

Abstract
Neuronal firing patterns and frequencies determine the nature of encoded information of the neurons. Here we discuss the molecular identity and cellular mechanisms of spike-frequency adaptation in central nervous system (CNS) neurons. Calcium-activated potassium (KCa) channels such as BKCa and SKCa channels have long been known to be important mediators of spike adaptation via generation of a large afterhyperpolarization when neurons are hyper-activated. However, it has been shown that a strong hyperpolarization via these KCa channels would cease action potential generation rather than reducing the frequency of spike generation. In some types of neurons, the strong hyperpolarization is followed by oscillatory activity in these neurons. Recently, spike-frequency adaptation in thalamocortical (TC) and CA1 hippocampal neurons is shown to be mediated by the Ca2+-activated Cl- channel (CACC), anoctamin-2 (ANO2). Knockdown of ANO2 in these neurons results in significantly reduced spike-frequency adaptation accompanied by increased number of spikes without shifting the firing mode, which suggests that ANO2 mediates a genuine form of spike adaptation, finely tuning the frequency of spikes in these neurons. Based on the finding of a broad expression of this new class of CACC in the brain, it can be proposed that the ANO2-mediated spike-frequency adaptation may be a general mechanism to control information transmission in the CNS neurons.
Keywords: spike-frequency adaptation, anoctamin-2, calcium-activated chloride channel, calcium-activated potassium channel, afterhyperpolarization, thalamocortical neuron
INTRODUCTION
In neurons, action potentials, also often referred to as spikes, play a central role in communication among a population of neurons by providing for the propagation of signals along the axon of the neurons towards the axon terminals, which can then connect with other neurons at synapses. A typical feature of neurons is their ability to encode neuronal information through controlling the generation of spikes. A dynamic change in the pattern and frequency of spikes in a neuron articulates its own story to the projecting neurons. The firing patterns and frequency of a neuron can be transformed according to external stimuli and intrinsic properties such as expression levels of various ion channels in the neuronal plasma membrane.
Spike adaptation refers to the progressive slowing of the frequency of discharge of action potentials following an initial high frequency of spikes during an extended period of excitation. This adaptation pattern of neurons can be clearly observed in the presence of sustained injections of depolarizing rectangular currents. In some types of neurons, the adaptation is so marked that despite the presence of a depolarizing current, only a small number of spikes are initiated. In most cases, spike adaptation appears with a slowly-developed hyperpolarizing current following an extended period of excitation of neurons [1,2,3]. Spike-frequency adaptation has been shown to play an important role in neural coding. Spike adaptation is also called spike accommodation. The accommodation index is a metric used to describe spike train data similar to other measures of accommodation such as local variance. Since its introduction by Shinomoto et al. [4], it has been modified to indicate the difference in the length of two consecutive interspike intervals (ISIs) in the initial stage to those in the last stages of neural spikes (Fig. 1A). For example, the accommodation index calculated from the ISI between the first two spikes over the ISI between the last two spikes would be smaller than 1. An index value closer to 1 would indicate that spike adaptation did not occur whereas an index value closer to 0 would indicate a distinct spike accommodation with prolongation in the ISIs. The detailed biophysical mechanisms that regulate spike adaptation in central nervous system (CNS) neurons are not fully understood. The extended period of excitation of neurons often accompanies an increase of intracellular calcium concentration via voltage-dependent calcium channels. Therefore, spike adaptation has been ascribed to the slow-type afterhyperpolarization (AHP) current activated by calcium-activated channels.
Fig. 1. Ca2+-dependent spike frequency adaptation and adaptation index. (A) TC neuron with a depolarizing current injection to induce tonic firing in 2.4 mM Ca2+ buffer displays the prolongation of inter-spike intervals (ISI). Adaptation index can be obtained from the ration of 1st ISI over nth ISI. (B) Replacement of extracellular buffer to Ca2+-free buffer abolishes the spike-frequency adaptation.

CALCIUM-ACTIVATED POTASSIUM CHANNELS
It has long been known that two types of potassium currents, i.e., the voltage-sensitive potassium current (I(M)) developed at the end of an action potential and calcium-sensitive potassium current slowly developed with prolonged excitation, shape spiking adaptation and thus the spiking pattern of neurons [5]. Slow AHP currents can be further categorized into medium AHP (mAHP) and very slow AHP (sAHP) currents, with decay kinetics of approximately hundreds of milliseconds and over seconds, respectively. Of these two types of currents, mAHP is known to be mediated by small conductance (SK) or large conductance (BK) Ca2+-activated K+ channels in several types of neurons (Fig. 2) [6,7,8,9].
Fig. 2. Ca2+-activated potassium channels mediate afterhyperpolarization. Ca2+-activated potassium channels such as BKCa and SKCa channels mediates afterhyperpolarization. BKCa channels are directly activated by Ca2+ binding where SKCa channels are activated by Ca2+-calmodulin. They are functionally linked to various voltage-gated Ca2+ channels depending on neuronal types.
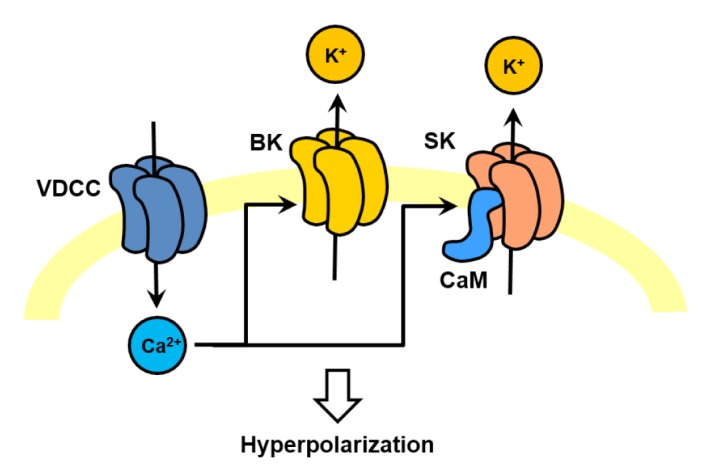
Calcium-activated potassium (KCa) channels activated by increase in intracellular calcium concentration [Ca2+]in, give rise to an efflux of potassium. The activation of KCa channels then hyperpolarizes the membrane potential to abolish the spikes and also feeds back onto [Ca2+]in by limiting the calcium influx either through deactivation of voltage-gated calcium channels because the reversal potential of potassium channels are near -90 to -100 mV, far-hyperpolarized than the resting membrane potential of neurons [10]. Based on their biophysical properties, KCa channels have been classified into two subtypes: BKCa, exhibiting large unitary conductance (200–400 pS) and gated by the cooperative action of membrane depolarization and [Ca2+]in, and SKCa, displaying small conductance (2–20pS) and gated solely by [Ca2+]in [11,12].
In CNS neurons, where they exhibit a broad expression pattern throughout most regions of the brain, the BKCa channels contribute to repolarization of action potentials, mediate the fast phase of AHP following an action potential in hippocampal [13] and cerebellar Purkinje [14] neurons, shape the dendritic calcium spikes, and influence the release of neurotransmitters. BKCa channels from mouse and rat brain assembled from BKalpha and BKbeta subunits 2 and 4 were found to be tightly associated with the voltagegated Ca2+ channels Cav1.2 (L-type channels), Cav2.1 (P/Q-type channels), and Cav2.2 (N-type channels) [15,16]. The formation of BKCa-Cav complex provided an insight into the molecular mechanisms. Activation of BKCa channels is rendered virtually independent of the global cellular calcium environment, but rather requires local delivery of micromolar [Ca2+]in that should not affect other calcium-dependent processes. The selective molecular coupling between Cav and BKCa channels may ensure specificity of calcium-mediated signaling in tissue specificity. For example, cerebellar Purkinje cells expressing BKCa channels tightly linked with Cav2.1 channels display relatively short action potentials allowing for higher firing rates [17,18], while hormone-secreting chromaffin cells have broad action potentials due to expression of Cav1.2 and Cav2.1 channels [19]. In hippocampal neurons, activation of BK channels was shown to underlie the falling phase of an action potential and generation of fast AHP whereas SK channel activation generates slow AHP after a burst of action potentials [20]. In this study, BKCa channels were found to be linked to CaV2.1 Ca2+ channels and SKCa channels to CaV1.2 Ca2+ channels [20].
The alpha subunit of SKCa channels, although shares the tetrameric six-transmembrane domain architecture of voltage-gated cation channels, lacks the typical features of the voltage-sensing S4 segments [21]. Consequently, the gating of SKCa channels is completely independent of the transmembrane voltage, in contrast to BKCa channels. Instead, opening and closing of SKCa channels is solely driven by changes in [Ca2+]in, with submicromolar concentrations being sufficient to effectively gate the channels. Calmodulin was identified as an exogenous calcium sensor that is constitutively associated with SKalpha [22]. Although a number of other structurally related calcium-binding proteins of the EF-hand protein family are expressed in the brain, proteomic studies have underlined the exclusive role of calmodulin as the calcium sensor of native SKCa channels.
SKCa channels appear to be selectively activated by L-type Cav channels in the somata of CA1 pyramidal cells [20], by R-type Cav channels in the dendritic spines of CA1 [23] and lateral amygdala neurons [24], or by T-type Cav channels and SERCAs in the dendrites of thalamic reticular nucleus (TRN) neurons [25]. SKCa channels have often been localized to the dendrites in neurons where they are also shown to be functionally linked to postsynaptic receptors such as NMDA receptors [26]. This may implicate the role of SKCa channels in controlling the general excitability of the neurons responding to the incoming excitable inputs. In TRN neurons, the coupling of T-type channels, SK2 channels, and SERCAs in dendrites comprises a specialized Ca2+ signaling triad to regulate the oscillatory dynamics of membrane potentials [25]. The lack of SK2 channels abolished the oscillatory spike discharges in TRN neurons and also led to a drastic reduction of low-frequency rhythms in the electroencephalogram of non-rapid-eye-movement sleep in animals.
CALCIUM-ACTIVATED CHLORIDE CHANNELS
Of the anoctamin family, also known as transmembrane protein (TMEM) 16 family, anoctamin 1 (ANO1, TMEM 16A) and 2 (ANO2, TMEM 16B) were identified as calcium-activated chloride channels (CACCs) relatively latter compared to the identification of KCa channels [27,28]. ANO1 and ANO2 are widely expressed in different tissues and are involved in various physiological conditions such as contraction of smooth muscles, control of blood pressure, control of cardiac excitability, signal transduction in olfactory and sensory neurons, and cell proliferation [29]. However, the expression and function of ANO1 and ANO2 in the brain have not been studied extensively. ANO1 has been reported not expressed in brain region [28], but recent study showed that ANO1 expressed in auditory brain stem nuclei and contributed to the high-frequency synaptic transmission of auditory signals [30], Also, it is confirmed that ANO2 expressed in various brain regions such as hippocampus [31], thalamus [32], cerebellum [33], and cortex [34].
Based on the intracellular chloride concentration ([Cl-]in), the activation of CACCs induces depolarization or hyperpolarization of cells. The relative ratio of Cl- transporter subtypes expressed on the cell membrane influences on the overall concentration of [Cl-]in and the corresponding driving force. Peripheral neurons and immature CNS neurons have the higher expression of Na+-K+-Cl- cotransporter, NKCC, which mediates Cl- influx and have higher [Cl-]in owing to the activity of NKCC2 [35]. Therefore, the activation of CACCs triggers an outward chloride flow, resulting in depolarization of the membrane potential. ANO2, expressed at the presynaptic end of the olfactory epithelium [36] and retinal photoreceptors [37], mediates transepithelial Cl- secretion and presynaptic Ca2+-activated depolarization of the membrane potential via the efflux of Cl-. In contrast, the high expression of K+/Cl- cotransporter (KCC) of mature CNS neurons could extrude Cl- and maintain low [Cl-]in [38]. The activation of CACCs results in an inward flow of Cl- into the CNS neurons, hyperpolarizing them. It was recently found that ANO2 in thalamocortical (TC) and CA1 hippocampal neurons mediates spike-frequency adaptation by generating a hyperpolarizing current (Fig. 2) [31,32]. TC neurons that respond to a long depolarizing current input displayed Ca2+-dependent prolongation of inter-spike intervals (Fig. 1A) [32]. Knockdown of ANO2 in TC neurons reduced this spike frequency adaptation, and significantly decreased the mAHP currents (Fig. 1B). Also, ANO2 in hippocampal pyramidal neurons could adjust the extent of synaptic excitation and control the waveform of the action potential [31]. This group showed that ANO2 regulated the duration of action potential by acting on repolarization steps whereas another study indicated that the activation of ANO2 channel by persistent stimulus modulated the probability of action potentials by shunting effect. However, both studies agreed that the opening of ANO2 channel could induce hyperpolarization of neurons by Cl- influx and play an important role for breaker of excitatory responses.
Several factors contribute to the different roles of peripheral and TC neuron CACCs. First, the reversal potential of Cl- in TC neurons with intact [Cl-]in is about -70 mV. This potential approximates the resting membrane potential of TC neurons, and is lower than the threshold required to activate voltage-gated Na+ channels. The endogenous [Cl-]in corrected with a relative permeability of 0.2, based on a 15 mM [HCO3-]in typical of CNS neurons, was 5.4 mM. This low [Cl-]in in TC neurons is in agreement with previous reports demonstrating that the KCC2 is highly expressed in TC neurons, and actively results in Cl- efflux [39], leading to hyperpolarization of EGABA [40]. Secondly, ANO2 generates an outwardly rectifying current, while ANO1 manifests a linear I-V curve. Therefore, ANO2 generates a relatively large outward current in neurons with an endogenous ionic content at the depolarized membrane. Moreover, ANO2 activation is relatively less sensitive to Ca2+ compared to that of ANO1, which confers a unique role on ANO2 in the control of excitability of TC neurons. When TC neurons generate spikes at a low frequency, the membrane potential of the neurons remains close to the reversal potential of Cl-, with low levels of Ca2+ influx. Therefore, ANO2 does not conduct substantial currents, and TC neurons generate spikes at regular intervals. When TC neurons generate a barrage of spikes at high frequencies, the membrane of the TC neurons gets depolarized, accompanied by high levels of Ca2+ influx near the soma. Hyperpolarization of the membrane potential then occurs via the ANO2 current, which elongates the inter-spike intervals (Fig. 3). Therefore, ANO2 functions to generate spike-frequency adaptation, resulting in interruption of the excessive firing in TC neurons. The endogenous Cl- reversal indicates that ANO2 mediates AHP current conduction in TC neurons, which might be assisted by the outwardly rectifying characteristic of the ANO2 channels. This phenotype was also observed in the knockdown of ANO2 in CA1 hippocampal neurons, providing further evidence that Ca2+-activated Cl- conductance via ANO2 channels hyperpolarizes the membrane potential in these CNS neurons.
Fig. 3. Ca2+-dependent spike frequency adaptation mediated by Ca2+-activated chloride channels in TC and CA1 neurons. When a neuron is highly activated, Ca2+influx via voltage gated Ca2+ channel would increase the local [Ca2+]in in turn activate ANO2. The influx of Cl- caused by the low [Cl-]in in CNS neurons would hyperpolarize membrane potential, which would decrease the spike generation probability.

The thalamus-specific ANO2 knockdown significantly increased visceral pain responses, reflecting the level of sensory information transmission from the thalamus to the cortex [32]. The role of ANO2-mediated spike adaptation, which can be considered as a type of self-inhibition in TC neurons, was emphasized on the basis of considerable increase in pain responses in mice with thalamic-restricted ANO2 knockdown. Interestingly, ANO2 currents restrict excessive spike generation but do not interfere with information transmission by TC neurons up to a certain level of spike-frequency. Spike frequency adaption in neurons has been suggested as a crucial contributor to stimulus encoding by neurons. Specifically, spike adaptation may enable neurons to respond more sensitively to coinciding inputs, or to provide a major contribution to network synchronization, suggesting that ANO2-mediated spike-frequency adaptation in TC neurons may facilitate synchronized TC activity.
The question remains whether spike adaptation mediated by CACC is the general mode of restricting spike generation in the CNS. The role of ANO2-mediated spike adaptation in TC and CA1 neurons can be considered as a type of self-inhibition in neurons. [Cl-]in in neurons could play a crucial role in determining whether the CACC in neurons elicits hyperpolarization or depolarization of membrane potential. Several mature neurons in the CNS are known to have relatively low [Cl-]in. In CNS neurons, the action of potassium-chloride cotransporter KCC2 leads to lower levels of [Cl-]in, and the activation of CACCs induces inward chloride flow to hyperpolarize neurons [38]. Therefore, in the CNS, the influx of chloride ions through CACCs could possibly modulate spike-frequency adaptation via the shunting effect.
Ca2+-MEDIATE SPIKE ADAPTATION IN CNS NEURONS
Spike adaptation is a self-inhibitory mechanism of neurons to control the amount of information transmission. KCa channels have long been known to be important mediators of spike adaptation via generation of large AHP when neurons are hyper-activated. However, the reversal potential under the general extracellular and intracellular potassium content in the CNS is far polarized to the negative value from the threshold potential for action potential generation. Hence, KCa channels such as SKCa and BKCa channels strongly hyperpolarize the membrane potential and thus cease the action potential generation. In some types of neurons, a strong hyperpolarization is followed by the activation of hyperpolarization-activated cation channels generating Ih current. In turn, neurons which express HCN and T-type Ca2+ channels together such as TRN neurons or cerebellar Purkinge neurons are shifted to low-threshold burst firing mode. Therefore, activation of these channels is often related to oscillatory activity in these neurons.
In contrast, ANO2, one of CACCs, mediates spike adaptation without completely abolishing spike generation or shifting the firing mode of neurons to oscillatory mode with a large hyperpolarization. Instead, ANO2 tunes the spike frequency with gradual prolongation of inter-spike intervals. This seems to be possible as the reversal potential for CACCs are not far from the resting membrane potential. Therefore, ANO2 mediate a genuine form of spike adaptation, which limits excessive information transmission to the subsequent neurons, without disabling spike generation by the neurons. In conclusion, it could be suggested that a slow type of spike adaptation via CACCs could be a general mechanism to regulate information in CNS neurons.
ACKNOWLEDGEMENTS
This research was supported by the National Research Foundation (NRF-2017R1A2B3011098 and NRF-2017M3C7A1023471) funded by the government of the Republic of Korea (Ministry of Science, ICT & Future Planning, MSIP), International Collaborative R&D Program, funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea), and the Brain Korea 21 (BK21) PLUS program. GEH is fellowship awardee by BK21 PLUS program.
References
- 1.Partridge LD, Stevens CF. A mechanism for spike frequency adaptation. J Physiol. 1976;256:315–332. doi: 10.1113/jphysiol.1976.sp011327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldissera F, Gustafsson B, Parmiggiani F. Saturating summation of the afterhyperpolarization conductance in spinal motoneurones: a mechanism for ‘secondary range’ repetitive firing. Brain Res. 1978;146:69–82. doi: 10.1016/0006-8993(78)90218-4. [DOI] [PubMed] [Google Scholar]
- 3.Wilson CJ, Weyrick A, Terman D, Hallworth NE, Bevan MD. A model of reverse spike frequency adaptation and repetitive firing of subthalamic nucleus neurons. J Neurophysiol. 2004;91:1963–1980. doi: 10.1152/jn.00924.2003. [DOI] [PubMed] [Google Scholar]
- 4.Shinomoto S, Shima K, Tanji J. Differences in spiking patterns among cortical neurons. Neural Comput. 2003;15:2823–2842. doi: 10.1162/089976603322518759. [DOI] [PubMed] [Google Scholar]
- 5.Velumian AA, Carlen PL. Differential control of three after-hyperpolarizations in rat hippocampal neurones by intracellular calcium buffering. J Physiol. 1999;517:201–216. doi: 10.1111/j.1469-7793.1999.0201z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- 7.Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 9.Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC, Crill WE. Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J Neurophysiol. 1988;59:424–449. doi: 10.1152/jn.1988.59.2.424. [DOI] [PubMed] [Google Scholar]
- 10.Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- 11.Marty A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 1981;291:497–500. doi: 10.1038/291497a0. [DOI] [PubMed] [Google Scholar]
- 12.Blatz AL, Magleby KL. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat skeletal muscle. Nature. 1986;323:718–720. doi: 10.1038/323718a0. [DOI] [PubMed] [Google Scholar]
- 13.Gu N, Vervaeke K, Storm JF. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol. 2007;580:859–882. doi: 10.1113/jphysiol.2006.126367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haghdoost-Yazdi H, Janahmadi M, Behzadi G. Iberiotoxin-sensitive large conductance Ca2+ -dependent K+ (BK) channels regulate the spike configuration in the burst firing of cerebellar Purkinje neurons. Brain Res. 2008;1212:1–8. doi: 10.1016/j.brainres.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 15.Grunnet M, Kaufmann WA. Coassembly of big conductance Ca2+-activated K+ channels and L-type voltage-gated Ca2+ channels in rat brain. J Biol Chem. 2004;279:36445–36453. doi: 10.1074/jbc.M402254200. [DOI] [PubMed] [Google Scholar]
- 16.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 17.Edgerton JR, Reinhart PH. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol. 2003;548:53–69. doi: 10.1113/jphysiol.2002.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Womack MD, Chevez C, Khodakhah K. Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci. 2004;24:8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prakriya M, Lingle CJ. BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J Neurophysiol. 1999;81:2267–2278. doi: 10.1152/jn.1999.81.5.2267. [DOI] [PubMed] [Google Scholar]
- 20.Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 21.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 22.Griffith T, Tsaneva-Atanasova K, Mellor JR. Control of Ca2+ influx and calmodulin activation by SK-channels in dendritic spines. PLOS Comput Biol. 2016;12:e1004949. doi: 10.1371/journal.pcbi.1004949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 24.Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- 25.Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- 26.Faber ES. Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. J Physiol. 2010;588:1281–1292. doi: 10.1113/jphysiol.2009.185645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 28.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 29.Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005;67:719–758. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 30.Cho SJ, Jeon JH, Chun DI, Yeo SW, Kim IB. Anoctamin 1 expression in the mouse auditory brainstem. Cell Tissue Res. 2014;357:563–569. doi: 10.1007/s00441-014-1897-6. [DOI] [PubMed] [Google Scholar]
- 31.Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY. Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron. 2012;74:179–192. doi: 10.1016/j.neuron.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ha GE, Lee J, Kwak H, Song K, Kwon J, Jung SY, Hong J, Chang GE, Hwang EM, Shin HS, Lee CJ, Cheong E. The Ca(2+)-activated chloride channel anoctamin-2 mediates spike-frequency adaptation and regulates sensory transmission in thalamocortical neurons. Nat Commun. 2016;7:13791. doi: 10.1038/ncomms13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, Schmelzeisen S, Parthier D, Frings S, Möhrlen F. Anoctamin calcium-activated chloride channels may modulate inhibitory transmission in the cerebellar cortex. PLoS One. 2015;10:e0142160. doi: 10.1371/journal.pone.0142160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vocke K, Dauner K, Hahn A, Ulbrich A, Broecker J, Keller S, Frings S, Möhrlen F. Calmodulin-dependent activation and inactivation of anoctamin calcium-gated chloride channels. J Gen Physiol. 2013;142:381–404. doi: 10.1085/jgp.201311015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasche S, Toetter B, Adler J, Tschapek A, Doerner JF, Kurtenbach S, Hatt H, Meyer H, Warscheid B, Neuhaus EM. Tmem16b is specifically expressed in the cilia of olfactory sensory neurons. Chem Senses. 2010;35:239–245. doi: 10.1093/chemse/bjq007. [DOI] [PubMed] [Google Scholar]
- 37.Stöhr H, Heisig JB, Benz PM, Schöberl S, Milenkovic VM, Strauss O, Aartsen WM, Wijnholds J, Weber BH, Schulz HL. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J Neurosci. 2009;29:6809–6818. doi: 10.1523/JNEUROSCI.5546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barthó P, Payne JA, Freund TF, Acsády L. Differential distribution of the KCl cotransporter KCC2 in thalamic relay and reticular nuclei. Eur J Neurosci. 2004;20:965–975. doi: 10.1111/j.1460-9568.2004.03562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ulrich D, Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci. 1997;17:2348–2354. doi: 10.1523/JNEUROSCI.17-07-02348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]