression in claudin-3,-5,-7,-8[60]. Furthermore, alterations in claudin expression resulted in similar outcomes with pronounced barrier dysfunction with aggravated mucosal damage and increased colonic permeability in DSS-induced colitis[60] .
Claudin-2 has been shown to provide a critical role in regulating colonic epithelial homeostasis and barrier function; therefore, regulating mucosal immune response and mucosal inflammation. A frequent regulatory step in inflammation is the increased expression of claudin-2 and it insertion into TJ strands. Upregulation of claudin-2 expression is found to start in the lower crypt and progress toward the surface epithelium[56]. The functional role of this modified expression and the consequential increase in intestinal permeability is still uncertain[56]. Consequently, recent human studies have examined potential alterations in claudin family functions n IBD patients and demonstrated a robust increase in claudin-2 expression[62]. Similarly, claudin-2 was found to assist the uptake of mucosal antigens[62]. A corollary finding in DSS-treated Cl-2TG mice revealed significant suppression of pro-inflammatory molecules with overexpression of claudin-2 suggesting its pivotal role in immune adaptation[62]. The paradoxical finding of increased regulatory CD4+ cells of unchallenged mice and decreases in immune cell infiltration of DSS-challenged mice suggests that claudin-2 induced epithelial permeability facilitates the interaction of host immune molecules and luminal antigens to promote adaptive tolerance and protection from colitis rather than increased sensitization[62]. Additionally, the DSS-challenged mice showed decreased apoptosis and increased epithelial proliferation further supporting the role of claudin-2 in intestinal epithelial cell regulation[62]. Conversely, claudin-2 knockout mice subjected to DSS exhibited severe colitis[62].
In addition to changes seen in claudin expression, other studies assessing barrier dysfunction assessed changes in ZO-1. Merely one day of DSS administration caused a statistically significant reduction of ZO-1[40]. Thus, the loss of TJ integrity led to an increase in permeability which occurred before any significant clinical or histological evidence of colitis[40]. Another study supporting this premise reported a redistribution of occludin and ZO-1, from the junctions in colonic epithelium after 4 d of administration of DSS[42]. As seen in the claudin studies, the ZO-1 data presented suggests that TJ complex changes are a prerequisite for the development of intestinal inflammation[40,42].
Another adhesion molecule with essential roles in the development and homeostasis of several tissues is the large group of proteins E-cadherin, that represent the major component of adherens junctions[63]. Studies suggest that E-cadherin may factor into the pathogenesis of UC as mice with E-cadherin deficiency had more severe colitis in the DSS model[63]. Further, similar to redistribution of occludin, ZO-1, E-cadherin and B-catenin also translocate from the junctions in colonic epithelium after 4 d of DSS treatment[2,42].
DSS EFFECTS ON MUCIN
Observations in several human studies found patients with colonic inflammation had alterations in colonic mucus and decreased effectiveness in its barrier function[64]. The gastrointestinal tract is quite remarkable with respect to the protective mucus barrier organization. The secretory, gel-forming mucins form the outer loose layer of mucus and the inner dense membrane-bound mucins covers and protects the surface epithelial cells[64,65]. Biochemically, mucins are usually very large, filamentous molecules with a large region within their polypeptides, which comprise relatively short tandemly repeated peptide domains which are highly O-glycosylated[66]. In humans, fifteen different mucins have been described and are assembled in the MUC gene family, with only a few encoded for activity in the colon[64,66]. Of the mucins, only Muc2 has been shown to be the principle secretory gel-forming mucin in both large and small intestines providing the functional barrier between epithelium and microbiota[64,65]. Muc2 is the predominant mucin produced by intestinal goblet cells, and is thickest in the healthy colon[67]. Whereas, the other membrane bound mucins with transmembrane regions are involved in cell signaling, adhesion, growth and modulation of the immune system[64,66].
As mucus is in direct contact with many microorganisms within the intestinal tract, defects in gene coding or protein folding of the mucins could lead to poor membrane integrity and ultimately a breach in the epithelial barrier or alterations in the mucosal-bacterial interactions. Prior in-vitro studies on intestinal cell lines revealed that the mucin expression and structure is influenced by cytokines, bacteria and their associated components[66,67]. Likewise, in UC the alteration of immunological or bacterial factors can influence mucin production[67]. Goblet cell depletion is a frequent histopathology finding in UC patients implying that the synthesis of Muc2 is decreased in association with smaller goblet cells thecae and histologic appearance of goblet cell depletion[67]. This, in turn, leads to further adverse effects resulting in chronic inflammation that is characteristic of IBD[64,66]. With these considerations in mind, it is still uncertain whether mucin decrease and goblet cell depletion are the primary contribution to IBD or the consequences of inflammation[67].
Mice deficient in Muc2 are characterized by a loss of mucus layer with bacteria not only in direct contact with epithelial cells but found deeper in the crypts (Table 2). These observations are absent in healthy animals. Furthermore, mice deficient in Muc2 ultimately are more susceptible to spontaneous severe colitis and eventually cause an increased risk of colon cancer development[52,68-70]. Beyond DSS mouse models, other models with defective mucus function all develop colitis[50,71,72]. In order to fully comprehend the role of mucins, specifically Muc2, in epithelial barrier protection and colitis, studies were conducted on Muc2 deficient mice. DSS administration to these mice produced fulminant colitis within days compared to the treated wild-type mice[68]. Further, a single missense mutation in Muc2 lead to spontaneous colitis with increased intestinal permeability and increased cytokine production in the distal colon[67]. Additionally, during DSS induction the thickness of the mucus gel decreased at the same time as the mice developed increasing colitis symptoms[64]. Analysis of IL-10-/- mice revealed thicker mucus layers than wild type, but in further analysis specific mucin properties were altered and the usually impenetrable inner layer was found to be penetrable to intestinal bacteria[52]. Therefore, mucus thickness alone was not shown to be a meaningful predictor or indicator of mucus barrier function[52].
Table 2.
Role of the different classes of mucins in dextran sodium sulfate induced colitis
| Mucin gene | Mucin class | Expression site (GI tract) | Chromosome | Pathological findings with DSS treatment | Ref. |
| Muc1 | Membrane bound | All epithelia | 3 | Muc1-/- protective when challenged with DSS | [64,66] |
| Increased thickness with adherent mucus | |||||
| Muc3 | Membrane bound | Intestine, enterocytes | 5 | Increased up-regulation of Muc3 gene | [65,66] |
| Muc4 | Membrane bound | All epithelia | n.d. | Large type I transmembrane glycoprotein | [65,66,73] |
| Muc4-/- more resistant to colitis due to up-regulation of Muc2 due to increase proliferation of cytokines | |||||
| Muc2 | Secretory | Intestine, goblet cells | 7 | Muc2-/- more susceptible to spontaneous colitis, increased risk of CAC | [66-68] |
| DSS produces fulminant colitis vs wild type | |||||
| Muc5ac | Secretory | Stomach | 7 | [66] | |
| Muc5b | Secretory | Tongue, sublingual glands | 7 | [66] |
CAC: Colitis-associated cancer; DSS: Dextran sodium sulfate.
Post-translations modifications on mucin are key to their functionality. Along these lines, an increased levels of Muc2 seen following DSS administration fails to control inflammation because of decreased sulphation in Muc2[66,67]. Further studies have shown that alteration in glycosylation of mucins resulted in decrease Muc2 synthesis and a diminished mucus barrier, thereby increasing the susceptibility to DSS-induced colitis[67]. The IL-10-/- germ free mice given normal enteric bacteria, presenting with a loss of sulphation of newly synthesized Muc2 molecules, were more prone to develop colitis and produced severe and chronic colitis[66]. Collectively, these observations underscore the importance of post-translational modification on mucin in its functional role in regulating intestinal barrier integrity[67].
In conjunction with the secretory mucins, the various cell surface mucins are considered to serve a critical function in mucosal protection[65,67]. Muc4 is a large type-I transmembrane glycoprotein component normally expressed on the surface of colonic epithelial cells. Muc4-/- mice were more resistant to colitis and CAC induced by AOM/DSS because of the compensatory upregulation of Muc2 expression in these mice[65]. Although not statistically significant, an upregulation of Muc3 (orthologue of human Muc17) was also observed[65]. Additionally, Muc4-/- mice induced with DSS had increased expression of pro-inflammatory cytokines (TNF-α and IL-1β) which could also be responsible for the observed upregulation of Muc2 and Muc3[65]. Although both Muc4 and Muc13 belong to the transmembrane mucin subtype, deletion of either of the two mucin genes results in opposing phenotypic response to DSS treatment. Muc13-/- mice had increased macrophage expression in the inflamed mucosa accompanied by increased expression of intestinal IL-1β and TNF-α mRNA and significantly increased loss of body weight, diarrhea score, fecal blood score and severe histologic damage including intestinal epithelial cell apoptosis following DSS exposure. This suggests Muc13 confers protection to colonic epithelial cells from apoptotic stimuli, preventing damage-induced cell death[67]. In contrast, Muc4-/- mice exhibit a protective phenotype (decreased loss of body weight, diarrhea score, fecal blood score and less severe histologic damage) which is related to a compensatory upregulation of Muc2 and Muc3[65,73].
Muc1, a non-gel forming mucin transmembrane mucin, when deleted resulted in protection of colonic epithelial cells similar to findings in Muc4-/- mice when challenged with DSS[64]. Muc1-/- deficient mice had increased thickness of adherent mucus resulting in mild colitis[64]. Additionally, germ-free mice had a thin colonic mucus barrier, but luminal exposure to the bacterial products lipopolysaccharides (LPS) and peptidoglycan quickly restores the firmly adherent mucus layer thickness to levels observed in conventionally housed mice[64]. Muc1-/- mice developed a more severe inflammatory response after exposure to H pylori compared with wild-type mice, demonstrating that cell surface mucins can modulate the inflammatory response to chronic infection[67]. As opposed to observations in Muc2 deficient mice resulting in severe colitis, the Muc1-/- mice developed a very mild colitis which is thought to be due to an increased colonic mucus barrier and a decreased ability to recruit T cells to the affected region[64].
DSS ALTERS THE MICROBIAL BALANCE
The intestinal microbiome plays an integral role in host immune development, tolerance and intestinal physiological processes. This has spurred large collaborative efforts aimed at identifying and characterizing the microorganisms which are associated with the health and disease in humans[25,32,74,75]. However, the sheer presence of microbiota in the intestinal tract alone is not enough to exert these physiological effects, rather variant microbe compositions, temporal changes in populations and relative abundance of specific microbes are important in homeostasis and specific disease states, namely IBD[66,74-76].
Effects on mucosal inflammation
Studies involving DSS-induced colitis have revealed the critical role that gut microflora play in the pathogenesis of mucosal inflammation and the related role of barrier function as a bulwark against extensive stimulation of the mucosal immune system. However, the exact mechanism through which bacteria induce inflammation has been elusive[22,76]. Indeed, a number of recent studies have identified compositional changes at the bacterial phyla and species levels that can influence phenotypic expression of both pro- and anti-inflammatory responses in humans and in murine models relevant to IBD[22,25,74,76].
As indicated earlier, microbiome composition alterations can result in normally underrepresented members of the microbiome to become dominant, leading to perturbations in structure and function of the microbiome. Various DSS-induced colitis studies have noted that murine microbiota alterations occurred early (within 3 d) and are characterized by diversity reduction and changes occurred prior to the clinical or biochemical evidence of inflammation[31,75,77]. Composition changes in DSS-induced murine colitis indicated a significant proliferation in Bacteroidaceae and Clostridiaceae families[38]. Further studies have corroborated these initial findings to suggest dramatic reductions in the genera of Bacteroidetes, Prevotella, Clostridium and Lactobacillus with corresponding increases in pro-inflammatory gut microbiotic components Bacillaceae, Enterococcales and Enterobacteriaceae[31,78,79].
Effects on barrier functions
Beyond the simple changes in microbiome composition that may herald alterations in physiologic homeostasis, other recent studies have examined the role that bacteria influence barrier function alteration. Evidence suggests that epithelial integrity is compromised in DSS-induced colitis leading to penetrance of microbes and associated antigens into the mucosa and[69,75,80,81]. One such study examining the anti-inflammatory effect of Faecalibacterium prausnitzii suggested that its immunomodulatory effects are mediated via decreasing the paracellular permeability to effectively reduce the severity of colitis and prevent colitis progression. Faecalibacterium prausnitzii decrease NF-κB activation and IL-8 secretion in vitro and impairs the colonic synthesis of pro-inflammatory cytokines while inducing the secretion of anti-inflammatory cytokine in vivo[19]. Likewise, various strains of bifidobacteria have shown promising anti-inflammatory effects[82]. Specifically, strains of Bifidobacterium bifidum have been reported to inhibit LPS-dependent NF-κB activation in IECs and induce anti-inflammatory macrophage, dendritic, and T cell populations[82].
Effects on mucin
Metagenomics studies following the administration of DSS revealed an elevation in Akkermansia muciniphila, a Verrucomicrobia member, which have been found to metabolize sulfur and lead to mucin degradation and correlate with disease activity in mice administered DSS[31,75,78]. Additionally, DSS induced colitis obliterated the difference in abundance and structure of bacterial communities between the two mucus layers[52,69]. There were fewer bacterial in the firmly adherent mucus but the abundance of bacteria in the mucus layer of the DSS treated animals was 10-100 times greater compared to the control group, suggesting DSS treatment increased the total count of infectious bacteria[22]. Antibiotic administration improved DSS-induced colitis akin to observations showing reduced inflammation in germ-free murine colitis model[31,32].
Changes during the recovery phase
Interestingly, there is a rapid shift of the gut microbial community toward a healthy profile in the recovery phase following the DSS treatment regimen. Within two days of stopping DSS administration, the mice gut microbiota showed relative abundance in Bacteroidetes/Prevotella, Bacillaceae, to levels comparable to those observed in healthy controls[31]. These data demonstrated the high degree of resilience of the gut microbiota and a rapid recovery of its healthy mutualistic profile after DSS-induced dysbiosis[31].
Probiotic treatment against dextran sodium sulfate-induced colitis
Probiotic therapies with Lactobacilli and Bifidobacteria produced favorable outcomes in murine colitis models[82,83]. Probiotic bacteria have been shown to decrease intestinal permeability and restore gut barrier integrity by modulation of tight junction proteins[83]. Pretreatment with Lactobacillus reuteri strains prevented the onset of DSS-induced colitis by reducing bacterial translocation and suppressing adherence of lactobacilli on colonic mucosa and[22]. Further, probiotic treatment suppressed upregulation of P-selectin in the colonic endothelium, which decreased leukocyte-endothelial cell interactions and concomitant leukocytes recruitment to tissue[22]. However, no significant changes in microbial composition were observed indicating that the protective effects are linked to strengthening of the epithelial barrier integrity to reduce bacterial translocation[22,69]. Other studies have also examined the effects of different bacterial species on epithelial TJ in the DSS-induced colitis model. Various strains of bifidobacteria have shown promising anti-inflammatory effects[83]. Bifidobacterium bifidum S17 has been found to adhere to cultured IEC and displays potent anti-inflammatory activity both in vitro and in two murine models of colitis[83]. Bacillus subtilis supplementation resulted in improved barrier function compared to the DSS group, as evident by upregulated expression of TJ proteins (claudin-1, occludin, JAM-A, and ZO-1) and downregulated cytokine expression (IL-6, IL-17, IL-23, and TNF-α)[84]. Additionally, only certain, limited strains of Bifidobacterium longum have indicated an increased expression of ZO-1 and occludin resulting in reduced severity of DSS-induced colitis[83].
DEXTRAN SODIUM SULFATE-INDUCED CHANGES IN METABOLOME
DSS colitis has been shown to produce disturbances in the metabolism of phospholipids depicted by decreased levels of phosphocholine and glycerophosphocholine in the colon of mice[1]. Phosphocholine and glycerophosphocholine are the most important metabolites of choline and the major cellular constituents required for the assembly of biological membranes and disturbance in the metabolism suggests the possibility of distorted membrane integrity in the presence of DSS[1].
The role of dietary fat intake in exacerbating intestinal inflammation and modulation of immune function has been investigated extensively[85]. There is supporting evidence to show that a high fat intake is associated with an increased risk of ulcerative colitis[85]. Of note, two factors determine the role of lipid nutrition in health and disease: (1) the composition and (2) the total amount of fat in the diet. Bile acids are produced in the liver and excreted into the duodenum as conjugated bile salts to facilitate homeostatic functions in the gastrointestinal tract. Once in the large intestine, various microbial species enable the transformation of the bile salts into secondary bile acids, such as deoxycholic acid and urosodeoxycholic acid (UDCA), which vary in hydrophobicity[86]. Increased luminal bile acid hydrophobicity reportedly leads to gut barrier dysfunction through the disruption of cell membranes causing cytotoxicity, production of reactive oxygen species, epithelial growth factor receptor activation and tight-junction redistribution[86]. Conversely, UDCA which is more hydrophilic leads to stabilization of lipid membranes[86]. Thus, changes in bile acid composition favoring increased hydrophobicity leads to increased gut permeability allowing for enhanced translocation of bacteria and associated antigens across the tight-junction barrier of the gut epithelium leading to intestinal inflammation. One such study identified animals with a lower concentration of fecal cholic acid had increased histologic damage[86,87]. Additionally, various types of dietary fats promote colitis via alterations in gallbladder bile and gut microbiota through changes in taurocholic acid levels in bile which facilitated the growth of the Bilophila wadsworthia, an inflammatory Gram-negative anaerobe[86,88]. Overall, fecal bile acid hydrophobicity found to be mediated through a higher proportion of deoxycholic acid positively correlates with the severity of DSS colitis[86].
A potential causal link between dietary fat, especially Omega-3 and Omega-6 fatty acids, and susceptibility to induced colitis has also been reported. A dose dependent effect of the changes in composition of omega fatty acids in exacerbating colitis in various models including the DSS colitis model has been reported[85,86]. Fat in the diet alone does not alter the susceptibility to DSS, rather the modifications to the fecal bile acid composition has been proven to be deleterious[85]. Omega-3 fatty causes decreased adiponectin mRNA expression acid leading to increased inflammation in colonic mucosa[85]. However, another study reported decreases adiponectin RNA expression with no changes in serum adiponectin levels by high fat diet[85]. There are also reports that suggest that the pro-inflammatory effect of adiponectin is mediated by increased IL-6 production[85].
In addition to studies examining primary effect of bile acids on inflammation, the effects that bile acids on cell surfaces have also been investigated[89]. A member of the G protein coupled receptor superfamily, GP-BAR1 is a cell surface bile acid-activated receptor that has been found to be highly expressed in the ileum and colon and activated by secondary bile acids, specifically lithocolic acid and tauro-LCA[89]. Cipriani et al[89] asserted that this receptor regulates intestinal barrier integrity since mice lacking the GP-BAR1 receptor developed alterations in colonic histopathology and mucous cell distribution and function[89]. GP-BAR1-/- mice challenged with DSS exhibited an exacerbation of colitis that was not correlated with major immunological abnormalities, but rather an increase in intestinal permeability[89]. Furthermore, they have discovered that oleanolic acid, a natural GP-BAR1 ligand, attenuated colon inflammation and these anti-inflammatory effects of ciprofloxacin were lost in GP-BAR1-/- mice[89].
The central argument that metabolome changes play a part in colitis development is highlighted by the critical role microbiome composition plays in bacterial-host immune response. Certain enzymes, such as 7α-dehydroxylase that are endemic to gut microbiota, can convert primary bile acids into their secondary forms and therefore result in pro-inflammatory versus anti-inflammatory conditions in the host gut lumen. Additionally, shifts in the community composition and structure can reduce potentially immunomodulatory mucosal-associated species, such as Faecalibacterium prausnitzii, Clostridium leptum and Clostridium coccoides, that act to maintain epithelial health through the production of short-chain fatty acids and stimulation of mucin production, thereby effecting the host’s inflammatory response[25,27,75,90].
CONCLUSION
The last several years have provided indispensable insights into the histopathological and morphological changes in intestinal barrier function that likely contribute to the development and progression of murine colitis and aids in the understanding to the pathogenesis of human IBD. As stated prior, no isolated model has proven to sufficiently represent the complex clinical and histopathological characteristics of human disease. But, the DSS-induced colitis model is the most commonly used model that provides an inexpensive, simple and reproducible model to study various aspects of the role of mucin in barrier integrity, alterations in microbial balance and changes in the metabolome that relate to the pathogenesis of IBD. It is our hope that our further understanding of the role of intestinal barrier function in activating the innate and adaptive immune system in the intestinal tract that may lead to new therapeutic targets for human IBD.
Footnotes
Manuscript source: Invited Manuscript
Specialty type: Gastroenterology and Hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C, C
Grade D (Fair): D
Grade E (Poor): 0
Conflict-of-interest statement: No potential conflicts of interest.
Peer-review started: May 12, 2017
First decision: June 5, 2017
Article in press: August 2, 2017
P- Reviewer: Boros M, Liu DY, Gassler N, Sergi CM, Touil-Boukoffa C S- Editor: Gong ZM L- Editor: A E- Editor: Huang Y
Contributor Information
Derrick D Eichele, Department of Internal Medicine, Nebraska Medical Center, Omaha, NE 68198, United States.
Kusum K Kharbanda, Research Service, Veterans Affairs Nebraska-Western Iowa Health Care System, Omaha, NE 68105, United States; Department of Internal Medicine, Nebraska Medical Center, Omaha, NE 68198, United States; Department of Biochemistry and Molecular Biology, Nebraska Medical Center, Omaha, NE 68198, United States. kkharbanda@unmc.edu.
References
- 1.Randhawa PK, Singh K, Singh N, Jaggi AS. A review on chemical-induced inflammatory bowel disease models in rodents. Korean J Physiol Pharmacol. 2014;18:279–288. doi: 10.4196/kjpp.2014.18.4.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiesler P, Fuss IJ, Strober W. Experimental Models of Inflammatory Bowel Diseases. Cell Mol Gastroenterol Hepatol. 2015;1:154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, Price VH, Grisham MB. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296:G135–G146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perše M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neurath M, Fuss I, Strober W. TNBS-colitis. Int Rev Immunol. 2000;19:51–62. doi: 10.3109/08830180009048389. [DOI] [PubMed] [Google Scholar]
- 6.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 7.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007;59:1073–1083. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104:Unit 15.25. doi: 10.1002/0471142735.im1525s104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boirivant M, Fuss IJ, Chu A, Strober W. Oxazolone colitis: A murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med. 1998;188:1929–1939. doi: 10.1084/jem.188.10.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population-based study. Gastroenterology. 2005;129:827–836. doi: 10.1053/j.gastro.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 13.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hviid A, Svanström H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. [DOI] [PubMed] [Google Scholar]
- 15.Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014;20:6–21. doi: 10.3748/wjg.v20.i1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham C, Cho JH. Functional consequences of NOD2 (CARD15) mutations. Inflamm Bowel Dis. 2006;12:641–650. doi: 10.1097/01.MIB.0000225332.83861.5f. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 18.Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. 2004;99:2393–2404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 19.Carlsson AH, Yakymenko O, Olivier I, Håkansson F, Postma E, Keita AV, Söderholm JD. Faecalibacterium prausnitzii supernatant improves intestinal barrier function in mice DSS colitis. Scand J Gastroenterol. 2013;48:1136–1144. doi: 10.3109/00365521.2013.828773. [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto J, Kadara H, Garcia MM, Kabbout M, Behrens C, Liu DD, Lee JJ, Solis LM, Kim ES, Kalhor N, et al. G-protein coupled receptor family C, group 5, member A (GPRC5A) expression is decreased in the adjacent field and normal bronchial epithelia of patients with chronic obstructive pulmonary disease and non-small-cell lung cancer. J Thorac Oncol. 2012;7:1747–1754. doi: 10.1097/JTO.0b013e31826bb1ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dicksved J, Schreiber O, Willing B, Petersson J, Rang S, Phillipson M, Holm L, Roos S. Lactobacillus reuteri maintains a functional mucosal barrier during DSS treatment despite mucus layer dysfunction. PLoS One. 2012;7:e46399. doi: 10.1371/journal.pone.0046399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Järnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854.e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 25.Mar JS, Nagalingam NA, Song Y, Onizawa M, Lee JW, Lynch SV. Amelioration of DSS-induced murine colitis by VSL#3 supplementation is primarily associated with changes in ileal microbiota composition. Gut Microbes. 2014;5:494–503. doi: 10.4161/gmic.32147. [DOI] [PubMed] [Google Scholar]
- 26.Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, Brostoff J, Parkhill J, Dougan G, Petrovska L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi O, Khan MT, Schwarzer M, Hudcovic T, Srutkova D, Duncan SH, Stolte EH, Kozakova H, Flint HJ, Samsom JN, et al. Faecalibacterium prausnitzii Strain HTF-F and Its Extracellular Polymeric Matrix Attenuate Clinical Parameters in DSS-Induced Colitis. PLoS One. 2015;10:e0123013. doi: 10.1371/journal.pone.0123013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martín R, Chain F, Miquel S, Lu J, Gratadoux JJ, Sokol H, Verdu EF, Bercik P, Bermúdez-Humarán LG, Langella P. The commensal bacterium Faecalibacterium prausnitzii is protective in DNBS-induced chronic moderate and severe colitis models. Inflamm Bowel Dis. 2014;20:417–430. doi: 10.1097/01.MIB.0000440815.76627.64. [DOI] [PubMed] [Google Scholar]
- 29.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Doré J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 30.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Fazio L, Cavazza E, Spisni E, Strillacci A, Centanni M, Candela M, Praticò C, Campieri M, Ricci C, Valerii MC. Longitudinal analysis of inflammation and microbiota dynamics in a model of mild chronic dextran sulfate sodium-induced colitis in mice. World J Gastroenterol. 2014;20:2051–2061. doi: 10.3748/wjg.v20.i8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gkouskou KK, Deligianni C, Tsatsanis C, Eliopoulos AG. The gut microbiota in mouse models of inflammatory bowel disease. Front Cell Infect Microbiol. 2014;4:28. doi: 10.3389/fcimb.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 34.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3–10. doi: 10.1016/j.autrev.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Jurjus AR, Khoury NN, Reimund JM. Animal models of inflammatory bowel disease. J Pharmacol Toxicol Methods. 2004;50:81–92. doi: 10.1016/j.vascn.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Gaudio E, Taddei G, Vetuschi A, Sferra R, Frieri G, Ricciardi G, Caprilli R. Dextran sulfate sodium (DSS) colitis in rats: clinical, structural, and ultrastructural aspects. Dig Dis Sci. 1999;44:1458–1475. doi: 10.1023/a:1026620322859. [DOI] [PubMed] [Google Scholar]
- 38.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 39.Kawada M, Arihiro A, Mizoguchi E. Insights from advances in research of chemically induced experimental models of human inflammatory bowel disease. World J Gastroenterol. 2007;13:5581–5593. doi: 10.3748/wjg.v13.i42.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poritz LS, Garver KI, Green C, Fitzpatrick L, Ruggiero F, Koltun WA. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J Surg Res. 2007;140:12–19. doi: 10.1016/j.jss.2006.07.050. [DOI] [PubMed] [Google Scholar]
- 41.Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating intestinal inflammation in DSS-induced model of IBD. J Vis Exp. 2012;(60):pii: 3678. doi: 10.3791/3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samak G, Chaudhry KK, Gangwar R, Narayanan D, Jaggar JH, Rao R. Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. Biochem J. 2015;465:503–515. doi: 10.1042/BJ20140450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 44.Giner E, Recio MC, Ríos JL, Giner RM. Oleuropein protects against dextran sodium sulfate-induced chronic colitis in mice. J Nat Prod. 2013;76:1113–1120. doi: 10.1021/np400175b. [DOI] [PubMed] [Google Scholar]
- 45.Seril DN, Liao J, Yang GY, Yang CS. Oxidative stress and ulcerative colitis-associated carcinogenesis: studies in humans and animal models. Carcinogenesis. 2003;24:353–362. doi: 10.1093/carcin/24.3.353. [DOI] [PubMed] [Google Scholar]
- 46.Snider AJ, Bialkowska AB, Ghaleb AM, Yang VW, Obeid LM, Hannun YA. Murine Model for Colitis-Associated Cancer of the Colon. Methods Mol Biol. 2016;1438:245–254. doi: 10.1007/978-1-4939-3661-8_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka T. Development of an inflammation-associated colorectal cancer model and its application for research on carcinogenesis and chemoprevention. Int J Inflam. 2012;2012:658786. doi: 10.1155/2012/658786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sussman DA, Santaolalla R, Strobel S, Dheer R, Abreu MT. Cancer in inflammatory bowel disease: lessons from animal models. Curr Opin Gastroenterol. 2012;28:327–333. doi: 10.1097/MOG.0b013e328354cc36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johansson ME, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10:352–361. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Low D, Nguyen DD, Mizoguchi E. Animal models of ulcerative colitis and their application in drug research. Drug Des Devel Ther. 2013;7:1341–1357. doi: 10.2147/DDDT.S40107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johansson ME, Gustafsson JK, Holmén-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjövall H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63:281–291. doi: 10.1136/gutjnl-2012-303207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morgan ME, Zheng B, Koelink PJ, van de Kant HJ, Haazen LC, van Roest M, Garssen J, Folkerts G, Kraneveld AD. New perspective on dextran sodium sulfate colitis: antigen-specific T cell development during intestinal inflammation. PLoS One. 2013;8:e69936. doi: 10.1371/journal.pone.0069936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwaya H, Maeta K, Hara H, Ishizuka S. Mucosal permeability is an intrinsic factor in susceptibility to dextran sulfate sodium-induced colitis in rats. Exp Biol Med (Maywood) 2012;237:451–460. doi: 10.1258/ebm.2011.011269. [DOI] [PubMed] [Google Scholar]
- 55.Kyoko OO, Kono H, Ishimaru K, Miyake K, Kubota T, Ogawa H, Okumura K, Shibata S, Nakao A. Expressions of tight junction proteins Occludin and Claudin-1 are under the circadian control in the mouse large intestine: implications in intestinal permeability and susceptibility to colitis. PLoS One. 2014;9:e98016. doi: 10.1371/journal.pone.0098016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luettig J, Rosenthal R, Barmeyer C, Schulzke JD. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers. 2015;3:e977176. doi: 10.4161/21688370.2014.977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48:137–143. doi: 10.1538/expanim.48.137. [DOI] [PubMed] [Google Scholar]
- 58.Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, Bruewer M. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1140–G1149. doi: 10.1152/ajpgi.90534.2008. [DOI] [PubMed] [Google Scholar]
- 59.Mähler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–G551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- 60.Yuan B, Zhou S, Lu Y, Liu J, Jin X, Wan H, Wang F. Changes in the Expression and Distribution of Claudins, Increased Epithelial Apoptosis, and a Mannan-Binding Lectin-Associated Immune Response Lead to Barrier Dysfunction in Dextran Sodium Sulfate-Induced Rat Colitis. Gut Liver. 2015;9:734–740. doi: 10.5009/gnl14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–273. doi: 10.1016/s0962-8924(99)01578-0. [DOI] [PubMed] [Google Scholar]
- 62.Ahmad R, Chaturvedi R, Olivares-Villagómez D, Habib T, Asim M, Shivesh P, Polk DB, Wilson KT, Washington MK, Van Kaer L, et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014;7:1340–1353. doi: 10.1038/mi.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grill JI, Neumann J, Hiltwein F, Kolligs FT, Schneider MR. Intestinal E-cadherin Deficiency Aggravates Dextran Sodium Sulfate-Induced Colitis. Dig Dis Sci. 2015;60:895–902. doi: 10.1007/s10620-015-3551-x. [DOI] [PubMed] [Google Scholar]
- 64.Petersson J, Schreiber O, Hansson GC, Gendler SJ, Velcich A, Lundberg JO, Roos S, Holm L, Phillipson M. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G327–G333. doi: 10.1152/ajpgi.00422.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Das S, Rachagani S, Sheinin Y, Smith LM, Gurumurthy CB, Roy HK, Batra SK. Mice deficient in Muc4 are resistant to experimental colitis and colitis-associated colorectal cancer. Oncogene. 2016;35:2645–2654. doi: 10.1038/onc.2015.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Einerhand AW, Renes IB, Makkink MK, van der Sluis M, Büller HA, Dekker J. Role of mucins in inflammatory bowel disease: important lessons from experimental models. Eur J Gastroenterol Hepatol. 2002;14:757–765. doi: 10.1097/00042737-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 67.Sheng YH, Hasnain SZ, Florin TH, McGuckin MA. Mucins in inflammatory bowel diseases and colorectal cancer. J Gastroenterol Hepatol. 2012;27:28–38. doi: 10.1111/j.1440-1746.2011.06909.x. [DOI] [PubMed] [Google Scholar]
- 68.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 69.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295:1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 71.Fu J, Wei B, Wen T, Johansson ME, Liu X, Bradford E, Thomsson KA, McGee S, Mansour L, Tong M, et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest. 2011;121:1657–1666. doi: 10.1172/JCI45538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoebler C, Gaudier E, De Coppet P, Rival M, Cherbut C. MUC genes are differently expressed during onset and maintenance of inflammation in dextran sodium sulfate-treated mice. Dig Dis Sci. 2006;51:381–389. doi: 10.1007/s10620-006-3142-y. [DOI] [PubMed] [Google Scholar]
- 74.Brinkman BM, Becker A, Ayiseh RB, Hildebrand F, Raes J, Huys G, Vandenabeele P. Gut microbiota affects sensitivity to acute DSS-induced colitis independently of host genotype. Inflamm Bowel Dis. 2013;19:2560–2567. doi: 10.1097/MIB.0b013e3182a8759a. [DOI] [PubMed] [Google Scholar]
- 75.Nagalingam NA, Kao JY, Young VB. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm Bowel Dis. 2011;17:917–926. doi: 10.1002/ibd.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 77.Deplancke B, Finster K, Graham WV, Collier CT, Thurmond JE, Gaskins HR. Gastrointestinal and microbial responses to sulfate-supplemented drinking water in mice. Exp Biol Med (Maywood) 2003;228:424–433. doi: 10.1177/153537020322800413. [DOI] [PubMed] [Google Scholar]
- 78.Håkansson Å, Tormo-Badia N, Baridi A, Xu J, Molin G, Hagslätt ML, Karlsson C, Jeppsson B, Cilio CM, Ahrné S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin Exp Med. 2015;15:107–120. doi: 10.1007/s10238-013-0270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rath HC, Schultz M, Freitag R, Dieleman LA, Li F, Linde HJ, Schölmerich J, Sartor RB. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun. 2001;69:2277–2285. doi: 10.1128/IAI.69.4.2277-2285.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ito R, Kita M, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Iwakura Y, Okanoue T, et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem Biophys Res Commun. 2008;377:12–16. doi: 10.1016/j.bbrc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 81.Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grimm V, Radulovic K, Riedel CU. Colonization of C57BL/6 Mice by a Potential Probiotic Bifidobacterium bifidum Strain under Germ-Free and Specific Pathogen-Free Conditions and during Experimental Colitis. PLoS One. 2015;10:e0139935. doi: 10.1371/journal.pone.0139935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Srutkova D, Schwarzer M, Hudcovic T, Zakostelska Z, Drab V, Spanova A, Rittich B, Kozakova H, Schabussova I. Bifidobacterium longum CCM 7952 Promotes Epithelial Barrier Function and Prevents Acute DSS-Induced Colitis in Strictly Strain-Specific Manner. PLoS One. 2015;10:e0134050. doi: 10.1371/journal.pone.0134050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gong Y, Li H, Li Y. Effects of Bacillus subtilis on Epithelial Tight Junctions of Mice with Inflammatory Bowel Disease. J Interferon Cytokine Res. 2016;36:75–85. doi: 10.1089/jir.2015.0030. [DOI] [PubMed] [Google Scholar]
- 85.Matsunaga H, Hokari R, Kurihara C, Okada Y, Takebayashi K, Okudaira K, Watanabe C, Komoto S, Nakamura M, Tsuzuki Y, et al. Omega-3 fatty acids exacerbate DSS-induced colitis through decreased adiponectin in colonic subepithelial myofibroblasts. Inflamm Bowel Dis. 2008;14:1348–1357. doi: 10.1002/ibd.20491. [DOI] [PubMed] [Google Scholar]
- 86.Stenman LK, Holma R, Forsgård R, Gylling H, Korpela R. Higher fecal bile acid hydrophobicity is associated with exacerbation of dextran sodium sulfate colitis in mice. J Nutr. 2013;143:1691–1697. doi: 10.3945/jn.113.180810. [DOI] [PubMed] [Google Scholar]
- 87.Araki Y, Andoh A, Tsujikawa T, Fujiyama Y, Bamba T. Alterations in intestinal microflora, faecal bile acids and short chain fatty acids in dextran sulphate sodium-induced experimental acute colitis in rats. Eur J Gastroenterol Hepatol. 2001;13:107–112. doi: 10.1097/00042737-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 88.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, Baldelli F, Donini A, Fiorucci S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One. 2011;6:e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000;46:493–499. doi: 10.1136/gut.46.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]