

Abstract
NADPH-oxidase (NOX) mediated superoxide originally found on leukocytes, but now recognized in several types of cells in the brain. It has been shown to play an important role in the progression of stroke and related cerebrovascular disease. NOX is a multisubunit complex consisting of 2 membrane-associated and 4 cytosolic subunits. NOX activation occurs when cytosolic subunits translocate to the membrane, leading to transport electrons to oxygen, thus producing superoxide. Superoxide produced by NOX is thought to function in long-term potentiation and intercellular signaling, but excessive production is damaging and has been implicated to play an important role in the progression of ischemic brain. Thus, inhibition of NOX activity may prove to be a promising treatment for ischemic brain as well as an adjunctive agent to prevent its secondary complications. There is mounting evidence that NOX inhibition in the ischemic brain is neuroprotective, and targeting NOX in circulating immune cells will also improve outcome. This review will focus on therapeutic effects of NOX assembly inhibitors in brain ischemia and stroke. However, the lack of specificity and toxicities of existing inhibitors are clear hurdles that will need to be overcome before this class of compounds could be translated clinically.
Keywords: ischemic stroke, NADPH oxidase, superoxide, NOX inhibition
INTRODUCTION
Stroke is caused by occlusion of or hemorrhage from a blood vessel supplying the brain. It is a cause of long-term disability and ranks as the third frequent cause of death following heart disease and cancer, yet despite high prevalence, the number of approved therapies remains low [1,2]. The major therapeutic strategy for treatment of acute ischemic stroke is rapid recanalization, either by pharmacological means through thrombolytic agents, or mechanical thrombectomy [3]. However, the time window for intervention limits these therapies to a small number of patients, and their inappropriate use can actually worsen outcome.
Stroke, via occlusion of cerebral artery(ies) brings on energy depletion and subsequent death of cells in the vascular territory. Currently, understanding of the pathophysiology of ischemic stroke indicates that the mechanism of neuronal injury can be characterized according to infarct progression over time. The acute stage appears within minutes up to a few hours after ischemic stroke onset, during which ionic homeostasis is disrupted by the decrease in cerebral blood flow. This phenomenon leads to increased intracellular calcium concentrations and stimulation of glutamate release, producing excitotoxicity and a spreading depression throughout the ischemic region [4,5,6]. Water enters the intracellular space due to osmotic gradients, causing cells swell, and the resulting vasogenic edema can give rise to intracranial pressure, vascular compression and herniation [5]. Furthermore, the generation of reactive oxygen species (ROS) can destroy membranes, mitochondria and DNA, leading to proteins misfolding and enzyme dysfunction. These factors are exacerbated in the event of reperfusion takes place. In the second-subacute phase occurs a few hours to a few days after ischemia. An inflammatory and cell death (apoptosis and necrosis) response develops as a result of the stimulatory influences of the acute phase [4,5,7]. In additional to these responses, high intracellular calcium concentrations built up during the acute phase lead to the overactivation of several proteolytic enzyme systems.
Recent studies have focused on the role of superoxide generating systems in immune cells and their consequences on reperfusion injury (RI), or that injury which results when the occluded vessel is reopened. Of the various systems by which cells can generate reactive oxygen species (ROS), the topic of this review will focus on superoxide produced by activation of the NADPH oxidase (NOX). Superoxide is soluble in solution and can combine with other ROS to form more toxic molecules after experimental ischemic stroke [8]. The importance of NOX in production of ROS and subsequent neuronal cell death has been the subject of somewhat recent investigations. Several other enzymes found throughout the cell are involved in ROS generation, including xanthine oxidase, lipoxygenase, cylcoocxygenases (COX) and substrate coupled nitric oxide synthetase, but none of these can produce the large amount of ROS observed in nonphagocytic cells in both normal and pathological conditions [9]. In the last decade, study into the NOX enzyme has made a major new turn with discovery of a large number of NOX homologs which may allow for a more targeted therapeutic approach. At present, no specific treatments exist for NOX induced ROS in central nervous system (CNS) diseases and most of these diseases have very few therapeutic options at all. This will be discussed in more detail in this review.
NADPH OXIDASE (NOX)
Oxidative stress leads to overload of ROS caused by excessive ROS production and/or impaired ROS reduction. Under conditions of oxidative stress, the overproduction of ROS becomes detrimental and possibly contributes to various acute and chronic diseases, such as myocardial infarction, hypertension, atherosclerosis, cancer, neurodegeneration, and inflammation. Consequently, oxidative stress has also been suggested as an important underlying mechanism of ischemic stroke [10].
Among many enzymes utilizing molecular oxygen as substrate, NOX is activated and can serve as a source of ROS in the CNS [11]. Seven NOX family members have been identified and isoforms are denoted as NOX1 to NOX5, dual oxidase (Duox)-1 and -2. NOX1, 2 and 4 have been detected in the CNS, with cells such as neurons and intracranial vessels as well as microglia [12,13]. NOX3 is almost exclusively expressed in the inner ear but not in blood vessels. Gene deletion of NOX5 in the rodent genome is the least functionally characterized isoform compared to others [14]. Nevertheless, several recent studies show that NOX5 is activated by calcium in human cells. Thus, it may be a key player in ROS generation in vascular cells [15,16]. Duox-1 and -2, also contain an oxidase domain, which appear to be expressed predominantly in epithelial cells [17]. Recently, in vitro studies have shown NOX expression in neurons, astrocytes, and microglia [11]. In vivo studies have also shown that NOX subunits are widely distributed in the cortex, the hippocampus, and in the cerebellum [13,18]. NOX is a multi-component enzyme comprising of cytoplasmic (p47phox, p67phox, p40phox and Rac) and membrane bound (p91phox and p22phox) subunits. Upon phosphorylation by specific protein kinases, these cytoplasmic subunits can form a complex and translocate to the plasma membrane to dock with the plasma membrane subunits [19]. Catalysis of NOX occurs in the p91phox subunit, which begins the transfer of electrons from molecular oxygen through redox coupling with NADPH, FAD and heme to generate superoxide [20] (Fig. 1). During activation, the p47phox component is phosphorylated and moves to the plasma membrane, where it assembles with the other subunits to generate an active enzyme complex. It produces superoxide by transferring electrons from NADPH inside the cell across the membrane and coupling these to molecular oxygen to produce superoxide. Superoxide can also be generated within phagosomes. Within phagosomes, superoxide can spontaneously form hydrogen peroxide, which is also a reactive species [20]. Vascular ROS are produced in endothelial, adventitial, and vascular smooth muscle (VSMC) and derived primarily from NOX, a multisubunit enzyme catalyzing a superoxide anion (O2•–) production by the 1 electron reduction of oxygen using NOX as the electron donor: 2O2+NADPH → 2O2-+NADP+H+ [21]. Superoxide is capable of killing bacteria and fungi by mechanisms that are not yet fully understood, but may inactivate critical metabolic enzymes, initiate lipid peroxidation and liberate redox active iron, which allows the generation of indiscriminate oxidants such as the hydroxyl radical. Superoxide probably kills bacteria directly as the virulence of many pathogens is dramatically attenuated when their superoxide dismutase (SOD) genes are deleted. However, downstream products of superoxide also include hydrogen peroxide and hypochlorous acid, the reactive agent in bleach. NOX activation depends on phosphorylation, especially of the p47phox subunit [22]. While other subunits can be phosphorylated, p47phox phosphorylation appears a key player in the membrane translocation of other subunits as well. Kinases known to phosphorylate p47 include several of protein kinase C isoforms (β, δ and ζ) as well as p38 and p21 mitogen activated kinases (MAPK) and protein kinase B. Further, it appears that NOX can be regulated by the inflammatory transcription factor, nuclear factor kappa B (NFκB). NFκB can induce gp91phox expression, as cells deficient in NFκB's p65 subunit express less gp91phox in response to lipopolysaccharide (LPS) stimulation [6].
Fig. 1. NADPH oxidase (NOX) activation. NOX comprises cytosolic (p47phox, p67 phox, p40 phox and Rac) and membrane subunits (gp91 phox and p22 phox). During activation of NOX, cytosolic subunits comprise a multi-component enzyme and migrate to the plasma membrane to dock with the membrane subunits. This multisubunit enzyme produces a superoxide anion (O2•–).

FUNCTION OF NOX IN REPERFUSION INJURY (RI) AFTER ISCHEMIC STROKE
NOX isoforms have been reported in the brain and its vasculature. Thus, cerebrovascular disease may benefit from NOX as a therapeutic target in the brain, as well as in its blood vessels. Recently, NOX may be a key player in the prevention of RI, an increasingly observed complication of delayed recanalization leading to brain edema or hemorrhage [23,24]. Thus, therapies to minimize RI might expand populations of stroke patients eligible for treatment. RI begins after ischemic stroke when the brain is quickly exposed to oxygenated blood, and ischemic mitochondria within the brain are rendered incapable of detoxifying reactive species [10].
A robust inflammatory reaction characterized by peripheral leukocyte influx into the cerebral parenchyma and activation of endogenous microglia follows ischemic stroke. While immune cells generate ROS through several enzyme systems, NOX is the major enzyme that generates superoxide in these cells. ROS elaborated by these cells can then activate other cells, even ischemic neurons, to secrete inflammatory factors. How NOX is activated in stroke is not entirely clear, but phosphorylation of the 47phox subunit appears key. This phosphorylation can occur through several kinases also upregulated and activated by brain ischemia, including several protein kinase C isoforms and the p38 and p21 been described [25], phagocytic NOX, also referred to as NOX2, is associated with immune cells. While NOX2 has been described in various cell types, NOX2 expression is greatest in myeloid cells [26], which, in the case of brain, are microglia [27]. A previous study showed that microglial cells isolated from the human fetal brain expressed Nox2, p22phox, p40phox, p47phox, and p67phox transcripts [28]. A large number of studies also demonstrated superoxide generating NOX systems in immune cells and their consequences on RI. Thus, inhibition of NOX can potentially reduce the amount of superoxide generated, and thus limit RI. This approach has the potential to not only treat acute ischemic stroke, but also reduce complications of recanalizing strategies by using it in combination with thrombolytics or mechanical thrombectomy devices.
NOX expression has been shown to increase in the brain after experimental stroke [29,30], and several groups have shown that mice deficient in the gp91phox subunit had smaller infarcts than wildtype mice [8,31,32,33,34]. Further, NOX appears to play a significant role in RI. While RI has been attributed to the restoration of blood oxygen leading to a surge in ROS, some studies indicate that the source of damaging ROS may, in fact, be due to the return of blood glucose, which then fuels NOX to generate more ROS. In this case, NOX serves as an electron donor to produce damaging levels of superoxide, and NOX activation is dependent on glucose [35]. NOX also appears to be a primary source of ROS generated by NMDA receptor activation, thus suggesting another mechanism of ROS generation [36].
The targeting of NOX does not need to be restricted to the brain itself. While NOX and its isoforms are certainly present in microglia and other brain cell types, NOX present on circulating leukocytes contributes substantially to stroke pathology [32]. In a prior study by our lab, the use of bone marrow chimeras allowed us to replace marrow derived cells lacking functional NOX2 into wild-type mice, and vice versa, replacing marrow with intact NOX2 into NOX2 deficient mice. After subjecting these chimeras to strokes, mice lacking circulating NOX2 but intact brain NOX2 had better outcomes compared to mice with intact circulating NOX2, but deficient brain NOX2. Vascular NOX is another relevant target. Aged mice have been found to have dysregulated cerebrovascular responses compared to similar aged mice deficient in NOX [37]. Vascular NOX also appears to contribute to cerebrovascular dysfunction and behavioral deficits in models of Alzheimer's disease [38,39]. Thus, the development of NOX inhibiting compounds may not necessary have to penetrate the blood brain barrier (BBB) in order to be effective against stroke and related conditions.
PHARMACOLOGICAL COMPOUNDS TO INHIBIT NOX ENZYMES
Several pharmacological compounds have been studied to target NOX enzymes in ischemic stroke. However, these are generally very low in specificity and selectivity. There has not been much clinical development since many are associated with toxicities that could hinder their use in humans. Yet, safer and more specific inhibitors are under development.
Diphenyleneiodonium (DPI)
DPI has been shown to protect the brain from stroke by blocking NOX [40]. It impairs NOX via flavoprotein inhibition to prevent superoxide production. Intravenous DPI administration reduced magnetic resonance imaging detected ischemic lesion size in an experimental stroke model suggesting that it is capable of penetrating the BBB. Further, it was shown to decrease expression of extracellular matrix proteins MMP-2 and MMP-9, proteins known to contribute to BBB disruption [40]. In a hypoxia model, DPI also improved the integrity of tight-junctions in the BBB by reducing ROS production [41]. DPI treatment has also been associated with the inhibition of the inflammatory response accompanying stroke [42]. However, DPI's usefulness is limited because it can also inhibit other flavoenzymes in vivo [31]. Flavoenzymes play a crucial role in many metabolic pathways.
Apocynin
Perhaps the most widely NOX inhibitor in experimental stroke is apocynin, also known as acetovanillone. Apocynin is a naturally occurring organic compound, originally derived from the root of Canadian hemp (Apocynum cannabinum) and Picrorhiza kurroa, a medicinal herb that has been used for centuries by the Chinese and South Asians to treat inflammatory diseases [43,44]. Its chemical name is 4-hydroxy-3-methoxy-acetophenone, and it is a methoxy-substituted catechol which was Apocynin is a commonly used anti-oxidant and anti-inflammatory capability, specifically, for blocking the activity of NOX in many experiment models involving phagocytic and nonphagocytic cells [45,46]. It also approached to NOX inhibitor with relatively low affinity (IC50: ~10 µmol/l) in neutrophils [47]. It does not seem to interfere with other polymorphonuclear neutrophil defense mechanisms, as it does not affect phagocytosis or intracellular killing [48]. Apocynin inhibits the release of superoxide through NOX by blocking migration of p47phox to the membrane, thus interfering with assembly of the functional NOX complex [49]. The inhibitory action of the compound, however, occurs after a lag time only. Some of its inhibitory activity at least initially may involve myeloperoxidase (MPO) because apocynin does not inhibit NOX or the generation of superoxide in cells devoid or deficient in MPO [48,50]. MPO together with hydrogen peroxide can facilitate apocynin dimerization, and these dimers can prevent assembly of an active enzyme complex (Fig. 2). Furthermore, agents such as zymosan that promote the release of MPO also enhance the efficacy of apocynin [47,48]. In cells with less MPO, apocynin can reduce oxidant stress through a non-specific oxidative scavenger effect instead of NOX inhibition [50]. However, besides MPO, other peroxidases, such as horseradish peroxidase, can also induce apocynin dimer formation with a consequent NOX inhibitory effect [49,51]. Recent study showed that MPO secreted by neutrophils can be taken up by endothelial cells, in which apocynin can then be metabolized to active dimers, thus inhibiting vascular NOX in vivo [51] (Fig. 3). In line with this concept, it was observed that supplementation with thiol provided either as glutathione or cysteine prevents the inhibitory effect of apocynin on the NOX. Apocynin dimer formation may be responsible for its delayed inhibitory property [52]. It has been suggested that this dimer is what blocks NOX activity [53].
Fig. 2. Mechanism of NOX inhibition by apocynin. Apocynin dimerization by myeloperoxidase (MPO) and hydrogen peroxide (H2O2).
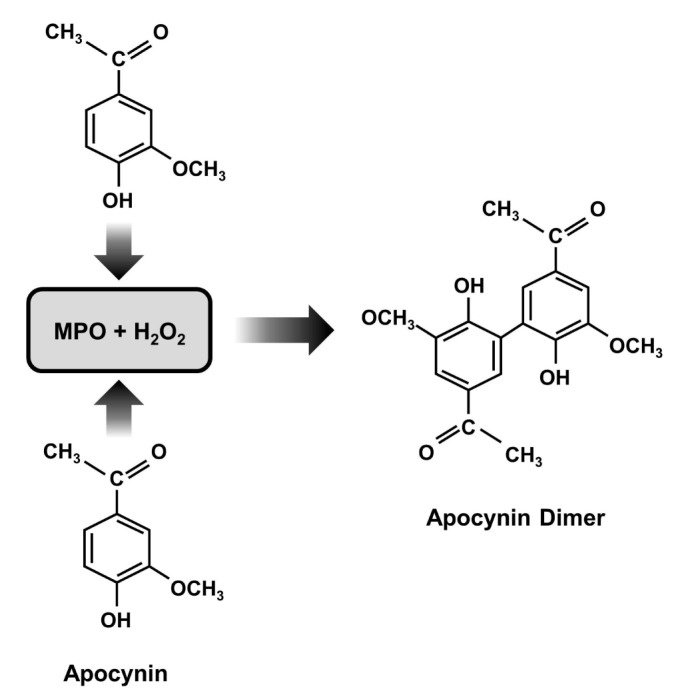
Fig. 3. The mechanism of inhibition of apocynin on NOX-induced superoxide production. In neutrophil, dimerized apocynin by MPO and H2O2 prevents assembly of an intact enzyme complex by preventing the association of cytosolic subunit p47 with the membrane subunit gp91. In vascular cell, apocynin could also be dimerized by MPO secreted from neutroophils. It inhibits vascular NOX, which is composed of NOX2 or its homologues (NOX1, NOX4, or NOX5) as the catalytic subunit. (Apo: Apocynin).

The effect of apocynin has been widely explored in experimental ischemic stroke. It has also been documented to penetrate into the brain following systemic delivery [54], and several groups have shown the salutary effects of treatment on lesion size, brain hemorrhage reduction and improvements in neurological deficits. Previous our lab reported that a dose of 2.5 mg/kg given just prior to reperfusion, or 1.5 hours after ischemic stroke, resulted in reduced infarct size and improved neurological outcome while markedly suppressing superoxide generation in the brain [54]. However, at higher doses (3.75 and 5 mg/kg), apocynin showed no benefit, and actually increased the severity of brain hemorrhage. Thus, this rather narrow therapeutic dose range may limit its translation to the clinical level. However, other groups have shown salutary effects of apocynin at doses as high as 40~50 mg/kg [31,55], while another group found no effect of apocynin using a dose of about 4 mg/kg [56]. In global cerebral ischemia, 5 mg/kg apocynin attenuated hippocampal injury when given prior to ischemia onset [57]. In safety studies of uninjured mice, apocynin was well tolerated in single oral doses of up to 1000 mg/kg [58]. Interestingly, apocynin failed to have any further beneficial effect in NOX2 deficient mice subjected to stroke, and might speak somewhat to its specificity or its dependence on NOX2 [32]. In addition to apocynin, Altered NOX function has been linked to neurological disorders such as Alzheimer's (ALS) and Parkinson's diseases (PD) [59]. Apocynin has been effective in ameliorating neuropathological damages in both in vivo and in vitro models of PD [60,61]. Apocynin also slowed disease progression and extended survival in a mouse ALS model [62]. There has clearly been some experience with apocynin in humans, considering the long history of its use in Eastern medicine, but a handful of studies in humans have been reported in unrelated conditions such as nebulized apocynin in asthmatics [63,64]. Under these circumstances, apocynin appears to be well tolerated clinically, but more work is still needed.
Honokiol and plumbagin
Other natural compounds of NOX inhibitor are honokiol and plumbagin. Honokiol was isolated from the herb Magnolia officinalis and has been of particular interest because this compound appears to inhibit superoxide production after the respiratory burst and not before the enzyme is activated as with other inhibitors [65]. Honokiol has also been shown to lesion size in experimental ischemic stroke followed by reperfusion and this decrease was correlated to a reduction in neutrophil infiltration and activation, and decreased lipid peroxidation [65,66]. The only inhibitor to date that directly interacts with a specific NOX homolog seems to be a plant derived naphthoquinone called plumbagin [67]. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) is a natural yellow pigment that comes from the roots of the black walnut plant Plumbago zeylanica. Plumbagin inhibits non-phagocytic NOX activity in HEK293 and LN229, a cell line that only express NOX4 and in a cell line transfected for NOX4 [67]. The regulation of NOX4 appears to be different from the other NOX homologs which require p47phox and p67phox, and it has been observed that NOX4 alone can produce superoxide activity [68,69,70]. The method by which it inhibits NOX4 is unknown but it is unlikely that it is due to cytotoxic effects as the cells were viable after 1 hour incubation with plumbagin [67]. It has been shown to have significant anti-cancer activity and may work by blocking superoxide production as many cancers have been shown to produce ROS and specifically express NOX homologs [71,72,73,74]. NOX4 is the dominant NOX homolog in vascular smooth muscle cells and its inhibition by plumbagin may well explain its anti-atheroscerotic effect.
Gp91ds-tat
The most selective NOX inhibitor to date is a chimeric peptide gp91ds-tat [75]. This peptide is constructed from the sequence of gp91phox that is known to be involved in the binding of gp91phox to p47phox and can inhibit the oxygen radical production in cell-free assays (gp91 docking sequence or gp91ds). In order to deliver this peptide into the cells, the gp91ds was linked to HIV coat peptide (HIVtat) that is known to be involved in internalization [75]. This gp91ds-tat specifically binds to p47phox and prevents the formation of the NOX complex. While this is the most specific inhibitor for NOX, it cannot distinguish between the phagocytic or non-phagocytic enzyme and it has little oral bioavailability as it is a peptide.
Serine protease inhibitors
One of a group of NOX inhibitors is the 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF) which an irreversible serine protease inhibitor [76]. AEBSF appears to have a direct effect on the plasma membrane components of NOX and interrupt with the binding of the cytosolic components p47phox and p67phox. AEBSF does not interfere with the electron transport and does not scavenge the oxygen radicals. Unfortunately, AEBSF modifies many proteins by covalent attachment preferentially on tyrosine, and to a lesser extent on lysine, histidine, and the amino-terminus. AEBSF is quite stable in aqueous solution and the extent to which the protein is modified continues to increase for several days and this significantly limits its use in vivo. Tosylphenylalanychloromethane is another irreversible serine protease inhibitor similar to AEBSF and has a similar mechanism of action [77]. However, no studies to date have been carried out in stroke models of these compounds.
Phenylarsine oxide and gliotoxin
Other NOX inhibitors include phenylarsine oxide and gliotoxin [78]. Phenylarsine oxide, via its interaction with cysteine residues, appears to prevent NADPH enzyme assembly. However, this inhibitor is quite non-specific. Due to its interactions with the cysteine groups it will also interact with other enzymes and proteins [79,80]. Gliotoxin is extracted from Aspergillus and it appears to inhibit the enzyme by blocking the phosphorylation of p47phox [53,81]. It also seems to react with the thiol groups, thus limiting its specificity. To date, there do not appear to be any studies examining the therapeutic value of these compounds in stroke or stroke models.
Ebselen
There are NOX inhibitors that have either been specifically developed by the pharmaceutical industry or are in clinical trials. Ebselen, 2-phenyl-1,2-benzisoselenazol-3(2H)-one, a mimic of glutathione peroxidase which also reacts with peroxynitrite, inhibits a variety of enzymes such as lipoxygenases, nitric oxide synthases, NOX, protein kinase C, and Hz/Kz-ATPase [82]. Ebselen is therefore quite non-specific, but is being used at some centers for the treatment of stroke in Japan. Ebselen has shown efficacy if the treatment is started within 24 hours of the stroke [83,84]. Currently, a multicenter phase 3 ebselen trial is ongoing (www.strokecenter.org).
VAS2870
A few other inhibitors have been developed by the pharmaceutical industry including VAS2870 (Vasopharm) [56,85]. The VAS2870 has been shown to be neuroprotective in preclinical stroke experiments [56]. It appears to a more specific to inhibitor of all NOX isoforms, and is neither general flavoprotein inhibitor nor ROS scavengers [85,86]. VAS2879 appears to be a pan-NOX inhibitor, as it prevents assembly or conformational change to active NOX complexes [87,88]. When VAS2870 was admitted before complex assembly induction, it inhibited a NOX2 activity [87]. Therefore, the order by which a NOX complex assembly activator and a NOX inhibitor are added in cell-free assays is crucial. A recent study showed that VAS2870 has potential off-target effects, suggesting that the compound alkylates cysteine thiol residues [89]. To date, there has been only one study examining this compound in a stroke model [56]. In this study, VAS2870 treatment led to superior lesion size reduction and improvement in neurological deficits compared to no treatment.
HMGCoA reductase inhibitors
tensin receptor-1 blockers (ARBs), and the HMGCoA reductase inhibitor (statins) drugs can all indirectly inhibit NOX activity [86,90]. Statins, ACE inhibitors and ARBs have all been shown to protect the brain from experimental stroke for various reasons. In particular, statins have been shown to improve outcome from experimental stroke via their ability to upregulate endothelial nitric oxide synthase [91]. All of these compounds have been shown to correlate to an anti-inflammatory effect, although whether this is directly due to NOX inhibition seems less likely, with these compounds acting as a related immune target.
CONCLUSION
In conclusion, NOX appear to an important player in ischemic stroke, particularly as it relates to reperfusion injury due to its generation of superoxide. Superoxide is well known to contribute to the progression of stroke and related cerebrovascular diseases as well as neurodegenerative conditions. By inhibiting NOX, several preclinical studies have reported improved neurological outcome, and reduced severity of brain injury including brain hemorrhage. While available inhibitors may be non-specific or have a relatively narrow therapeutic window, we suggest that it is still worthwhile to investigate NOX inhibitors, and to carry out studies to develop safe and selective drugs for treatment of clinical stroke, its complications and for related conditions as well. Further, there is rapidly increasing knowledge of the role of the various NOX isoforms. Whether one form versus another is relevant to one type of pathology compared to another is still unknown. Such knowledge could also drive the development of appropriate therapies as well. Clearly, several laboratory and preclinical animal model studies have shown the salutary effects of several NOX isoforms as viable targets for treatment of ischemic stroke. While limiting elaboration of damaging ROS into the ischemic brain during reperfusion, its inhibition leads to the inhibition of the local inflammatory response and, as a result, may prevent secondary complications of stroke such as BBB disruption and brain hemorrhage. Since reperfusion injury, including hemorrhagic transformation and the development of a frank parenchymal hematoma, are significant complications of thrombolysis, NOX inhibitors have the potential to be used in conjunction with recombinant tissue plasminogen activator (rt-PA), in addition to being used as a stand alone neuroprotectant. However, pharmacological development of a specific and safe inhibitor is still lacking. Several inhibitors have been studied by various labs and, while shown to be effective in laboratory models, are either non-specific in their inhibition, or may have off target toxicities that could limit translational development. The issue of specificity may not be as critical, since several NOX isoforms have been shown to contribute to stroke pathophysiology, and off target effects, such as non-specific anti-oxidant properties, may contribute to neuroprotection. More importantly, it will be important to develop therapies with less toxicity. Finally, the issue of whether such compounds may pass the BBB may be less important compared to other therapies, since NOX is present on circulating leukoctyes, which could be targeted without requiring a NOX inhibitor to enter the brain.
ACKNOWLEDGEMENTS
This study was funded by grants from the National Institutes of Health (RO1 NS40516), Department of Defense and the Veteran's Merit Award (I01 BX000589) to MAY, Basic Science Research Program through the National R esearch Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1D-1A1B03933017) to JYK and a National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT & Future Planning (NRF-2016M3C7A1905098) to JEL. Grants to MAY was administered by the Northern California Institute for Research and Education, and supported by resources of the Veterans Affairs Medical Center, San Francisco, California.
References
- 1.Kriz J, Lalancette-Hébert M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009;117:497–509. doi: 10.1007/s00401-009-0496-1. [DOI] [PubMed] [Google Scholar]
- 2.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jahan R. Hyperacute therapy of ischemic stroke: intravenous thrombolysis. Tech Vasc Interv Radiol. 2005;8:81–86. doi: 10.1053/j.tvir.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 5.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 7.Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr Med Chem. 2014;21:2076–2097. doi: 10.2174/0929867321666131228205146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- 10.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 12.Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, Wakisaka M, Kawahara T, Rokutan K, Ibayashi S, Iida M. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 13.Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- 14.Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal. 2009;11:2443–2452. doi: 10.1089/ars.2009.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res. 2010;106:1363–1373. doi: 10.1161/CIRCRESAHA.109.216036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedard K, Jaquet V, Krause KH. NOX5: from basic biology to signaling and disease. Free Radic Biol Med. 2012;52:725–734. doi: 10.1016/j.freeradbiomed.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 17.Bedard K, Lardy B, Krause KH. NOX family NADPH oxidases: not just in mammals. Biochimie. 2007;89:1107–1112. doi: 10.1016/j.biochi.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Kim MJ, Shin KS, Chung YB, Jung KW, Cha CI, Shin DH. Immunohistochemical study of p47Phox and gp-91Phox distributions in rat brain. Brain Res. 2005;1040:178–186. doi: 10.1016/j.brainres.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 19.Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci. 2003;28:502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 20.Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr Med Chem. 2014;21:2076–2097. doi: 10.2174/0929867321666131228205146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lassègue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 22.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Kuroda S, Siesjö BK. Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. Clin Neurosci. 1997;4:199–212. [PubMed] [Google Scholar]
- 25.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 26.Kahles T, Kohnen A, Heumueller S, Rappert A, Bechmann I, Liebner S, Wittko IM, Neumann-Haefelin T, Steinmetz H, Schroeder K, Brandes RP. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol Dis. 2010;40:185–192. doi: 10.1016/j.nbd.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 27.Harrigan TJ, Abdullaev IF, Jourd’heuil D, Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem. 2008;106:2449–2462. doi: 10.1111/j.1471-4159.2008.05553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green SP, Cairns B, Rae J, Errett-Baroncini C, Hongo JA, Erickson RW, Curnutte JT. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J Cereb Blood Flow Metab. 2001;21:374–384. doi: 10.1097/00004647-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Choi DH, Kim JH, Lee KH, Kim HY, Kim YS, Choi WS, Lee J. Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PLoS One. 2015;10:e0116814. doi: 10.1371/journal.pone.0116814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 31.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 32.Tang XN, Zheng Z, Giffard RG, Yenari MA. Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann Neurol. 2011;70:606–615. doi: 10.1002/ana.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Wei X, Liu K, Zhang X, Yang F, Zhang H, He Y, Zhu T, Li F, Shi W, Zhang Y, Xu H, Liu J, Yi F. NOX2 deficiency ameliorates cerebral injury through reduction of complexin II-mediated glutamate excitotoxicity in experimental stroke. Free Radic Biol Med. 2013;65:942–951. doi: 10.1016/j.freeradbiomed.2013.08.166. [DOI] [PubMed] [Google Scholar]
- 35.Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol. 2008;64:654–663. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 38.Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagel S, Genius J, Heiland S, Horstmann S, Gardner H, Wagner S. Diphenyleneiodonium and dimethylsulfoxide for treatment of reperfusion injury in cerebral ischemia of the rat. Brain Res. 2007;1132:210–217. doi: 10.1016/j.brainres.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 41.Zehendner CM, Librizzi L, Hedrich J, Bauer NM, Angamo EA, de Curtis M, Luhmann HJ. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 2013;8:e82823. doi: 10.1371/journal.pone.0082823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagel S, Hadley G, Pfleger K, Grond-Ginsbach C, Buchan AM, Wagner S, Papadakis M. Suppression of the inflammatory response by diphenyleneiodonium after transient focal cerebral ischemia. J Neurochem. 2012;123(Suppl 2):98–107. doi: 10.1111/j.1471-4159.2012.07948.x. [DOI] [PubMed] [Google Scholar]
- 43.Sun AY, Wang Q, Simonyi A, Sun GY. Botanical phenolics and brain health. Neuromolecular Med. 2008;10:259–274. doi: 10.1007/s12017-008-8052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lafeber FP, Beukelman CJ, van den Worm E, van Roy JL, Vianen ME, van Roon JA, van Dijk H, Bijlsma JW. Apocynin, a plant-derived, cartilage-saving drug, might be useful in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 1999;38:1088–1093. doi: 10.1093/rheumatology/38.11.1088. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Chan MM, Andrews MC, Mori TA, Croft KD, McKenzie KU, Schyvens CG, Whitworth JA. Apocynin but not allopurinol prevents and reverses adrenocorticotropic hormone-induced hypertension in the rat. Am J Hypertens. 2005;18:910–916. doi: 10.1016/j.amjhyper.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 47.Simons JM, Hart BA, Ip Vai, Van Dijk H, Labadie RP. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic Biol Med. 1990;8:251–258. doi: 10.1016/0891-5849(90)90070-y. [DOI] [PubMed] [Google Scholar]
- 48.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 49.Touyz RM. Apocynin, NADPH oxidase, and vascular cells: a complex matter. Hypertension. 2008;51:172–174. doi: 10.1161/HYPERTENSIONAHA.107.103200. [DOI] [PubMed] [Google Scholar]
- 50.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 51.Astern JM, Pendergraft WF, 3rd, Falk RJ, Jennette JC, Schmaier AH, Mahdi F, Preston GA. Myeloperoxidase interacts with endothelial cell-surface cytokeratin 1 and modulates bradykinin production by the plasma Kallikrein-Kinin system. Am J Pathol. 2007;171:349–360. doi: 10.2353/ajpath.2007.060831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ximenes VF, Fernandes JR, Bueno VB, Catalani LH, de Oliveira GH, Machado RG. The effect of pH on horseradish peroxidase-catalyzed oxidation of melatonin: production of N1-acetyl-N2-5-methoxykynuramine versus radicalmediated degradation. J Pineal Res. 2007;42:291–296. doi: 10.1111/j.1600-079X.2007.00419.x. [DOI] [PubMed] [Google Scholar]
- 53.Van den Worm E, Beukelman CJ, Van den Berg AJ, Kroes BH, Labadie RP, Van Dijk H. Effects of methoxylation of apocynin and analogs on the inhibition of reactive oxygen species production by stimulated human neutrophils. Eur J Pharmacol. 2001;433:225–230. doi: 10.1016/s0014-2999(01)01516-3. [DOI] [PubMed] [Google Scholar]
- 54.Tang XN, Cairns B, Cairns N, Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience. 2008;154:556–562. doi: 10.1016/j.neuroscience.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang LL, Ye K, Yang XF, Zheng JS. Apocynin attenuates cerebral infarction after transient focal ischaemia in rats. J Int Med Res. 2007;35:517–522. doi: 10.1177/147323000703500411. [DOI] [PubMed] [Google Scholar]
- 56.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8:e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 58.Pandey A, Kour K, Bani S, Suri KA, Satti NK, Sharma P, Qazi GN. Amelioration of adjuvant induced arthritis by apocynin. Phytother Res. 2009;23:1462–1468. doi: 10.1002/ptr.2803. [DOI] [PubMed] [Google Scholar]
- 59.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anantharam V, Kaul S, Song C, Kanthasamy A, Kanthasamy AG. Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. Neurotoxicology. 2007;28:988–997. doi: 10.1016/j.neuro.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao HM, Hong JS, Zhang W, Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. J Neurosci. 2003;23:1228–1236. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boillée S, Cleveland DW. Revisiting oxidative damage in ALS: microglia, Nox, and mutant SOD1. J Clin Invest. 2008;118:474–478. doi: 10.1172/JCI34613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stefanska J, Sarniak A, Wlodarczyk A, Sokolowska M, Pniewska E, Doniec Z, Nowak D, Pawliczak R. Apocynin reduces reactive oxygen species concentrations in exhaled breath condensate in asthmatics. Exp Lung Res. 2012;38:90–99. doi: 10.3109/01902148.2011.649823. [DOI] [PubMed] [Google Scholar]
- 64.Stefanska J, Sokolowska M, Sarniak A, Wlodarczyk A, Doniec Z, Nowak D, Pawliczak R. Apocynin decreases hydrogen peroxide and nitrate concentrations in exhaled breath in healthy subjects. Pulm Pharmacol Ther. 2010;23:48–54. doi: 10.1016/j.pupt.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 65.Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. Honokiol protects rat brain from focal cerebral ischemiareperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res. 2003;992:159–166. doi: 10.1016/j.brainres.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 66.Chen CM, Liu SH, Lin-Shiau SY. Honokiol, a neuroprotectant against mouse cerebral ischaemia, mediated by preserving Na+, K+-ATPase activity and mitochondrial functions. Basic Clin Pharmacol Toxicol. 2007;101:108–116. doi: 10.1111/j.1742-7843.2007.00082.x. [DOI] [PubMed] [Google Scholar]
- 67.Ding Y, Chen ZJ, Liu S, Che D, Vetter M, Chang CH. Inhibition of Nox-4 activity by plumbagin, a plant-derived bioactive naphthoquinone. J Pharm Pharmacol. 2005;57:111–116. doi: 10.1211/0022357055119. [DOI] [PubMed] [Google Scholar]
- 68.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE(-/-) mice. J Clin Invest. 2001;108:1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem. 2001;276:1417–1423. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 70.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 71.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 72.Hazra B, Sarkar R, Bhattacharyya S, Ghosh PK, Chel G, Dinda B. Synthesis of plumbagin derivatives and their inhibitory activities against Ehrlich ascites carcinoma in vivo and Leishmania donovani Promastigotes in vitro. Phytother Res. 2002;16:133–137. doi: 10.1002/ptr.867. [DOI] [PubMed] [Google Scholar]
- 73.Parimala R, Sachdanandam P. Effect of Plumbagin on some glucose metabolising enzymes studied in rats in experimental hepatoma. Mol Cell Biochem. 1993;125:59–63. doi: 10.1007/BF00926835. [DOI] [PubMed] [Google Scholar]
- 74.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 75.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 76.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem. 1997;272:13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- 77.Gillibert M, Dehry Z, Terrier M, El Benna J, Lederer F. Another biological effect of tosylphenylalanylchloromethane (TPCK): it prevents p47phox phosphorylation and translocation upon neutrophil stimulation. Biochem J. 2005;386:549–556. doi: 10.1042/BJ20041475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 79.Doussiere J, Poinas A, Blais C, Vignais PV. Phenylarsine oxide as an inhibitor of the activation of the neutrophil NADPH oxidase--identification of the beta subunit of the flavocytochrome b component of the NADPH oxidase as a target site for phenylarsine oxide by photoaffinity labeling and photoinactivation. Eur J Biochem. 1998;251:649–658. doi: 10.1046/j.1432-1327.1998.2510649.x. [DOI] [PubMed] [Google Scholar]
- 80.Kutsumi H, Kawai K, Johnston RB, Jr, Rokutan K. Evidence for participation of vicinal dithiols in the activation sequence of the respiratory burst of human neutrophils. Blood. 1995;85:2559–2569. [PubMed] [Google Scholar]
- 81.Yoshida LS, Abe S, Tsunawaki S. Fungal gliotoxin targets the onset of superoxide-generating NADPH oxidase of human neutrophils. Biochem Biophys Res Commun. 2000;268:716–723. doi: 10.1006/bbrc.2000.2192. [DOI] [PubMed] [Google Scholar]
- 82.Cotgreave IA, Duddy SK, Kass GE, Thompson D, Moldéus P. Studies on the anti-inflammatory activity of ebselen. Ebselen interferes with granulocyte oxidative burst by dual inhibition of NADPH oxidase and protein kinase C? Biochem Pharmacol. 1989;38:649–656. doi: 10.1016/0006-2952(89)90211-6. [DOI] [PubMed] [Google Scholar]
- 83.Parnham M, Sies H. Ebselen: prospective therapy for cerebral ischaemia. Expert Opin Investig Drugs. 2000;9:607–619. doi: 10.1517/13543784.9.3.607. [DOI] [PubMed] [Google Scholar]
- 84.Yamaguchi T, Sano K, Takakura K, Saito I, Shinohara Y, Asano T, Yasuhara H. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke. 1998;29:12–17. doi: 10.1161/01.str.29.1.12. [DOI] [PubMed] [Google Scholar]
- 85.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Bäumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 86.Warner DS, Sheng H, BatiniXMLLink_XYZ-Haberle I. Oxidants, antioxidants and the ischemic brain. J Exp Biol. 2004;207:3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- 87.Altenhöfer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wingler K, Altenhoefer SA, Kleikers PW, Radermacher KA, Kleinschnitz C, Schmidt HH. VAS2870 is a pan-NADPH oxidase inhibitor. Cell Mol Life Sci. 2012;69:3159–3160. doi: 10.1007/s00018-012-1107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun QA, Hess DT, Wang B, Miyagi M, Stamler JS. Offtarget thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870) Free Radic Biol Med. 2012;52:1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wagner AH, Köhler T, Rückschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–69. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- 91.Li Q, Zhuang QK, Yang JN, Zhang YY. Statins excert neuroprotection on cerebral ischemia independent of their lipid-lowering action: the potential molecular mechanisms. Eur Rev Med Pharmacol Sci. 2014;18:1113–1126. [PubMed] [Google Scholar]