

Abstract
AIM
To investigate the molecular mechanisms of gastric carcinogenesis.
METHODS
We used label-free quantification technology integrated with liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to identify differentially expressed proteins in 160 specimens of normal gastric mucosa, gastric mucosa with mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal gastric cancer (GC) collected at the Second Hospital of Lanzhou University from 2010 to 2015. Immunohistochemistry was used to verify the differentially expressed proteins detected by LC-MS/MS.
RESULTS
With a threshold of a 1.2-fold change and a P-value < 0.05 between mild dysplasia, moderate dysplasia, severe dysplasia or early mucosal GC and matched normal gastric mucosa tissues, proteomic analysis identified 365 significantly differentially expressed proteins. ERGIC1 expression decreased, while DNA-PKcs expression increased gradually along with different stages of GC initiation based on the tendency of fold change. The expression patterns of ERGIC1 and DNA-PKcs revealed by immunohistochemistry were consistent with the LC-MS/MS results.
CONCLUSION
The results suggest that aberrant ERGIC1 and DNA-PKcs expression may be involved in GC initiation.
Keywords: DNA-PKcs, ERGIC1, Dysplasia, Proteomics, Gastric cancer
Core tip: Using label-free combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS), the expression of 365 proteins based on the tendency of fold change was revealed to be statistically different between the various stages of gastric cancer (GC) initiation. Furthermore, we observed that ERGIC1 expression decreased, while DNA-PKcs expression increased gradually along with different stages of GC initiation based on the tendency of fold change. The expression patterns of ERGIC1 and DNA-PKcs revealed by immunohistochemistry were consistent with the LC-MS/MS results. These data indicate that abnormal ERGIC1 and DNA-PKcs expression may play an important role in GC initiation.
INTRODUCTION
Gastric cancer (GC) accounts for a large quantity of cancer-related deaths[1]. Over the decades, the prognosis of GC patients has improved, while overall survival rate of patients with GC has not been improved significantly yet, especially for advanced GC patients[2-4], which might be attributed to the poor understanding of mechanisms underlying the initiation and progression of GC. It is widely accepted that GC is a complex, heterogeneous, and multistep disease caused by various factors encompassing Helicobacter pylori infection, heredity, living habits, etc., which trigger series of molecular alterations, such as inactivation of tumor suppressor genes, activation of oncogenes, telomerase activation, DNA damage and even genome instability[5-8]. DNA-PKcs is the catalytic subunit of the DNA-dependent protein kinase and can combine with the Ku 70/Ku 80 heterodimer (Ku 70/80) into the DNA-dependent protein kinase (DNA-PK), which plays a crucial role in the activation of the non-homologous end joining (NHEJ) pathway[9,10]. Moreover, ERGIC1 has been identified as a cycling protein and participates in membrane trafficking and selective transport of cargo between the endoplasmic reticulum (ER), the intermediate compartment (ERGIC), and the Golgi apparatus in HepG2 cells, suggesting a possible role in protein secretion out of the ER[11].
Currently, high-throughput sequencing technologies, such as microarray assay and next-generation sequencing, have helped researchers obtain much more insight into molecular alterations[12]. These new tools have given a huge impetus to the discovery of novel intracellular molecular pathways and molecular subtypes of GC. For instance, some researchers have recommended that GC should be classified into Epstein-Barr virus-positive subtype, microsatellite instability subtype, genomically stable subtype, and chromosomally unstable subtype on the basis of the spectra of genetic alterations related with relevant clinical features, which is a totally new classification method[7,12].
However, it has been reported that GC is characterized by subtype heterogeneity and a lack of consistent genomic alterations across different individuals[13]. Furthermore, due to different translation regulation, posttranslational modifications and stability of proteins, genetic conditions cannot always provide reliable protein expression patterns, suggesting that it may be not sufficient to take advantage of the gene sequence-targeted tools to explore the molecular mechanisms underlying the GC initiation and progression[14]. Thus, proteomics has been thrust into the spotlight for its accurate and direct presentation of protein expression patterns that can provide much more information about the cellular function or dysfunction compared with genetic analysis[15]. It has been widely accepted that GC initiation can be divided into five stages including normal gastric mucosa, atrophic gastritis with mild dysplasia, atrophic gastritis with moderate dysplasia, atrophic gastritis with severe dysplasia, and mucosal GC. To date, abnormal expression of many proteins has been reported to be involved in GC carcinogenesis in several GC proteomic studies[12]. However, all of the previous studies only focused on identifying the differences between normal gastric epithelial tissues and GC rather than investigating the differences of protein among mild dysplasia, moderate dysplasia, severe dysplasia, and mucosal GC, which may contribute to more accurate insight into the molecular mechanisms underlying GC initiation. Thus, in this study we collected specimens of normal gastric mucosa, gastric mucosa with mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal GC, and utilized label-free quantification technology integrated with liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to identify differentially expressed proteins in the five kinds of specimens in order to determine the molecular mechanisms underlying GC initiation, which may promote potential preventive and therapeutic strategies for GC. We also focused on ERGIC1 and DNA-PKcs proteins for their inverse expression patterns identified in the proteomic analysis.
MATERIALS AND METHODS
Clinical specimens
Clinical specimens were collected at the Second Hospital of Lanzhou University from 2010 to 2015. A total of 160 patients were enrolled in this study, including 30 patients with a normal gastric mucosa, 30 patients with atrophic gastritis accompanied by mild dysplasia, 30 patients with atrophic gastritis accompanied by moderate dysplasia, 30 patients with atrophic gastritis accompanied by severe dysplasia, and 40 patients with early intestinal-type GC (Table 1 and Figure 1). All patients underwent upper gastroduodenoscopy or gastrectomy and were pathologically reviewed and examined. This study was approved ethically by the Second Hospital of Lanzhou University. All of the patients provided written informed consent to participate in the study.
Table 1.
Basic demographic data of the study subjects
| Group | n | Age (range) (yr) | Male/female | |
| Healthy | 30 | 43 (38-51) | 16/14 | |
| Mild dysplasia | 30 | 54 (42-67) | 22/18 | |
| Moderate dysplasia | 30 | 58 (40-65) | 14/16 | |
| Severe dysplasia | 30 | 63 (43-76) | 19/11 | |
| Gastric cancer | 40 | 61 (39-68) | 23/17 | |
Figure 1.
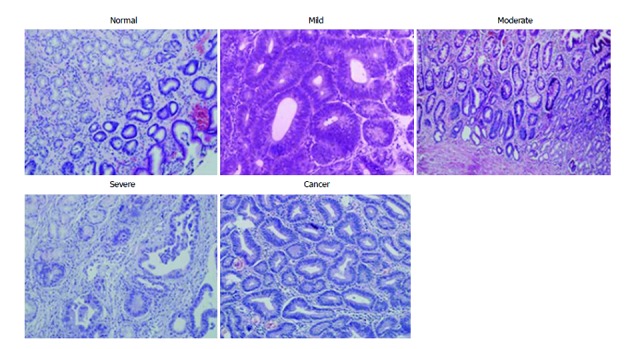
Representative graphs of HE staining (20 ×) of various gastric tissues including dysplasia.
Proteomic analysis of gastric samples
Sample preparation: The five kinds of formalin-fixed, paraffin-embedded (FFPE) specimens were divided into three groups, respectively. Each FFPE specimen was cut into six pieces at a thickness of at least 4 μm, in order to ensure that the protein quantity to detect was no less than 40 μg. Fifteen groups of FFPE specimens were deposited in liquid nitrogen after conventional dewaxing, gradient alcohol, and hydration for proteomic analysis.
After 2 μL of SDT buffer was added, the sample was ground with a pestle and a mortar. The homogenate was sonicated and then boiled for 15 min. After centrifugation at 14000 g for 40 min, the supernatant was filtered with 0.22 μm filters. The protein content of the filtrate was quantified with the BCA Protein Assay Kit (Bio-Rad, the United States), and the sample was then stored at -80 °C.
SDS-PAGE separation: Samples (40 μg proteins) were mixed with 5 × loading buffer and boiled for 5 min. The proteins were separated on a 12.5% SDS-PAGE gel at a constant current of 14 mA for 90 min. Protein bands were visualized by Coomassie Brilliant Blue R-250 staining.
Filter-aided sample preparation (FASP digestion): A total of 100 μg proteins for each sample were added into 30 μL SDT buffer (4% SDS, 100 mmol/L DTT, 150 mmol/L Tris-HCl, pH 8.0). The detergent, DTT and other low-molecular-weight components were removed using UA buffer (8 mol/L urea, 150 mmol/L Tris-HCl pH 8.0) by repeated ultrafiltration (Microcon units, 10 kDa). Then, 100 μL of iodoacetamide (100 mmol/L IAA in UA buffer) was added to block reduced cysteine residues and the samples were incubated for 30 min in darkness. The filters were washed with 100 μL of UA buffer three times and then 100 μL of 25 mmol/L NH4HCO3 buffer twice. Finally, the protein suspensions were digested with 4 μg of trypsin (Promega) in 40 μL of 25 mmol/L NH4HCO3 buffer overnight at 37 °C, and the resulting peptides were collected as a filtrate. The peptides of each sample were desalted on C18 Cartridges (Empore SPE Cartridges C18 (standard density), bed I.D. 7 mm, volume 3 mL, Sigma), concentrated by vacuum centrifugation, and reconstituted in 40 μL of 0.1% (v/v) formic acid. The peptide content was estimated by UV light spectral density at 280 nm using an extinction coefficient of 1.1 of 0.1% (g/L) solution that was calculated on the basis of the frequency of tryptophan and tyrosine in vertebrate proteins.
Mass spectrometry: Each fraction was injected for nanoLC-MS/MS analysis. The peptide mixture was loaded onto a reverse phase trap column (Thermo Scientific Acclaim PepMap100, 100 μm × 2 cm, nanoViper C18) connected to a C18-reversed phase analytical column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin) in buffer A (0.1% formic acid) and separated with a linear gradient of buffer B (84% acetonitrile and 0.1% Formic acid) at a flow rate of 300 NL/min controlled by IntelliFlow technology (one hour gradient: 0%-35% buffer B for 50 min, 35%-100% buffer B for 5 min, and held in 100% buffer B for 5 min).
LC-MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Scientific) that was coupled to Easy nLC (Proxeon Biosystems, now Thermo Fisher Scientific) for 120 min. The mass spectrometer was operated in positive ion mode. MS data were acquired using a data-dependent top10 method that dynamically chooses the most abundant precursor ions from the survey scan (300-1800 m/z) for high energy collision dissociation (HCD) fragmentation. Automatic gain control target was set at 3 × 106, and maximum inject time set at 10 ms. Dynamic exclusion duration was 40.0 s. Survey scans were acquired at a resolution of 70000 at m/z 200. The resolution for HCD spectra was set at 17500 at m/z 200, and isolation width was 2 m/z. Normalized collision energy was 30 eV and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled (Figure 2).
Figure 2.
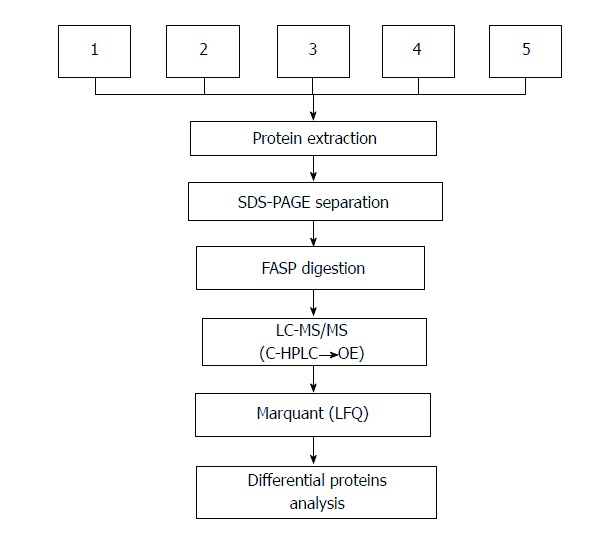
Outline of proteomic experimental workflow. SDS-PAGE: Sodium dodecyl sulfate polyacrylamide gel electrophoresis; C-HPLC: Capillary-high performance liquid chromatography; QE: Q-exactive; LFQ: Label free quantitative.
Data analysis: The MS data were analyzed using MaxQuant software version 1.3.0.5 (Max Planck Institute of Biochemistry in Martinsried, Germany).
Immunohistochemistry
Primary antibodies were used in this study as follows: mouse monoclonal anti-human DNA-PKcs (GR190339-3; Abcam, Cambridge, United Kingdom) and anti-human ERGIC1 (GR104221-6; Anbobio, San Francisco, CA, the United States). The anti-human DNA-PKcs antibody was diluted 1:400, and the anti-human ERGIC1 antibody diluted 1:3500. The paraffin sections were mounted on slides, dewaxed in xylene, and sequentially dehydrated in 100%, 95% and 85% ethanol. The sections were stained using the PV-6000 Polymer Detection System (Zhongshan Goldenbridge, Beijing, China). Initially, endogenous peroxidase was blocked using 3% H2O2. After the sections were incubated with the primary antibody overnight at 4 °C, they were washed with PBS and then incubated with polymer helper for 30 min and poly peroxidase-anti-mouse/rabbit IgG for 30 min. After that, the sections were washed with PBS, and then incubated with 3,3-diaminobenzidine (DAB, Zhongshan Goldenbridge). The sections incubated with PBS without primary antibodies were used as negative controls. Finally, the sections were counterstained with hematoxylin and examined under a light microscope. The cells that were stained a yellow or brown color in the nucleus and/or cytoplasm were defined as positive. Five randomly selected fields per section were analyzed. In a randomly selected field from representative areas, the immunoreactive cells among 100 cells were assessed and quantified by percentage. Then, the average percentage of the five fields was used to assess the area of immunostaining (0 = 0%-5%; 1 = 6%-25%; 2 = 26%-50%; 3 =51%-75%; 4 = 76%-100%). In addition, the intensity of immunostaining was also semi-quantitatively assessed (0 = negative, 1 = weak, 2 = moderate, 3 = intense). Then, the scores from the ‘‘area × intensity’’ were calculated and used to describe the overall staining intensity that semi-quantitatively reflects the overall expression levels of proteins. The overall staining intensity was scored as follows: negative (-): 0-2; mild (+): 3-5; moderate (++): 6-8; and strong (+++): 9-12. All the sections were assessed and scored by two pathologists who were blinded to the clinical data of the patients.
Statistical analysis
All the data are expressed as mean ± SD or percentage. The χ2 test (SPSS v.16.0 for Windows; SPSS, Inc, Chicago, IL, United States) was used to evaluate the difference between categorical variables. A P-value less than 0.05 was considered statistically significant.
RESULTS
Differentially expressed proteins detected by LC-MS/MS
To identify differentially expressed proteins in normal gastric mucosa, atrophic gastritis with mild dysplasia, atrophic gastritis with moderate dysplasia, atrophic gastritis with severe dysplasia, and early mucosal GC, we performed a study with label-free quantification technology integrated with LC-MS/MS. A total of 17443 peptides matching 2807 proteins were identified from tissue analysis. The expression of a total of 365 proteins, with a threshold of a 1.2-fold change and a P-value less than 0.05 between mild dysplasia, moderate dysplasia, severe dysplasia or early mucosal GC and matched normal gastric mucosa tissues, were considered to be statistically significantly different.
Aberrant expression of DNA-PKcs and ERGIC1 revealed by proteomic analysis
The fold changes in mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal GC compared to matched normal gastric mucosa tissues were 1.12, 1.54, 1.6, and 2.3 for DNA-PKcs, and 1.18, 0.84, 0.73, and 0.46 for ERGIC1, respectively (Table 2).
Table 2.
Results of DNA-PKcs and ERGIC1 from proteomic analysis
Aberrant expression of DNA-PKcs and ERGIC1 revealed by immunohistochemistry
In order to confirm the results of DNA-PKcs and ERGC1 from proteomic analysis, we further examined the expression of DNA-PKcs and ERGC1 by immunohistochemistry. The strongly positive (+++) rates in normal gastric mucosa, mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal GC were 80% (24/30), 73% (22/30), 0% (0/30), 0% (0/30), and 0% (0/40), respectively, for ERGC1, and for DNA-PKcs the strongly positive (+++) staining was observed only in the sections of early mucosal GC. The moderately positive (++) rates in normal gastric mucosa, mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal GC were 6.6% (2/30), 6.6% (3/30), 83% (25/30), 67% (20/30), and 10% (4/40), respectively, for ERGC1, and 0% (0/30), 0% (0/30), 76%(23/30), 83% (25/30), and 20% (6/30), respectively, for DNA-PKcs (Figures 3 and 4). The details on the rates of different-grade overall staining intensity are presented in Table 3.
Figure 3.

Immunohistochemical staining of DNA-PKcs (40 ×). A: Normal; B: Mild dysplasia; C: Moderate dysplasia; D: Severe dysplasia; E: Early mucosal cancer.
Figure 4.

Immunohistochemical staining of ERGIC1 (40 ×). A: Normal; B: Mild dysplasia; C: Moderate dysplasia; D: Severe dysplasia; E: Early mucosal cancer.
Table 3.
Results of DNA-PKcs and ERGIC1 from immunohistochemistry
| Group | n |
DNA-PKcs |
n |
ERGIC1 |
||||||
| - | + | ++ | +++ | - | + | ++ | +++ | |||
| Normal | 30 | 12 | 18 | 0 | 0 | 30 | 2 | 2 | 2 | 24 |
| Mild | 30 | 14 | 16 | 0 | 0 | 30 | 2 | 3 | 3 | 22 |
| Moderate | 30 | 5 | 3 | 23 | 0 | 30 | 2 | 3 | 25 | 0 |
| Severe | 30 | 3 | 2 | 25 | 0 | 30 | 5 | 5 | 20 | 0 |
| Cancer | 30 | 0 | 2 | 6 | 22 | 40 | 9 | 27 | 4 | 0 |
In general, the average immunohistochemistry scores of the DNA-PKcs protein significantly increased, while those of ERGC1 protein significantly decreased along the sequence of normal gastric mucosa, mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal GC.
DISCUSSION
Using label-free quantification technology combined with LC-MC/MC, the expression of 365 proteins based on the tendency of fold change was revealed to be statistically different between various stages of GC initiation. Furthermore, we observed that ERGIC1 expression decreased, while DNA-PKcs expression increased gradually along with different stages of GC initiation based on the tendency of fold change, suggesting that abnormal ERGIC1 and DNA-PKcs expression may play an important role in GC initiation.
It has been reported that DNA-PKcs plays an important role in the activation of the NHEJ pathway[9,10]. When a DNA damage signal in cells is sent out, DNA-PKcs will recognize it and initiate a damage response in the first place, followed by the event that Ku 70/80 binds to the damaged DNA ends and induces DNA-PKcs to form DNA-PK that triggers NHEJ repair activities[16]. NHEJ is an error-prone and non-specific DNA repair mechanism and can be induced before homologous recombination, whose excessive activation has the capability of regulating cell cycle arrest, cell apoptosis, chromosome recombination, and genome instability, all of which are closely related with carcinogenesis[17,18]. Dysregulation of DNA-PKcs has been reported to be associated with pathological processes in various cancers[19]. Consistent with the previous study indicating that DNA-PKcs expression in GC was up-regulated compared with normal gastric mucosa, and associated with GC progression[20], up-regulation of DNA-PKcs expression was also found in our study. Furthermore, our study showed more information that DNA-PKcs expression increased along with different stages of GC initiation. Hence, it is possible to hypothesize that enhanced NHEJ resulting from the overexpression of DNA-PKcs may contribute to GC initiation and progression.
ERGIC1, a cycling protein, plays a part in protein secretion out of the ER by participating in membrane trafficking and selective transport of cargo between the ER, the intermediate compartment, and the Golgi apparatus[11]. ER stress is defined as a disequilibrium between protein folding ability of the ER and protein load, causing the accumulation of misfolded and unfolded proteins[21]. Many factors, including hypoxia, starvation, infections, changes in secretary needs and so on, throw a challenge to the folding capacity of the cell and subsequently trigger ER stress[22]. ER stress has been speculated to be involved in many cancers including GC[23]. ER stress is capable of activating NF-κB, ROS, and JNK signal pathways[24-27], resulting in chronic inflammatory response that contributes to carcinogenesis. Besides, accumulating evidence indicated that ER stress response could facilitate cancer progression probably by promoting tumor angiogenesis and malignant cell autophagy, and mitigating apoptosis[28-31]. In this study, we observed that ERGIC1 expression decreased gradually along with different stages of GC initiation. Down-regulation of ERGIC1 expression may affect the protein secretion function of the ER and then disturb ER homeostasis, which may cause the accumulation of unfolded and misfolded proteins, finally resulting in ERS. Thus, it is possibly reasonable to speculate that down-regulation of ERGIC1 expression may contribute to GC initiation by promoting ERS in gastric mucosal epithelial cells. Besides, some studies suggested that ER stress response can also induce the apoptosis of malignant cells[32-34], indicating that ER stress response may play a dual role in cancers. Furthermore, a study by Vainio et al[35] indicated that ERGIC1 expression was up-regulated in prostate cancer samples and knockdown of ERGIC1 could inhibit the ERG oncogene expression in prostate cancer cells in vitro, which suggested that the change tendency of ERGIC1 expression in cancers may be different, depending on the genomic or biological function of cancer cells. Therefore, another possible molecular mechanism responsible for the role of down-regulation of ERGIC1 in the GC initiation is that down-regulation of ERGIC1 perhaps disturbs the expression of some oncogenes or anti-oncogenes. Additionally, despite that there are some specific relationships between ERGIC1 and DNA-PKcs based on the inverse expression patterns observed in the current study, we did not provide adequate evidence to confirm that. Thus, further studies are required to elucidate the mechanisms by which down-regulation of ERGIC1 contributes to the initiation and progression of GCs and the internal link between ERGIC1 and DNA-PKcs.
In conclusion, our study conducted a systematic proteomic analysis of normal gastric mucosa, gastric mucosa with mild dysplasia, gastric mucosa with moderate dysplasia, gastric mucosa with severe dysplasia and early mucosal GC, which covered the whole sequential process of GC initiation, and suggested that the aberrant DNA-PKcs and ERGIC1 expression may be involved in the initiation of GC.
COMMENTS
Background
Gastric cancer (GC) is a global malignant disease with high incidence and mortality. The carcinogenesis of GC has been defined as a biological process involving polygene-driven, multi-step, and multi-stage events. However, the molecular mechanisms underlying the carcinogenesis of GC remain unknown.
Research frontiers
Using proteomics analysis, the current study aimed to investigate the molecular mechanisms of GC carcinogenesis.
Innovations and breakthroughs
The authors first report that aberrant ERGIC1 and DNA-PKcs expression may be involved in GC initiation.
Applications
Using label-free quantification technology combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS), the authors observed that ERGIC1 expression decreased, while DNA-PKcs expression increased gradually along with different stages of GC initiation based on the tendency of fold change. The expression patterns of ERGIC1 and DNA-PKcs revealed by immunohistochemistry were consistent with the LC-MS/MS results.
Peer-review
The authors demonstrated that that ERGIC1 expression decreased, while DNA-PKcs expression increased gradually along with different stages of GC initiation. A total of 160 specimens were enrolled in this study to identify differentially expressed proteins in normal gastric mucosa, gastric mucosa with mild dysplasia, moderate dysplasia, severe dysplasia, and early mucosal GC. These results are interesting and very valuable to verify the molecular mechanisms of GC carcinogenesis.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was approved by the Second Hospital of Lanzhou University.
Informed consent statement: All specimens from the patients were taken after informed consent and ethical permission were obtained for participation in the study.
Conflict-of-interest statement: To the best of our knowledge, no conflict of interest exists.
Data sharing statement: No additional data are available.
Peer-review started: April 8, 2017
First decision: May 12, 2017
Article in press: August 2, 2017
P- Reviewer: Aoyagi K S- Editor: Gong ZM L- Editor: Wang TQ E- Editor: Huang Y
Contributor Information
Fu-Rong Wang, School of Life Sciences, Lanzhou University, Lanzhou 730000, Gansu Province, China; Department of Pathology, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China.
Yu-Cai Wei, Department of General Surgery, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China.
Zhi-Jian Han, The Key Laboratory of the Digestive System Tumors of Gansu Province, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China.
Wen-Ting He, The Key Laboratory of the Digestive System Tumors of Gansu Province, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China.
Xiao-Ying Guan, Department of Pathology, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China.
Hao Chen, Department of General Surgery, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China.
Yu-Min Li, School of Life Sciences, Lanzhou University, Lanzhou 730000, Gansu Province, China; Department of General Surgery, Second Hospital of Lanzhou University, Lanzhou 730000, Gansu Province, China. liym@lzu.edu.cn.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009;374:477–490. doi: 10.1016/S0140-6736(09)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miao RL, Wu AW. Towards personalized perioperative treatment for advanced gastric cancer. World J Gastroenterol. 2014;20:11586–11594. doi: 10.3748/wjg.v20.i33.11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koessler T, Roth A, Cacheux W. [Early gastric cancer: epidemiology, diagnostic and management] Rev Med Suisse. 2014;10:1118–1122. [PubMed] [Google Scholar]
- 5.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 6.Bosetti C, Bertuccio P, Malvezzi M, Levi F, Chatenoud L, Negri E, La Vecchia C. Cancer mortality in Europe, 2005-2009, and an overview of trends since 1980. Ann Oncol. 2013;24:2657–2671. doi: 10.1093/annonc/mdt301. [DOI] [PubMed] [Google Scholar]
- 7.Riquelme I, Saavedra K, Espinoza JA, Weber H, García P, Nervi B, Garrido M, Corvalán AH, Roa JC, Bizama C. Molecular classification of gastric cancer: Towards a pathway-driven targeted therapy. Oncotarget. 2015;6:24750–24779. doi: 10.18632/oncotarget.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shrivastav M, Miller CA, De Haro LP, Durant ST, Chen BP, Chen DJ, Nickoloff JA. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst) 2009;8:920–929. doi: 10.1016/j.dnarep.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieber MR, Gu J, Lu H, Shimazaki N, Tsai AG. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell Biochem. 2010;50:279–296. doi: 10.1007/978-90-481-3471-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breuza L, Halbeisen R, Jenö P, Otte S, Barlowe C, Hong W, Hauri HP. Proteomics of endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membranes from brefeldin A-treated HepG2 cells identifies ERGIC-32, a new cycling protein that interacts with human Erv46. J Biol Chem. 2004;279:47242–47253. doi: 10.1074/jbc.M406644200. [DOI] [PubMed] [Google Scholar]
- 12.Leal MF, Wisnieski F, de Oliveira Gigek C, do Santos LC, Calcagno DQ, Burbano RR, Smith MC. What gastric cancer proteomic studies show about gastric carcinogenesis? Tumour Biol. 2016;37:9991–10010. doi: 10.1007/s13277-016-5043-9. [DOI] [PubMed] [Google Scholar]
- 13.Cui J, Yin Y, Ma Q, Wang G, Olman V, Zhang Y, Chou WC, Hong CS, Zhang C, Cao S, et al. Comprehensive characterization of the genomic alterations in human gastric cancer. Int J Cancer. 2015;137:86–95. doi: 10.1002/ijc.29352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li JJ, Qi RZ, Ng GK, Xie D. Proteomics in gastric cancer research: Benefits and challenges. Proteomics Clin Appl. 2009;3:185–196. doi: 10.1002/prca.200800151. [DOI] [PubMed] [Google Scholar]
- 15.Leal ML, Assumpção PP, Smith MAC, Burbano RR. Searching for gastric cancer biomarkers through proteomic approaches. J Gastroenterol Hepatol Res. 2014;3:989–995. [Google Scholar]
- 16.Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, Ballarati L, Vergult S, Giardino D, Hansson K, et al. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep. 2012;1:648–655. doi: 10.1016/j.celrep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 18.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 19.Abe T, Ishiai M, Hosono Y, Yoshimura A, Tada S, Adachi N, Koyama H, Takata M, Takeda S, Enomoto T, et al. KU70/80, DNA-PKcs, and Artemis are essential for the rapid induction of apoptosis after massive DSB formation. Cell Signal. 2008;20:1978–1985. doi: 10.1016/j.cellsig.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Li W, Xie C, Yang Z, Chen J, Lu NH. Abnormal DNA-PKcs and Ku 70/80 expression may promote malignant pathological processes in gastric carcinoma. World J Gastroenterol. 2013;19:6894–6901. doi: 10.3748/wjg.v19.i40.6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly E, Greene CM, Carroll TP, McElvaney NG, O’Neill SJ. Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant alpha1-antitrypsin deficiency. J Biol Chem. 2009;284:16891–16897. doi: 10.1074/jbc.M109.006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283:2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 23.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen WT, Zhu G, Pfaffenbach K, Kanel G, Stiles B, Lee AS. GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene. 2014;33:4997–5005. doi: 10.1038/onc.2013.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013;332:249–264. doi: 10.1016/j.canlet.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Joe Y, Jeong SO, Zheng M, Back SH, Park SW, Ryter SW, Chung HT. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 2014;20:799–815. doi: 10.1177/1753425913508593. [DOI] [PubMed] [Google Scholar]
- 28.Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25:563–573. doi: 10.1016/j.ccr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D, Diehl JA. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–3895. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nör JE, Polverini PJ. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012;72:5396–5406. doi: 10.1158/0008-5472.CAN-12-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P, Ashkenazi A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue H, Lu J, Yuan R, Liu J, Liu Y, Wu K, Wu J, Du J, Shen B. Knockdown of CLIC4 enhances ATP-induced HN4 cell apoptosis through mitochondrial and endoplasmic reticulum pathways. Cell Biosci. 2016;6:5. doi: 10.1186/s13578-016-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei Y, Henderson BR, Emmanuel C, Harnett PR, deFazio A. Inhibition of ANKRD1 sensitizes human ovarian cancer cells to endoplasmic reticulum stress-induced apoptosis. Oncogene. 2015;34:485–495. doi: 10.1038/onc.2013.566. [DOI] [PubMed] [Google Scholar]
- 34.Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013;18:537–546. doi: 10.1007/s10495-013-0818-6. [DOI] [PubMed] [Google Scholar]
- 35.Vainio P, Mpindi JP, Kohonen P, Fey V, Mirtti T, Alanen KA, Perälä M, Kallioniemi O, Iljin K. High-throughput transcriptomic and RNAi analysis identifies AIM1, ERGIC1, TMED3 and TPX2 as potential drug targets in prostate cancer. PLoS One. 2012;7:e39801. doi: 10.1371/journal.pone.0039801. [DOI] [PMC free article] [PubMed] [Google Scholar]