

Abstract
Postconditioning has been shown to protect the mouse brain from ischemic injury. However, the neuroprotective mechanisms of postconditioning remain elusive. We have found that toll-like receptor 5 (TLR5) plays an integral role in postconditioning-induced neuroprotection through Akt/nuclear factor kappa B (NF-κB) activation in cerebral ischemia. Compared to animals that received 30 min of transient middle cerebral artery occlusion (tMCAO) group, animals that also underwent postconditioning showed a significant reduction of up to 60.51% in infarct volume. Postconditioning increased phospho-Akt (p-Akt) levels and NF-κB translocation to the nucleus as early as 1 h after tMCAO and oxygen-glucose deprivation. Furthermore, inhibition of Akt by Akt inhibitor IV decreased NF-κB promoter activity after postconditioning. Immunoprecipitation showed that interactions between TLR5, MyD88, and p-Akt were increased from postconditioning both in vivo and in vitro. Similar to postconditioning, flagellin, an agonist of TLR5, increased NF-κB nuclear translocation and Akt phosphorylation. Our results suggest that postconditioning has neuroprotective effects by activating NF-κB and Akt survival pathways via TLR5 after cerebral ischemia. Additionally, the TLR5 agonist flagellin can simulate the neuroprotective mechanism of postconditioning in cerebral ischemia.
Keywords: Neuroprotection, Postconditioning, Cerebral ischemia, Toll-like receptor 5, Nuclear factor kappa B
INTRODUCTION
Stroke is the most common neurological disorder and cause of severe disability in adults. Although numerous animal studies have shown that exogenously administered drugs should have neuroprotective effects on cerebral ischemia, tissue plasminogen activator (tPA) is the only agent that has been successfully translated from animal models to clinical trials [1]. Over the last few decades, the failure of exogenously administered drugs to act as stroke neuroprotectants has increased research to identify endogenously-activated mechanisms in cerebral ischemia [2,3,4]. Sublethal hypoxic or ischemic events can improve the tolerance of cells or tissues. This phenomenon is referred to as “hypoxic” or “ischemic” “preconditioning” or “postconditioning” [5]. Preconditioning has limitations, since it must be executed before the stroke onset. Postconditioning, on the other hand, could prevent ischemia and reperfusion (I/R) injury after stroke through a series of mechanical interruptions of reperfusion [6]. Several experiments to study the neuroprotective mechanisms of postconditioning are underway, such as those demonstrating the role of phosphoinositide 3-kinase (PI3K) and AMPK in this process [7]. Through activation of PI3K, postconditioning has been shown to attenuate cerebral ischemia and induce recovery of behavioral deficits in mice [7]. PI3K contributed to an increase in ɛPKC activity, part of a survival-promoting pathway, and a reduction in MAPK during postconditioning in the rat brain [8]. Postconditioning improved functional recovery and decreased oxidative damage, which were abrogated in the presence of an AMPK inhibitor [9]. Although many studies are underway, the mechanisms that account for postconditioning's effects in the brain are largely unknown.
The activation of nuclear factor kappa B (NF-κB) has been shown to induce multiple factors that contribute to cell protection and promote tissue regeneration, including apoptosis inhibitors, reactive oxygen species scavengers, and cytokines, by regulating genes involved in inflammation, immunity, and processes [10]. However, establishing what role NF-κB plays in the nervous system (neuroprotection vs. neurodegeneration) has been controversial. It has been argued that during cerebral ischemia, the activation of NF-κB induces proapoptotic as well as antiapoptotic mechanisms [11,12]. From the literature on endogenously neuroprotective mechanisms, it has been reported that NF-κB activation is implicated in neuroprotective symptoms, such as brain tolerance, after cerebral ischemic preconditioning [13]. It was demonstrated that the NF-κB inhibitor, diethyl dithiocarbamic acid, attenuated the effects of postconditioning-induced reduction of cerebral I/R injury [14]. To dissect the NF-κB-mediated neuroprotective mechanisms of postconditioning, further studies are needed to demonstrate the role of NF-κB in postconditioning after cerebral ischemia.
Akt, a serine/threonine protein kinase, plays a critical role in controlling the balance between apoptosis and cell survival by promoting growth factor-mediated cell survival and preventing apoptotic cell death [15,16,17]. Several studies have asserted that the activation of the PI3K-Akt pathway plays an important role in cardiac and cerebral protection [7,18]. In postconditioning, it has been reported that activation of Akt induces neuroprotection [19,20]. The activation of Akt-phosphorylated IκB kinase α/β causes the translocation of NF-κB from the cytoplasm to the nucleus [21]. In mild transient focal cerebral ischemia, Akt signaling plays a neuroprotective role by activating NF-κB [22]. It is essential to clarify how the Akt/NF-κB pathway is involved in the neuroprotective effects of postconditioning.
In addition to Akt, one of the signaling pathways that activates NF-κB is Toll-like receptor (TLR) 5 [23,24]. TLRs are a part of the type-1 transmembrane protein receptor family, which recognizes diverse pathogen-associated molecules, such as lipopolysaccharides, peptidoglycans, lipoteichoic acid, bacterial CpG DNA, and bacterial flagellin [25,26]. TLRs are composed of an extracellular, leucine-rich repeat domain that recognizes a specific agonist and are responsible for mediating downstream signaling that plays a critical role in TLR-associated innate immunity and proinflammatory responses [27]. Among the 13 members of the TLR family characterized to date, only TLR5 specifically recognizes flagellin. Myeloid differentiation primary response gene 88 (MyD88) is required for the induction of NF-κB by TLR5 [28,29]. It has been demonstrated that, in colonic epithelial cells, the adaptor molecule MyD88 binds to TLR5 in response to flagellin, and MyD88 activates the PI3K pathway [30]. Nevertheless, it has not been determined whether TLR5 is associated with the Akt/NF-κB pathway in mediating the neuroprotective effects of postconditioning in the brain.
In this study, we tested whether postconditioning has neuroprotective effects through the activation of NF-κB via the Akt and TLR5-MyD88 pathways after stroke in mice. Experiments were conducted using a 30 min tMCAO model in mice and oxygen glucose deprivation (OGD) in mouse brain-derived cells. To clarify the role of Akt in TLR5 signaling, Akt inhibitor IV and the TLR5 agonist, flagellin, were used. These results provide a neuroprotective mechanism of postconditioning after tMCAO, whereby TLR5 activation leads to activation of NF-κB via Akt phosphorylation.
MATERIALS AND METHODS
Animals
Experiments with animals were performed in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care of Sookmyung Women's University. CD male mice (37~40 g, 8 weeks old) were used (Samtako Biokorea, Osan, South Korea). The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Rectal temperature was controlled at 37℃ with a thermostatically-controlled heating pad.
Primary neuron culture
Primary neurons were prepared from the cortex of embryonic day 16 ICR mice. In brief, brains were harvested, and the cortical layer was dissected under a microscope. Cortical neurons were dissociated in growth medium (Modified Eagle Medium [MEM; Gibco, 11090], GlutaMAX, 0.02 M glucose, and 5% horse serum). The cells were seeded into 6-well plates coated with poly-D-lysine (Sigma-Aldrich, St. Louis, MO, USA) followed by a change to Neurobasal medium (Gibco, 21103; plus GlutaMAX and antibiotics) with B27 (Gibco, 26050) supplement the following day. Neurobasal Medium was replaced every 2~3 days, and the cultures were maintained at 37℃ in a humidified incubator with 5% CO2.
Cell culture
Mouse brain-derived endothelial (bEnd.3) and microglial (BV-2) cells were used for in vitro studies. These cells were purchased from the American Type Culture Collection (ATCC, Manassas, MA, USA). Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal calf serum until confluent. OGD was applied using an anaerobic chamber (PlasLabs, Lansing, MI, USA) filled with a combination of 5% H2 and 95% N2, and cells were cultured in DMEM without glucose (ThermoFisher Scientific, Waltham, MA, USA). After 120 min of OGD, bEnd.3 and BV-2 cells were further cultured in DMEM with 30 mM glucose under normoxia for 1, 4, 6, or 24 h. Control cells were cultured in DMEM with 30 mM glucose under normoxia for 4 h.
Transient middle cerebral artery occlusion
CD mice were subjected to 30 min of tMCAO and reperfusion. Transient focal cerebral ischemia was induced by occlusion of the left MCA using a 5-0 suture (monofilament nylon). Briefly, the left common carotid artery was exposed, and the external carotid artery (ECA) was dissected distally. The internal carotid artery (ICA) was isolated, and a blunted suture was introduced into the ECA lumen and then gently advanced into the ICA lumen to block MCA blood flow. After 30 min of tMCAO, cerebral blood flow was restored by suture withdrawal. The incision was closed, and the mice recovered on a heating pad.
Oxygen-glucose deprivation
Primary neurons or cell lines underwent OGD by transfer to an anaerobic chamber (PlasLabs, Lansing, MI, USA) with 5% H2 and 95% N2. The cells were placed in a humidified 37℃ incubator within the anaerobic chamber. The culture medium was replaced with a buffered salt solution without glucose. bEnd.3 and BV-2 cells were subjected to OGD for 120 min, followed by 10 min of recovery and then 10 min of OGD. Under these conditions, pO2 in the chamber was 0 mmHg after 10 min, and pO2 in the medium was 15~20 mmHg. OGD ended by replacing the culture medium with medium containing 30 mM glucose and returning the cultures to the normoxic incubator. The bEnd3 cells were reoxygenated for 1, 4, 6, or 24 h before sampling to determine lactate dehydrogenase (LDH) activity. The BV-2 cells were reoxygenated for 1, 4, or 6 h.
Schematic representation of postconditioning
1) In vivo model
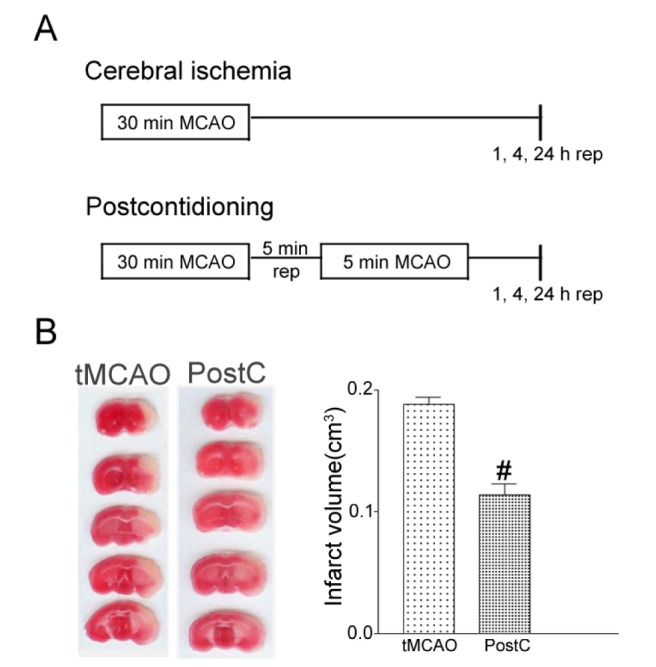
The experimental protocol is shown in Fig. 1A. In the sham group, mice did not undergo surgery. In the I/R group, mice underwent 30 min of tMCAO. In the ischemic postconditioning group, after 30 min of tMCAO, reperfusion was established for 5 min, after which the MCA was occluded again for 5 min. Animals then recovered for 1, 4, or 24 h.
Fig. 1. Postconditioning reduced cerebral ischemia infarct volume after tMCAO in mice. (A) Schematic diagram showing the time window of postconditioning after tMCAO in mice. (B) Animals that underwent 30 min of tMCAO had a significant infarct, as determined by TTC staining after 24 h of reperfusion (rep). Postconditioning, consisting of 5 min of reperfusion and 5 min of occlusion after severe 30 min tMCAO, reduced infarct size. The graph displays the statistical analysis of infarct volume. The postconditioning group had decreased infarct volume, up to 60.51%, compared to the tMCAO-only group. Data are mean±SE. n=5. Data were analyzed by ANOVA (#p<0.05 compared with tMCAO).
2) In vitro model
To model ischemia in vitro, primary neurons, bEnd.3 cells, and BV-2 cells were challenged by OGD. The cells were divided into normoxia, OGD, and postconditioning groups (Fig. 2A). Cells in the normoxia group were incubated in glucose-containing DMEM. Cells undergoing OGD were cultured in DMEM lacking glucose and placed in an anaerobic chamber for 120 min. Anaerobic conditions in the chamber were monitored using an anaerobic indicator (Oxoid, Basingstoke, UK). In the postconditioning group, cultured cells were subjected to 120 min of OGD and allowed to for 10 min before undergoing a further 10 min of OGD.
Fig. 2. Neuroprotective effects of postconditioning. (A) Schematic diagram showing the time window of postconditioning after OGD. (B) In primary neurons, BV-2 cells, and bEnd.3 cells, cell cytotoxicity levels were measured by an LDH assay after OGD. Postconditioning reduced cell cytotoxicity, which was increased by OGD. Data are mean±SE. n=4. Data were analyzed by ANOVA (#p<0.05, ##p<0.01 compared with normoxia [Nor]; **p<0.01 compared with the OGD group at the same time point).

2,3,5-Triphenyltetrazolium Chloride (TTC) staining
After 24 h of reperfusion, the brains were removed and sliced coronally into five 1-mm sections in a mouse brain matrix. The slices were then incubated in 2% TTC in saline for 10 min at 37°C, followed by 4% formalin. Stained sections were scanned with a scanner (HP Scanjet 2300c). The infarct area was measured on each section using Image J (Version 1.6.0; National Institutes of Health, Bethesda, MD, USA). Infarct volume was calculated by multiplying the area and thickness of the brain slice (1 mm). Results are expressed as mean±SE. Statistical differences in infarct volumes between the groups were established by analysis of variance followed by Student's Neuman-Keuls test.
Drug treatment
To examine the role of the TLR5/MyD88 pathway after tMCAO, we injected flagellin (InvivoGen, San Diego, CA, USA) intracerebroventricularly (i.c.v.) 5 min after tMCAO. Flagellin was dissolved in sterile, endotoxin-free water. The scalp was cut along the midline, and the skull was exposed. Flagellin (50 or 100 ng/5 µl) or vehicle (endotoxin-free water) were injected i.c.v. (bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). Akt inhibitor IV (Calbiochem, La Jolla, CA, USA; dissolved in 0.5% dimethyl sulfoxide) was added to bEnd.3 cell media in control, OGD, or postconditioning groups to examine the role of the Akt pathway after ischemia. bEnd.3 cells received Akt inhibitor IV treatment and then underwent the NF-κB reporter assay described below.
Western blotting
Animals were decapitated after reperfusion. Subcellular fractions were obtained from the tMCAO-affected brain regions on the ischemic side, including the striatum and cortex, and were quickly frozen in powdered dry ice and maintained at -80℃ until use. Cytosolic and nuclear fractions were prepared from the ischemic brains using ProteoExtract (Calbiochem, La Jolla, CA, USA). Protein concentration was determined using a bicinchoninic acidbased method. Equal amounts of sample were loaded across lanes and were electrophoretically separated on sodium dodecyl sulfate (SDS) gels. The resolved proteins were electrophoretically transferred to a polyvinylidene difluoride membrane. Blots were incubated with primary antibodies overnight at 4℃ and secondary antibodies for 60 min at room temperature. The primary antibodies that were used were polyclonal antibodies against phospho-Akt (Cell Signaling Technology, Danvers, MA, USA), NF-κB p65 (Cell Signaling Technology, Danvers, MA, USA), TLR5 (Santa Cruz Biotechnology, Delaware, CA, USA), MyD88 (Cell Signaling Technology, Danvers, MA, USA), TFIID (Santa Cruz Biotechnology, Santa Cruz, CA, USA), α-tubulin (Sigma-Aldrich, St. Louis, MO, USA), or β-actin (monoclonal, Sigma-Aldrich, St. Louis, MO, USA). After washing, membranes were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G (Amersham International, Little Chalfont, UK) for 60 min. The signal was then detected with an enhanced chemiluminescent kit (Thermo Fisher Scientific, Rockford, IL, USA). Protein quantitation was analyzed by scanning the films of the blots and calculating the intensity as measured by TINA.
Immunoprecipitation Assay (IP)
Sample lysates were prepared in lysis buffer (20 mM Tris, 10% Glycerol, 5 nM MgCl2, 0.1% Tween 20, 0.3 M KCl, 1/1000 2-mercaptoethanol) containing phosphatase inhibitors and protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA). Total protein (500 µg) was mixed with a primary antibody against MyD88 (Cell Signaling Technology, Danvers, MA, USA) at 4℃ overnight. Protein-G beads (Amersham Biosciences, Sunnyvale, CA, USA) were added and incubated at 4℃ for 2~3 h. The precipitates were washed three times with lysis buffer containing phosphatase and protease inhibitors as described above. The precipitates were fractionated on a SDS-PAGE gel followed by the immunoblotting procedure described above.
LDH assay
Cell viability was quantified by a standard measurement of LDH release using an LDH assay kit (BioVision, Milpitas, CA, USA). In this assay, NAD+ is reduced to NADH/H+ by the LDH-catalyzed conversion of lactate to pyruvate. In turn, yellow tetrazolium dye is reduced to a red formazan dye by the H/H+ resulting from NADH/H+. The amount of extracellular LDH was measured in a sample of the cell culture medium. The medium was sampled at 1, 4, 6 and 24 h after reoxygenation. A general procedure was followed using the manufacturer's guidelines.
NF-κB reporter assay
bEnd.3 cells were plated in 12-well plates (1×105 cells/well). Cells were transfected with the appropriate NF-κB plasmid DNA (Promega, Madison, WI, USA), including a β-galactosidase expression plasmid (HSP70-β gal), which served as an internal control, using Lipofectamine LTX reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. One day after transfection, cells underwent 120 min of OGD and were allowed to for 10 min before undergoing a further 10 min of OGD. Next, cells were reoxygenated for 1 or 4 h. The relative luciferase activity was determined by normalization with β-galactosidase activity, as described by Rhee et al. [31]. The total amount of NF-κB DNA was kept consistent by adding the empty vector for each transfection. All assays were performed in duplicate, and a single representative experiment is shown. Data are expressed as mean values±SE.
Statistical analysis
The data are expressed as mean±SE. Comparisons among multiple groups were performed by one-way ANOVA with appropriate Student Newman-Keuls test for multiple comparisons (GraphPad Prism, San Diego, CA, USA). Differences were considered statistically significant if p-values were less than 0.05, 0.01, and 0.001.
RESULTS
Postconditioning reduces cerebral ischemic damage
Through oxidative stress, cerebral ischemia can give rise to cerebral infarction. It has been reported that the protective effects of rapid postconditioning after I/R injury in myocardial injury and focal ischemia [32,33]. To demonstrate the neuroprotective effects of postconditioning, 30 min of tMCAO with 24 h of reperfusion was performed in the control group of mice. Postconditioning groups were given an additional 5 min of reperfusion and 5 min of tMCAO after 30 min of tMCAO and 24 h of reperfusion. Mice in the control group showed significant infarct volumes as determined by TTC staining (Fig. 1B). This tMCAO model generated ischemic damage in the cortex and striatum. In the postconditioning group, infarct size was significantly smaller than in the tMCAO group; postconditioning reduced the mean infarct size by approximately 60.51%.
To confirm the neuroprotective effect of postconditioning in response to oxidative stress, OGD was carried out in primary neurons, bEnd.3 and BV-2 cells. Cells underwent 120 min of OGD followed by 10 min of reperfusion and 10 min of OGD for postconditioning. As illustrated in Fig. 2B, postconditioning significantly reduced cell cytotoxicity, which had been increased by hypoxia and 4 h of reperfusion. These results provide strong evidence that postconditioning has neuroprotective effects on oxidative stress, which can arise from tMCAO or OGD-induced hypoxia.
NF-κB activation and Akt phosphorylation are increased after postconditioning in tMCAO and OGD
It has also been shown that phosphorylated Akt activates IκB kinase (IKK) α/β [21]. In turn, activated IKKα/β leads to phosphorylation of IκBα/β and increased nuclear translocation of NF-κB-dependent prosurvival gene. tMCAO increased nuclear translocation of p65 at 1 and 4 h after reperfusion (Fig. 3A). Postconditioning induced higher translocation of p65, as early as 1 h after reperfusion, compared to tMCAO. Our results show that postconditioning increases NF-κB activation after tMCAO in the mouse brain. After 30 min of tMCAO, Akt was activated in the ipsilateral hemisphere at 1, 4, and 24 h after reperfusion. Postconditioning induced greater phosphorylation of Akt protein kinases than tMCAO. In accordance with increased NF-κB activation after postconditioning, quantitative analysis showed that postconditioning increased phosphorylation of Akt after tMCAO in the mouse brain (Fig. 3B).
Fig. 3. NF-κB activation and Akt phosphorylation were increased after postconditioning in mice. (A) The effects of postconditioning on the NF-κB pathway were confirmed using western blot analysis. Postconditioning increased nuclear translocation of p65, especially at 1 and 4 h reperfusion following tMCAO. Postconditioning increased the phosphorylation of Akt after tMCAO in mice. (B) Relative changes in the amount of pAkt and p65 protein after tMCAO and postconditioning; difference from the sham group (#p<0.05, ##p<0.01) and from the tMCAO group (*p<0.05, ***p<0.001). Data are mean±SE. n=5.

OGD was used to confirm the activation of Akt and NF-κB in response to oxidative stress. Phosphorylation of Akt and translocation of p65 were evaluated after 120 min of OGD and postconditioning. In BV-2 cells, OGD increased NF-κB translocation and Akt activation (Fig. 4). Postconditioning increased phosphorylation of Akt and nuclear translocation of NF-κB compared to OGD. These trends were also observed in bEnd.3 cells (Fig. 5). Nuclear translocation of p65 and phosphorylation of Akt were increased in the OGD group. In the postconditioning group, greater activation of Akt and translocation of p65 to the nucleus, especially at early time points after reperfusion, was observed, which was consistent with findings in the mouse brain. Our results demonstrate that NF-κB activation through Akt is involved in the neuroprotective mechanism of postconditioning.
Fig. 4. Postconditioning activated Akt and p65 after OGD in BV-2 cells. (A) Nuclear translocation of p65 was increased in postconditioning after OGD. Postconditioning also prolonged the phosphorylation of Akt. (B) Relative changes in the amount of p-Akt and p65 protein after OGD and postconditioning. Data are mean±SE. n =4. Data were analyzed by ANOVA (#p<0.05, ##p<0.01, ###p<0.001 compared with the normoxia group; *p<0.05, ***p<0.001 compared with the OGD group at the same time point).

Fig. 5. Postconditioning activated Akt and p65 after OGD in bEnd.3 cells. (A) Postconditioning increased the translocation of p65 from the cytoplasm to the nucleus after OGD. Western blot analysis probed the phosphorylated form of Akt, which indicated that postconditioning increased the phosphorylation of Akt at an early time point after reperfusion. (B) Relative changes in the protein levels of p-Akt and p65 after OGD and postconditioning. Data mean±SE. n=3. Data were analyzed by ANOVA (#p<0.05 compared with the normoxia group; *p<0.05 compared with the OGD group at the same time point).

Akt inhibition reduces postconditioning-induced NF-κB activation
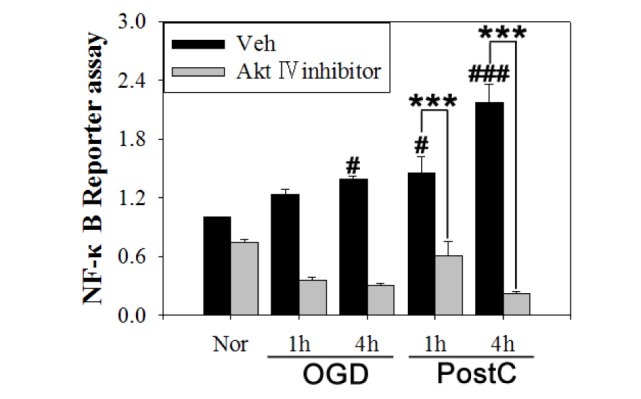
To verify the mechanism by which postconditioning has a neuroprotective effect, a NF-κB luciferase reporter assay was performed in mouse endothelial cells after OGD (Fig. 6). NF-κB activity was detected by the reporter gene assay in OGD and postconditioning groups 1 and 4 h after reperfusion in bEnd.3 cells. NF-κB promoter activity was increased by 23% in the OGD group compared to the normoxia group after 4 h reperfusion. NF-κB promoter activity was increased by 11% and 34% in the postconditioning group compared to the OGD group at 1 and 4 h reperfusion, respectively. In the presence of Akt inhibitor IV, this increase was significantly reduced in the OGD and postconditioning groups but was unaffected in the normoxia group. Our results show that postconditioning activates the NF-κB promoter, which is blocked by Akt inhibitor IV. These results indicate that postconditioning induces NF-κB-mediated neuroprotection through Akt activation.
Fig. 6. Akt inhibitor IV reduced the activity of the NF-κB promoter in postconditioning. NF-κB activity was detected by a reporter gene assay in bEnd.3 cells at 1 and 4 h reperfusion. In the OGD group, NF-κB promoter activity was increased compared to the normoxia group, and activity was higher in the postconditioning group than the OGD group. When Akt inhibitor IV was applied, the activity of the NF-κB promoter was reduced in the OGD group and was even lower in the postconditioning group. Data are mean±SE. n=4. Data were analyzed by ANOVA (#p<0.05 and ###p<0.001 compared with normoxia group; ***p<0.001 compared with the vehicle group at the same time point).
TLR5-MyD88, upstream of NF-κB, mediates the neuroprotective effect of postconditioning via Akt activation
It has been shown that activation of the TLR5 receptor mobilizes NF-κB in intestinal epithelial cells. Additionally, it has been demonstrated in epithelial cells that PI3K is involved in TLR5 signaling, and that MyD88 is necessary for the association of TLR5 with PI3K [34,35]. We investigated whether TLR5 might be associated with activation of NF-κB and Akt after postconditioning in the brain. TLR5 levels were not significantly altered after tMCAO (Fig. 7A). However, compared with the tMCAO group, MyD88 was increased in the postconditioning group at 1 and 4 h after reperfusion. Since there was not a significant change in the levels of TLR5 after tMCAO, we focused on the recruitment of MyD88 and p-Akt by TLR5. To confirm whether TLR5 interacts with MyD88 after postconditioning, we captured MyD88-bound proteins by IP with an anti-MyD88 antibody. Western blot analysis showed increases in TLR5 interactions with p-Akt with MyD88 after tMCAO at 1 h after reperfusion. The postconditioning group showed increased recruitment of TLR5 and p-Akt compared to the tMCAO group.
Fig. 7. Changes in TLR5 and MyD88 after postconditioning. (A) There was no difference in the amount of TLR5 between tMCAO and postconditioning groups. Compared with after tMCAO, MyD88 was slightly increased after postconditioning at 1 and 4 h reperfusion. MyD88 was captured by IP with an anti-MyD88 antibody in the total fraction. Results were detected by immunoblotting (IB) using antibodies to TLR5, p-Akt, and Akt. Coimmunoprecipitation analysis for TLR5/p-Akt showed an increase in the postconditioning group compared with the tMCAO group. Input lysates showed little difference between groups. Data are mean±SE. n=5 (#p<0.05, ##p<0.01, and ###p<0.001 compared with normoxia group). (B) Changes in TLR5 and MyD88 after OGD were in accordance with findings in vivo. Postconditioning increased the interaction of these proteins. Data are mean±SE. n=4 (#p<0.05 and ###p<0.001 compared with normoxia group; *p<0.05 compared with the OGD group).

To confirm the interaction between TLR5 and MyD88, IP was performed in BV-2 and bEnd.3 cells (Fig. 7B). After 120 min of OGD, the recruitment of TLR5 and p-Akt was increased in BV-2 cells. In the postconditioning group, interactions between TLR5, MyD88 and p-Akt were greater than in the OGD group 1 h after reperfusion. These changes were also observed in bEnd.3 cells. The activation of MyD88 recruited TLR5 and p-Akt after OGD. Postconditioning further increased interactions between MyD88 and TLR5 or p-Akt. Our results demonstrated that TLR5 recruited MyD88 and p-Akt in response to postconditioning.
Flagellin, a TLR5 agonist, has a neuroprotective effect via NF-κB activation in cerebral ischemic damage
To investigate the role of TLR5 signaling in postconditioning after cerebral ischemia, we treated mice with the TLR5-specific agonist flagellin. Flagellin was injected i.c.v. (50 or 100 ng/5 µl) 5 min after the onset of 30 min of tMCAO. Compared to the vehicle group, flagellin administration significantly reduced ischemic infarct volume (Fig. 8B). Additionally, administration of 50 or 100 ng of flagellin induced increased nuclear translocation of NF-κB and Akt phosphorylation compared to the vehicle group at 1h after reperfusion following tMCAO (Fig. 8C). Quantitative analysis showed that flagellin increased Akt phosphorylation and nuclear translocation of p65 after tMCAO in mice.
Fig. 8. Administration of flagellin reduced ischemic infarct volume and increased the activation of NF-κB and p-Akt after tMCAO in mice. (A) Schematic diagram showing the time window of flagellin injection after tMCAO in mice. (B) Animals that underwent a 30-min period of tMCAO had a significant infarct after 24 h of reperfusion. Administration of flagellin, especially at 50 and 100 ng/5 µl i.c.v., reduced these infarct volumes. This graph displays the statistical analysis of infarct volume. Data are mean±SE. n=3. Data were analyzed by ANOVA (#p<0.05 compared with tMCAO group). (C) Effects of flagellin on NF-κB and Akt signaling using western blotting. Flagellin administration, especially at 50 and 100 ng/5 µl i.c.v., increased the translocation of p65 to the nucleus in mice. Phosphorylation of Akt was also increased in mice treated with flagellin. These results suggest that flagellin at low doses and early time points has neuroprotective effects on cerebral ischemia. The graphs illustrate the relative changes in the amount of phosphorylated Akt and p65. Data are mean±SE. n=4.

In microglial and endothelial cells, flagellin treatment (0.1, 0.25, or 0.5 ng/ml) reduced LDH release after OGD (Fig. 9A). To confirm that flagellin regulates NF-κB via TLR5 signaling in the brain, a NF-κB luciferase reporter assay was performed in mouse endothelial cells (Fig. 9B). NF-κB activity was increased in the vehicle group compared to the normoxia group. In the flagellin-treated group, luciferase activity was increased 47%, 31%, and 23% at 0.10, 0.25, and 0.50 ng/ml, respectively, compared with the vehicle group. A lower dose of flagellin (0.1 ng/ml) is more effective than a higher dose in reducing cell death and NF-κB activation. In BV-2 cells, p-Akt activation and translocation of NF-κB were also increased in the flagellin-treated group compared with the vehicle group after 120 min of OGD (Fig. 9C). Similar results were shown in bEnd.3 cells. In the flagellin-treated group, nuclear translocation of p65 and Akt activation were increased compared to the vehicle group. These results provide evidence that flagellin has a neuroprotective effect on cerebral ischemia and hypoxia in mice. The neuroprotective effects of flagellin were mediated through the NF-κB/TLR5 pathway, which is one of the neuroprotective mechanistic pathways associated with postconditioning. These data may lead to the development of pharmacological postconditioning agents, such as low doses of flagellin, which mimic the actions of postconditioning.
Fig. 9. Treatment of flagellin increased cell survival in hypoxic conditions. (A) After 2 h OGD, cell cytotoxicity levels were measured by a LDH assay at 1 h reperfusion. The flagellin-treated groups had lower cell cytotoxicity than the vehicle group. Up to 0.5 ng/ml, flagellin had protective effects on BV-2 and bEnd.3 cells. Data are mean±SE. n =4 (###p<0.001 compared with normoxia group; *p<0.05 and ***p<0.001 compared with the vehicle group). (B) NF-κB activity was detected by a reporter gene assay in bEnd.3 cells 1 h after reperfusion. In the vehicle group, the expression of the NF-κB promoter was increased compared to the normoxia group. When cells were treated with flagellin, the activity of NF-κB was increased compared to the vehicle group. Data are mean±SE. n=3. (C) Western blot analysis revealed that flagellin treatment increased the phosphorylation of Akt compared to vehicle treatment after 1 h reperfusion. Flagellin treatment increased the nuclear translocation of p65 compared to vehicle at the same time point. Data are mean±SE. n=4 (#p<0.05, ##p<0.01, and ###p<0.001 compared with normoxia group; *p<0.05 and **p<0.01 compared with the vehicle group).

DISCUSSION
Many studies on postconditioning have been performed in cardiac research and with clinical application over the last few years [36,37,38,39]. However, little is known about the mechanisms mediating the neuroprotective effects of postconditioning against cerebral ischemia. It has been demonstrated that postconditioning has neuroprotective effects and reduces infarct size after cerebral ischemia and neurological deficits [7,33]. Postconditioning in the brain involves activation of survival signals, such as Akt and ERK [8,19]. Additionally, it has been demonstrated that the expression of endogenous antioxidant enzymes, such as Mn-SOD, is increased in response to postconditioning after cerebral ischemia [40]. In this study, we showed that postconditioning reduced infarct size when 5 min of tMCAO was again executed after 30 min of tMCAO. After 120 min of OGD, postconditioning protected mouse primary neurons, microglia, and endothelial cells from cell death. Although many efforts have concentrated on studying postconditioning after cerebral ischemia and hypoxia, the neuroprotective mechanisms of postconditioning remain elusive.
It has been demonstrated that NF-κB regulates inflammation and immunity and induces anti-apoptosis [10]. In myocardial ischemia, preconditioning with sevoflurane upregulated NF-κB and anti-apoptotic proteins and downregulated inflammatory ICAM-1, TNFα, and caspase-3 [41]. The activation of NF-κB was found to be the key mechanism by which ischemic preconditioning acted in the brain, and NF-κB was implicated in postconditioning-induced neuroprotection from cerebral ischemia [13]. This report supports our results that postconditioning after 30 min of tMCAO activated the nuclear translocation of NF-κB and phosphorylation of Akt, especially at 1 and 4 h reperfusion. Additionally, postconditioning reduced cell death and increased the activation of NF-κB and Akt after OGD in bEnd.3 and BV-2 cells. To uncover a correlation between Akt signaling and NF-κB, a NF-κB reporter assay was performed after postconditioning with Akt inhibitor IV. Our results showed that postconditioning increased NF-κB activity compared to OGD. We supposed that differences in luciferase activity between postconditioning and OGD groups were derived from the role of Akt/NF-κB in postconditioning. Blocking Akt activation by its specific inhibitor, Akt inhibitor IV, significantly reduced NF-κB reporter activity in postconditioning and OGD groups. The decrease in NF-κB was greater in the postconditioning group than in the OGD group, which suggested that Akt signaling was involved in the neuroprotective action of postconditioning. Interestingly, NF-κB activity was not reduced in normoxia after treatment with Akt inhibitor IV. This suggests that other signaling pathways, aside from Akt, may mediate the activation of NF-κB in normoxia. Therefore, treatment with Akt inhibitor IV might differentially influence NF-κB activity in normoxic and hypoxic conditions.
Upstream of Akt/NF-κB, TLR5's role in ischemic postconditioning has not been studied. It has been reported that TLR5 induced rapid PI3K activation, as evidenced by Akt phosphorylation, and TLR5 mediated PI3K negative regulation of proinflammatory gene expression in intestinal epithelial cells [42]. TLR5 delayed neutrophil apoptosis, which was dependent on the activation of NF-κB and phosphorylation of Akt, a major target of PI3K [43,44]. Our results showed that the expression level of TLR5 varied little between the tMCAO and postconditioning groups. However, the interactions between TLR5, MyD88, and p-Akt were increased when postconditioning was executed after tMCAO. Increasing the recruitment of MyD88 and p-Akt to TLR5 was consistent in mouse brain microglia and endothelial cells. We focused on the interactions captured by MyD88, because it had been reported that recruitment of the adaptor molecule MyD88 to TLR5 induced PI3K, and silencing MyD88 blocked PI3K activation in colonocytes [35]. Downregulated TLR5 reduced the differences of p-Akt and MyD88 between OGD and postconditioning, demonstrating that TLR5 had a relevant role in postconditioning-induced neuroprotection.
It was demonstrated that TLR5-activated NF-κB had radioprotective activity in lethally irradiated rhesus monkeys, and antiapoptotic mechanisms were involved in this radioprotection [45]. A TLR5 agonist given after ischemic kidney reperfusion had a protective effect in acute renal ischemic failure [46]. In agreement with these studies, our results demonstrate that the neuroprotective effects of postconditioning are mediated by the TLR5 pathway after tMCAO in the brain. Treatment with flagellin, a TLR5 agonist, increased nuclear translocation of NF-kB and phosphorylation of Akt, which were similar actions observed in response to postconditioning after tMCAO in mice and to OGD in primary neurons and cell lines. Flagellin significantly reduced cell cytotoxicity and increased NF-kB promoter activity. Although these results provided flagellin-induced protection against focal cerebral ischemia, further development of the reagent and its administration will be necessary to carry into future clinical treatment. For instance, it is difficult for flagellin to cross the blood-brain barrier, because it is a protein and has a high mass (30~60 kDa). Much effort will be needed to construct a delivery system for it to reach the brain. Since flagellin has shown immunogenicity and toxicity, there has been an attempt to reduce its immunogenic properties by using CBLB502 instead [45]. CBLB502 is a recombinant protein of Salmonella enterica flagellin and is less toxic than flagellin, with a maximum tolerated dose of 25 mg/kg in mice compared to 12 mg/kg for flagellin [47]. Using pharmacological postconditioning, such as treatment with detoxified flagellin, would be a more likely avenue to carry flagellin into clinical trials for the treatment of cerebral ischemia.
In conclusion, we have demonstrated the importance of NF-κB in mediating neuroprotection in response to postconditioning and suggest that TLR5 is an upstream neuroprotective factor during postconditioning after tMCAO. In addition, a pharmacological postconditioning treatment, such as a low dose of flagellin, has the potential to be developed as a therapy for stroke.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea grant (NRF-2016K1A1A8A01939090, 2017M3C7A1031102) funded by the Government of Korea (MSIP).
References
- 1.Hoyte L, Kaur J, Buchan AM. Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol. 2004;188:200–204. doi: 10.1016/j.expneurol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- 3.Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 4.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 5.Li S, Hafeez A, Noorulla F, Geng X, Shao G, Ren C, Lu G, Zhao H, Ding Y, Ji X. Preconditioning in neuroprotection: from hypoxia to ischemia. Prog Neurobiol. doi: 10.1016/j.pneurobio.2017.01.001. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- 7.Rehni AK, Singh N. Role of phosphoinositide 3-kinase in ischemic postconditioning-induced attenuation of cerebral ischemia-evoked behavioral deficits in mice. Pharmacol Rep. 2007;59:192–198. [PubMed] [Google Scholar]
- 8.Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermann R, Marina Prendes MG, Torresin ME, Vélez D, Savino EA, Varela A. Effects of the AMP-activated protein kinase inhibitor compound C on the postconditioned rat heart. J Physiol Sci. 2012;62:333–341. doi: 10.1007/s12576-012-0209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 11.Irving EA, Hadingham SJ, Roberts J, Gibbons M, Chabot-Fletcher M, Roshak A, Parsons AA. Decreased nuclear factor-kappaB DNA binding activity following permanent focal cerebral ischaemia in the rat. Neurosci Lett. 2000;288:45–48. doi: 10.1016/s0304-3940(00)01203-9. [DOI] [PubMed] [Google Scholar]
- 12.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 13.Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci. 2001;21:4668–4677. doi: 10.1523/JNEUROSCI.21-13-04668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehni AK, Bhateja P, Singh N. Diethyl dithiocarbamic acid, a possible nuclear factor kappa B inhibitor, attenuates ischemic postconditioning-induced attenuation of cerebral ischemia-reperfusion injury in mice. Can J Physiol Pharmacol. 2009;87:63–68. doi: 10.1139/Y08-100. [DOI] [PubMed] [Google Scholar]
- 15.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 16.del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 17.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res. 2004;95:230–232. doi: 10.1161/01.RES.0000138303.76488.fe. [DOI] [PubMed] [Google Scholar]
- 19.Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab. 2008;28:232–241. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- 20.Gao X, Zhang H, Steinberg G, Zhao H. The Akt pathway is involved in rapid ischemic tolerance in focal ischemia in rats. Transl Stroke Res. 2010;1:202–209. doi: 10.1007/s12975-010-0017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 22.Song YS, Narasimhan P, Kim GS, Jung JE, Park EH, Chan PH. The role of Akt signaling in oxidative stress mediates NF-kappaB activation in mild transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1917–1926. doi: 10.1038/jcbfm.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 24.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 25.O'Neill LA. TLRs: Professor Mechnikov, sit on your hat. Trends Immunol. 2004;25:687–693. doi: 10.1016/j.it.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Akira S. Mammalian Toll-like receptors. Curr Opin Immunol. 2003;15:5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 27.Rhee SH, Hwang D. Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase. J Biol Chem. 2000;275:34035–34040. doi: 10.1074/jbc.M007386200. [DOI] [PubMed] [Google Scholar]
- 28.Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SL, Cookson BT, Aderem A. Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol. 2003;4:1247–1253. doi: 10.1038/ni1011. [DOI] [PubMed] [Google Scholar]
- 29.Vijay-Kumar M, Aitken JD, Sanders CJ, Frias A, Sloane VM, Xu J, Neish AS, Rojas M, Gewirtz AT. Flagellin treatment protects against chemicals, bacteria, viruses, and radiation. J Immunol. 2008;180:8280–8285. doi: 10.4049/jimmunol.180.12.8280. [DOI] [PubMed] [Google Scholar]
- 30.Rhee SH, Kim H, Moyer MP, Pothoulakis C. Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J Biol Chem. 2006;281:18560–18568. doi: 10.1074/jbc.M513861200. [DOI] [PubMed] [Google Scholar]
- 31.Rhee SH, Keates AC, Moyer MP, Pothoulakis C. MEK is a key modulator for TLR5-induced interleukin-8 and MIP3alpha gene expression in non-transformed human colonic epithelial cells. J Biol Chem. 2004;279:25179–25188. doi: 10.1074/jbc.M400967200. [DOI] [PubMed] [Google Scholar]
- 32.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 33.Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- 34.Ivison SM, Khan MA, Graham NR, Shobab LA, Yao Y, Kifayet A, Sly LM, Steiner TS. The p110α and p110β isoforms of class I phosphatidylinositol 3-kinase are involved in toll-like receptor 5 signaling in epithelial cells. Mediators Inflamm. 2010;2010:652098. doi: 10.1155/2010/652098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhee SH, Kim H, Moyer MP, Pothoulakis C. Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J Biol Chem. 2006;281:18560–18568. doi: 10.1074/jbc.M513861200. [DOI] [PubMed] [Google Scholar]
- 36.Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 37.Yellon DM, Hausenloy DJ. Realizing the clinical potential of ischemic preconditioning and postconditioning. Nat Clin Pract Cardiovasc Med. 2005;2:568–575. doi: 10.1038/ncpcardio0346. [DOI] [PubMed] [Google Scholar]
- 38.Fan Q, Yang XC, Liu Y, Wang LF, Liu SH, Ge YG, Chen ML, Wang W, Zhang LK, Irwin MG, Xia Z. Postconditioning attenuates myocardial injury by reducing nitro-oxidative stress in vivo in rats and in humans. Clin Sci (Lond) 2011;120:251–261. doi: 10.1042/CS20100369. [DOI] [PubMed] [Google Scholar]
- 39.Na HS, Kim YI, Yoon YW, Han HC, Nahm SH, Hong SK. Ventricular premature beat-driven intermittent restoration of coronary blood flow reduces the incidence of reperfusion-induced ventricular fibrillation in a cat model of regional ischemia. Am Heart J. 1996;132:78–83. doi: 10.1016/s0002-8703(96)90393-2. [DOI] [PubMed] [Google Scholar]
- 40.Nemethova M, Danielisova V, Gottlieb M, Burda J. Post-conditioning exacerbates the MnSOD immune-reactivity after experimental cerebral global ischemia and reperfusion in the rat brain hippocampus. Cell Biol Int. 2008;32:128–135. doi: 10.1016/j.cellbi.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 41.Wang C, Xie H, Liu X, Qin Q, Wu X, Liu H, Liu C. Role of nuclear factor-kappaB in volatile anaesthetic preconditioning with sevoflurane during myocardial ischaemia/reperfusion. Eur J Anaesthesiol. 2010;27:747–756. doi: 10.1097/EJA.0b013e32833bb3ba. [DOI] [PubMed] [Google Scholar]
- 42.Yu Y, Nagai S, Wu H, Neish AS, Koyasu S, Gewirtz AT. TLR5-mediated phosphoinositide 3-kinase activation negatively regulates flagellin-induced proinflammatory gene expression. J Immunol. 2006;176:6194–6201. doi: 10.4049/jimmunol.176.10.6194. [DOI] [PubMed] [Google Scholar]
- 43.François S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol. 2005;174:3633–3642. doi: 10.4049/jimmunol.174.6.3633. [DOI] [PubMed] [Google Scholar]
- 44.Salamone GV, Petracca Y, Fuxman Bass JI, Rumbo M, Nahmod KA, Gabelloni ML, Vermeulen ME, Matteo MJ, Geffner JR, Trevani AS. Flagellin delays spontaneous human neutrophil apoptosis. Lab Invest. 2010;90:1049–1059. doi: 10.1038/labinvest.2010.77. [DOI] [PubMed] [Google Scholar]
- 45.Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D, Kurnasov OV, Fort FL, Osterman AL, Didonato JA, Feinstein E, Gudkov AV. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science. 2008;320:226–230. doi: 10.1126/science.1154986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuzawa N, Petro M, Baldwin WM, 3rd, Gudkov AV, Fairchild RL. A TLR5 agonist inhibits acute renal ischemic failure. J Immunol. 2011;187:3831–3839. doi: 10.4049/jimmunol.1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eaves-Pyles T, Murthy K, Liaudet L, Virág L, Ross G, Soriano FG, Szabó C, Salzman AL. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J Immunol. 2001;166:1248–1260. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]