

Abstract
Uptake of low-density lipoprotein (LDL) particles by macrophages represents a key step in the development of atherosclerotic plaques, leading to the foam cell formation. Chemical modification of LDL is however necessary to induce this process. Proatherogenic LDL modifications include aggregation, enzymatic digestion and oxidation. LDL oxidation by one-electron (free radicals) and two-electron oxidants dramatically increases LDL affinity to macrophage scavenger receptors, leading to rapid LDL uptake and fatty streak formation.
Circulating high-density lipoprotein (HDL) particles, primarily small, dense, protein-rich HDL3, provide potent protection of LDL from oxidative damage by free radicals, resulting in the inhibition of the generation of pro-inflammatory oxidized lipids. HDL-mediated inactivation of lipid hydroperoxides involves their initial transfer from LDL to HDL and subsequent reduction to inactive hydroxides by redox-active Met residues of apolipoprotein A-I. Several HDL-associated enzymes are present at elevated concentrations in HDL3 relative to large, light HDL2 and can be involved in the inactivation of short-chain oxidized phospholipids. Therefore, HDL represents a multimolecular complex capable of acquiring and inactivating proatherogenic lipids.
Antioxidative function of HDL can be impaired in several metabolic and inflammatory diseases. Structural and compositional anomalies in the HDL proteome and lipidome underlie such functional deficiency. Concomitant normalization of the metabolism, circulating levels, composition and biological activities of HDL particles, primarily those of small, dense HDL3, can constitute future therapeutic target.
Highlights
-
•
LDL oxidation dramatically increases LDL affinity to macrophage scavenger receptors, leading fatty streak formation.
-
•
HDL particles, primarily small, dense HDL3, provide potent protection to LDL from oxidative damage by free radicals.
-
•
HDL represents a multimolecular complex capable of acquiring and inactivating of proatherogenic oxidized lipids.
-
•
Antioxidative function of HDL is impaired in metabolic diseases; normalization of HDL functions can be a therapeutic target.
1. Introduction
Atherosclerosis is a complex process which begins in areas where disturbed blood flow induces partial endothelial activation. Activated endothelial cells produce adhesion molecules, including E-selectin, P-selectin, and vascular cell adhesion molecule-1 (VCAM-1), causing monocytes to attach to the endothelium and to move into the arterial intima. Monocytes are then transformed into macrophages and further activated by encounters with pathogen- and damage-associated molecular patterns, as well as by various proinflammatory cytokines [1]. A key step in the development of the atherosclerotic plaque is the engulfment by endocytosis of low-density lipoprotein (LDL) particles by macrophages, leading to the formation of foam cells, which together with activated endothelial cells secrete a host of proinflammatory cytokines, matrix metalloproteinases, and cathepsins, fragilising the plaque [1]. However, in vitro experiments have shown that macrophages present low expression of the LDL receptor and possess little affinity for unmodified LDL particles [2]. Chemical modification of LDL is necessary to induce the formation of foam cells. Such proatherogenic modifications of LDL include aggregation, enzymatic digestion and oxidation. LDL oxidation dramatically increases the affinity of the particle to macrophage scavenger receptors, leading to rapid uptake of the modified lipoproteins [3]. This process begins when, following transcytosis to the subendothelial space [4], LDL particles are retained in the arterial wall through specific interactions between apolipoprotein (apo) B and arterial proteoglycans [5], [6], [7], [8]. This interaction is mediated by specific apo B residues at the positions 3359 to 3369 [9].
Oxidative stress in the subendothelial space is responsible for the formation of oxidized LDL (oxLDL) via oxidants of both enzymatic and non-enzymatic origin [3] (Fig. 1). Indeed, LDL oxidation is negligible in the circulation, reflecting the presence of multiple antioxidant defense systems which involve antioxidant enzymes, such as superoxide dismutase, catalase and glutathione-peroxidase, but equally water-soluble antioxidants, such as ascorbate, urate and bilirubin [10], [11], [12], [13]. Additionally, lipid-soluble vitamin E, mainly in the form of α-tocopherol, is transported by LDL particles together with other lipophilic antioxidants, such as ubiquinol-10, which protect LDL from oxidation not only in plasma but also in the arterial wall [14]. Exposure to cell-derived oxidants is necessary for the oxidative modification of LDL. These oxidants can be of free-radical (one-electron) and non-free-radical (two-electron) nature, and mainly originate from reactions catalyzed by 12/15-lipoxygenase, myeloperoxidase, nitric oxide synthases and NADPH oxidases, as well as from those mediated by transition metals, heme and hemoglobin [15]. Two-electron oxidants (hypochlorite, peroxynitrite) modify LDL almost instantly, primarily attacking apo B. The process of LDL oxidation by one-electron oxidants (free radicals) is assumed to occur in two main steps. During initial stages, oxidative modifications of LDL lipids occur in the absence of changes in apo B. Such modified lipoprotein is termed minimally oxidized LDL. It retains the affinity for the LDL receptor, does not possess elevated negative charge, activates anti-apoptotic signaling, and induces inflammatory pathways, involving increased chemokines and cytokine secretion [16]. Recruitment of inflammatory cells may result in enhanced production of a variety of cytokines and further oxidation of LDL. Subsequently, LDL lipids are oxidized to a greater extent, generating both free and core aldehydes, and ketones that covalently modify ε-amino groups of lysine residues present in apo B, which finally suffers a process of fragmentation. These events lead to a loss of recognition by the LDL receptor and to a shift to recognition by scavenger receptors, resulting in the formation of macrophage-derived foam cells [17]. Such highly oxidized LDL is commonly termed oxLDL; however, LDL may undergo various degrees of oxidation. Importantly, such a diversity of LDL oxidation states provides distinct biological effects on vascular cells [16]. For example, tissue factor expression in endothelial cells is exclusively induced by mildly oxidized LDL, whilst only lipids present in highly oxidized LDL are cytotoxic and pro-apoptotic [16]. Accordingly, oxLDL contains multiple products of free radical-induced lipid peroxidation, including lipid hydroperoxides (LOOH) as primary products, secondary LOOH-derived short-chain oxidized phospholipids (oxPL) and oxidized sterols [3].
Fig. 1.

Mechanisms of LDL oxidation in the arterial wall. Upon entry in the subendothelial space LDL particles are exposed to local oxidative stress arising from the presence of cell-associated enzymes, including NADPH oxidase, lipoxygenases and myeloperoxidase, as well as from transition metal ions. Oxidants produced by these entities may oxidize LDL to different degrees. Whereas NADPH oxidase and lipoxygenases only lead to the formation of minimally oxidized LDL, myeloperoxidase and NADPH oxidase combined with NO synthase oxidize LDL extensively. The minimally oxidized LDL is predominantly characterized by the presence of oxidized lipids, whilst extensive oxidation of both protein and lipid components constitutes the hallmark of extensively oxidized LDL. Minimally oxidized LDL displays low affinity to macrophage scavenger receptors and can readily return to the bloodstream. Locally, minimally oxidized LDL induces inflammatory activation involving chemokine and cytokine production and recruitment of inflammatory cells, which in turn increases chemokine and cytokine accumulation. As a result, lipid oxidation proceeds ending in the formation of heavily oxidized and fragmented apo B. Such extensively oxidized LDL are readily taken up by macrophages via scavenger receptors leading to the formation of foam cells.
LDL, low density lipoprotein; oxLDL, oxidized LDL; apo, apolipoprotein; oxPL, oxidized phospholipid; LOOH, lipid hydroperoxide.
Nevertheless, it is important to note that characterization of naturally occurring oxLDL remains challenging. There is convincing evidence that oxLDL is present in atherosclerotic lesions of both humans and experimental animals [18]. Indeed, modified LDL particles with properties similar to those of in vitro highly oxidized LDL have been isolated from atherosclerotic lesions of both humans and rabbits [19]. In addition, levels of minimally oxidized LDL have been shown, through specific monoclonal antibodies, to be increased in the circulation of individuals suffering from coronary artery disease [20].
High density lipoprotein (HDL) has been postulated to exert an important antioxidative function, among other antiatherogenic activities. Such antoxidative capacity can be crucial for the impairment of atherosclerotic plaque formation and development. There is abundant evidence showing that atheroprotective actions of HDL involve its ability to protect LDL from oxidation by free radicals. Indeed, HDL potently protects both lipid and protein moieties of LDL, inhibiting accumulation of primary and secondary peroxidation products [21].
HDL particles are however highly heterogenic and can be separated using different techniques into distinct subclasses according to their physicochemical properties. Thus, HDL particles can be fractionated by sequential ultracentrifugation on the basis of density into large, light, lipid-rich HDL2 (d 1.063–1.125 g/ml) and small, dense, protein-rich HDL3 (d 1.125–1.21 g/ml) [22] (Table 1). HDL2 and HDL3 can be further subfractionated using non-denaturing polyacrylamide gradient gel electrophoresis into five subpopulations of increasing density and decreasing size, which include HDL2b, HDL2a, HDL3a, HDL3b and HDL3c [22]. Equivalent HDL subpopulations can be quantitatively isolated by isopycnic density gradient ultracentrifugation [22].
Table 1.
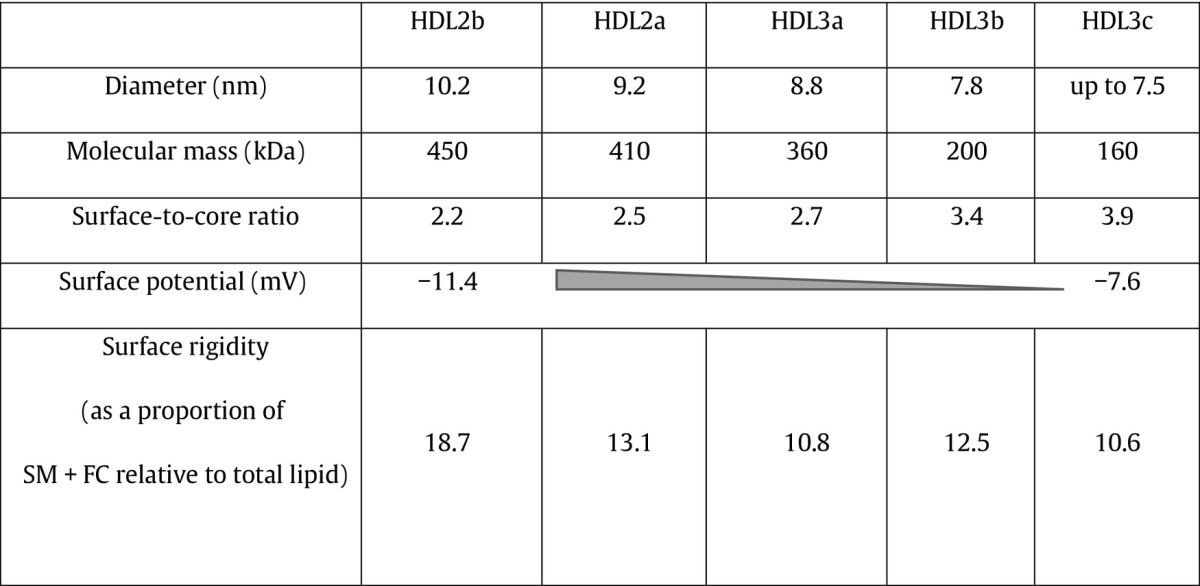
Surface-to-core ratio is calculated as a mass ratio of the sum of surface components (phospholipid, free cholesterol, protein) to the sum of core components (triglyceride, cholesteryl ester). HDL, high density lipoprotein; SM, sphingomyelin; FC, free cholesterol.
As expected, the diversity in the composition of HDL particles results in differences in the biological activity and atheroprotective potential of these subpopulations [23], [24], [25]. Unveiling specific mechanisms underlying the antioxidative role played by HDL particles can reveal novel pathways, which if manipulated, can help to combat atherosclerosis.
2. Antioxidative activity of HDL
In vitro experiments have provided evidence of HDL-mediated prevention of LDL oxidation (Fig. 2). HDL prevents the accumulation of lipid hydroperoxides in LDL [26], [27], without a parallel increase in HDL lipid hydroperoxide content when both lipoproteins are co-incubated under oxidizing conditions. The inhibition of oxidation by HDL can last for several hours and is not derived from the action of chain-breaking antioxidants or from chelation of transition metals but mainly due to protein-based activity [27], [28], [29]. It is worth noting that these effects of HDL are already observed using in vitro experiments carried out at similar protein concentrations of LDL and HDL. When HDL concentrations however exceed those of LDL, as they do in the subendothelial space, even greater suppression of LDL oxidation is to be expected.
Fig. 2.

Mechanisms of HDL-mediated protection against LDL oxidation in the arterial wall. HDL present in the intima space can protect LDL against oxidation by several mechanisms. HDL directly protects LDL from oxidation induced by one-electron oxidants (free radicals) and removes oxidized lipids from LDL. Altogether, these activities can decrease local concentrations of oxLDL; as a consequence, LDL can retain the capacity to re-enter the bloodstream for longer time periods, diminishing inflammatory impact of LDL oxidation. Antioxidative potential of HDL particles originates both from activities of their proteins and from lipid components. Noteworthy, HDL displays much lower protective activity (if any) against two-electron oxidants as compared to one-electron oxidants.
LDL, low density lipoprotein; HDL, high density lipoprotein; oxLDL, oxidized LDL; apo, apolipoprotein; oxPL, oxidized phospholipid; LOOH, lipid hydroperoxide; PON 1, paraoxonase 1; GPX3, Glutathione selenoperoxidase 3; PAF-AH, platelet-activating factor acetyl hydrolase; PS, phophatidylserine; S1P, sphingosine-1 phosphate.
Both chemical and physical properties of HDL determine the efficiency of this inhibition. We have previously established a role for the rigidity of HDL surface phospholipid monolayer as a modulator of the transfer efficiency of phosphatidylcholine hydroperoxides (PCOOH) from LDL to HDL [30]. In this study, both the PCOOH transfer rate from oxLDL to HDL and the capacity of HDL to delay LDL oxidation were inversely correlated with surface lipid rigidity in reconstituted HDL (rHDL). Furthermore, HDL can also remove LOOH from membranes of erythrocytes and astrocytes [31], [32], [33], a process also dependent on cellular membrane phospholipid rigidity [34]. Small, dense HDL3 particles may be superior to large, light HDL2 in terms of their capacity to prevent LDL oxidation [35], [36], [37]. Structural defects in the packing of surface lipids, which allow insertion of exogenous molecules, become more pronounced with decreasing HDL particle size and may account for this property [38]. Consistent with this finding, small discoid rHDL complexes appear to remove negatively charged lipids from oxidized LDL to a greater degree than native, spherical HDL [39]. Furthermore, antioxidative activity of HDL can increase in parallel to electronegativity [40]. However, it is important to note that these small HDL subpopulations also display distinct proteome and lipidome [24], [41], [42].
Chemical composition of HDL particles heavily influences their antioxidative properties [43]. HDL ability to decrease LDL oxidation is inversely correlated to its free cholesterol and sphingomyelin content, whilst positively to sphingosine-1-phosphate levels [42]. Furthermore, several HDL protein components, such as apo A-I [44], [45], apo A-II [46], apo A-IV [47], apo E [48], apo M [49], apo D, apo F, apo J, apo L-1 and serum amyloid A [41], can display antioxidative properties. In addition to these proteins, HDL-associated hydrolases, including paraoxonase (PON) 1, lecithin-cholesterol acyltransferase (LCAT) and platelet activating factor acetyl hydrolase (PAF-AH) [50], [51], can contribute to antioxidative activity of HDL. Lipid transfer proteins, notably cholesteryl ester transfer protein (CETP) and phospholipid transfer protein (PLTP), are another group of proteins which may be involved in the inhibition of LDL oxidation mediated by HDL [41], [52], [53]. Finally, HDL particles carry small amounts of lipophilic antioxidants, primarily tocopherols, which may provide a minor contribution to their antioxidative properties [54].
Transfer of LOOH can occur directly between lipoproteins, although evidence suggests that transfer proteins such as CETP can accelerate the process [52], [53].Consequently, HDL particles constitute a major carrier of LOOH in human plasma and have therefore been proposed to function as a ‘sink’ for oxidized lipids [55], which can accumulate when LOOH-inactivating capacity of HDL is overwhelmed. Consistent with this hypothesis, HDL has been shown to represent the major carrier of plasma F2-isoprostanes, stable end products of lipid peroxidation [56]. From the physiological point of view, such “sink-like” function of HDL may ensure efficient elimination of oxidized lipids from the circulation through the liver [52].
3. Molecular components of HDL contributing to its antioxidative activity
Different HDL-associated apolipoproteins, lipid transfer proteins and enzymes have been shown to contribute to antioxidative capacity of HDL (Table 2).
-
•
Apo A-I is the major HDL apolipoprotein which is synthesized by the liver and small intestine. Apart from being essential for HDL structure and for activation of HDL-associated enzymes such as PON1 and LCAT [57], it is involved in the HDL ability to neutralize one-electron free-radical oxidants. Indeed, Met residues located in the positions 112 and 148 of apo A-I can reduce LOOH into redox-inactive lipid hydroxides (LOH) by oxidation of their thiol groups, thereby terminating chain reactions of lipid peroxidation [44], [45]. Recent evidence suggests that Tyr 115 is also involved in such redox reactions [58]. Concentrations of redox-active Met residues in apo A-I and of PLOOH suggest a 1:1 reaction stoichiometry [30], [44], [45]. Furthermore, lipid-free apo A-I has been shown to remove seeding LOOH molecules from LDL [59]. Indeed, rHDL containing only purified apo A-I and phospholipids, show similar capacity to inhibit LDL oxidation as compared to native, normolipidemic small, dense HDL3b and 3c [30]. Thus, the redox state of apo A-I Met residues seems to represent a key determinant of HDL antioxidative capacity, following the transfer of LOOH to HDL particles.
-
•
Apo A-II is another apolipoprotein of hepatic origin preferentially associated with large HDL particles. These particles typically possess diminished antioxidative activity in comparison with small HDL subpopulations [60]. Nevertheless, Met residue-dependent antioxidative properties have previously been also attributed to apo A-II [46]. However, the finding that transgenic mice over-expressing human apo A-II present increased aortic accumulation of oxLDL and possess HDL with diminished antioxidative activity directly contradicts this assertion [61]. Indeed, apo A-II displaces both apo A-I and the antioxidant enzyme PON1 from HDL particles in these mice [61]. Furthermore, in vitro experiments reveal that apo A-II suppresses binding of PON1 to HDL [48], [62].
-
•
Apo A-IV shares several structural characteristics with apo A-I [63]. It is synthesized by the intestine and secreted on chylomicrons. In the circulation, apo A-IV dissociates from chylomicrons following triglyceride hydrolysis and associates to HDL by approximately 25% [64], [65]. Earlier studies carried out in mice have attributed antioxidative properties to apo A-IV [47]. A recent in vitro study suggested that only its lipid-free form possessed anthiaterogenic properties similar to those of apo A-I [66].
-
•
Apo E is another apolipoprotein component of HDL of hepatic origin which displays allele-specific antioxidative properties [67]. It can bind PON1 and stimulate its activity in a manner similar to apo A-I, though to a lesser extent [48]. However, a recent study carried out in transgenic mice suggested that apo E-containing HDL particles are markedly less effective in their antioxidative activity than apo A-I-containing particles [68].
-
•
Apo M is secreted by the liver and kidney and initially remains bound to cell membranes [69]. In the circulation, it is mainly found associated to HDL [70]. Recent evidence suggests that apo M can bind oxidized phospholipids and enhance antioxidative activity of HDL [49].
-
•
Other apolipoproteins which have been implicated in antoxidant activity of HDL include apo C-I, apo C-II, apo F, apo J, apo L-1 and apo D [41]. Apo C is a family of four proteins (apo C-I, apo C-II, apo C-III and apo C-IV) which belong to surface components of HDL, chylomicrons and VLDL. HDL particles are the main carrier of apo Cs in the fasting state [71]. Apo D is secreted by the brain and testes; in plasma, it is carried by HDL in association with LCAT, mediating interaction between HDL and LDL particles [72], [73]. Apo D contributes to antioxidative activity of HDL by reducing LOOH molecules [74]. Apo F is a modulator of CETP activity responsible for enhancing lipid transfer between VLDL and HDL, whilst inhibiting the exchange between VLDL and LDL [75]. Apo J is associated with HDL where it serves as a chaperone for misfolded proteins [76]. Several functions have been attributed to apo J, including protecting cell membranes and sensing oxidative status [76]. Apo L-1 binds in plasma to HDL3; the protein has been proposed to play a role in cell death and innate immunity [77], [78]. Interestingly, all these apolipoproteins were shown to be present in the proteome of HDL3c, the HDL subfraction which is highly potent in the prevention of LDL oxidation. Moreover, apo F, apo J and apo L-1 were exclusively found in HDL3, with HDL3c being their main carrier. Furthermore, HDL3c content of apo J, apo L-1 and apo D correlated positively with its antioxidative activity [41].
-
•
CETP is a plasma protein primarily secreted by the liver and adipose tissue. In plasma, it mediates the exchange of cholesteryl esters and triglycerides between apo B-containing lipoproteins and HDL [79]. Different studies have shown that CETP can facilitate the passage of both phospholipid and cholesteryl ester hydroperoxides from LDL to HDL particles [52], [53]. As a corollary, Hines et al. [80] showed that addition of CETP to HDL increases its ability to inhibit LDL oxidation.
-
•
PLTP is another lipid transfer protein involved in the remodeling of HDL particles [81]. It mediates the transfer of phospholipids from triglyceride-rich lipoproteins to HDL as well as phospholipid exchange between HDL subspecies [81]. It has been reported that PLTP can contribute to antioxidative activity of HDL, though no specific mechanisms have been revealed [41].
-
•
Serum amyloid A (SAA) is a family of liver-secreted proteins which are in the circulation predominantly found on HDL [82]. Three of the SAA isoforms (SAA 1, SAA 2 and SAA 3) are expressed during the acute phase of inflammation [83] and the fourth isoform (SAA 4) is expressed constitutively [84]. Earlier studies found a positive correlation between SAA 4 concentration and HDL antioxidative activity [41]. Furthermore, recent studies revealed that rHDL which contained SAA displayed higher capacity to inhibit LDL oxidation as compared to normal HDL [85], [86]. Indeed, HDL particles isolated from individuals presenting high SAA levels in the circulation showed potent antioxidative activity [85]. It is noteworthy in this regard that unbound SAA can bind lipid hydroperoxides, in a manner similar to Apo AI, though to a lesser extent [86].
-
•
PON1 is a glycoprotein enzyme which in the circulation exclusively associates with HDL. It is mainly secreted by the liver, though local synthesis occurs in several tissues [87], [88]. PON1 is capable of hydrolyzing a wide variety of substrates such as lactones, glucuronide drugs, thiolactones, arylesters, cyclic carbonates, organophosphorus pesticides and nerve gases [89], [90]. Its name is derived from the ability to hydrolyze pesticide paraoxon [91]. PON1 activity towards some substrates, including paraoxon, is influenced by genetic polymorphisms, the most studied of which is the R192Q substitution [92]. Traditionally, hydrolysis of oxPLs has been considered to represent the major function of this enzyme [21]. This activity involves hydrolysis of phosphatidylcholine-based oxPL and generation of bioactive lysophosphatidylcholine [89]. Furthermore, several studies have provided evidence linking PON1 to atherosclerosis. In vitro experiments first showed that the PON1-containing fraction of HDL was highly efficient in the prevention of the accumulation of lipid peroxides in LDL [26], [28]. Animal models have provided further evidence for the antioxidative role of PON1. Knockout mice, which lacked PON1, presented impaired antioxidative activity in HDL, in addition to increased susceptibility to organophosphate poisoning and atherosclerosis [93], [94]. Consistent with this finding, mice over-expressing human PON1 showed reduced generation of reactive oxygen species and decreased foam cell formation [95]. Different epidemiological studies have reported an inverse association between serum PON1 activity and coronary events [96], [97]. In particular, the largest single cohort study performed to date (Cleveland Clinic GeneBank study) involving 3668 patients following coronary angiography revealed a greater than two-fold risk of new cardiovascular events in the lowest quartile of serum PON1 activity compared with the highest one [98]. Regarding genetic evidence, studies have shown that PON1 192QQ homozygotes display HDL possessing elevated antioxidative activity [99], [100]. Nevertheless, other studies have questioned the relevance of PON1 as an antioxidant enzyme. Thus, PON1 failed to hydrolyze oxPLs in several in vitro experiments [101], [102], [103]. Moreover, PON1 is weakly reactive towards LOOH [104]. Indeed, Khersonsky et al. suggested that lactones and not oxPLs constitute physiological substrates of PON1 [90]. An interesting possibility derives from a recent work by Huang et al. [105] who propose that PON1 can form a tertiary complex with HDL and myeloperoxidase which can modulate the activity of the latter.
-
•
PON3 mainly functions as a lactonase and is also present in HDL particles, but to a lesser extent than PON1 [102]. Transgenic mice lacking PON3 are prone to atherosclerosis [106]. However, relevance of PON3 with respect to lipid oxidation remains unknown [41].
-
•
LCAT is another hydrolase associated with HDL. This enzyme catalyzes the conversion of free cholesterol into cholesteryl ester, thereby increasing its hydrophobicity [107]. LCAT can also hydrolyze oxidized acyl chains of oxPLs, generating lysophosphatidylcholine and oxidized free fatty acids [108]. Furthermore, LCAT can work as a chain-breaking antioxidant via its Cys residues at the positions 31 and 184 [109]. There is evidence that mutations in LCAT may reduce antioxidative capacity of HDL [110]. However, these mutations were not associated with increased concentrations of lipid peroxidation products in plasma [110]. Furthermore, inhibition of LCAT activity did not affect HDL capacity to neutralize lipid hydroperoxides derived from LDL [30].
-
•
PAF-AH, another hydrolase present in HDL, can hydrolyze oxidized short-chain phospholipids; however, it is inactive against long-chain non-oxidized phospholipids [111]. Macrophages represent the most important source of this enzyme [112]. Plasma PAF-AH circulates in the association with LDL and HDL particles, with the majority of the enzyme bound to small, dense LDL and to lipoprotein (a) [113]. Several studies suggest that PAF-AH is the main oxPL hydrolase in HDL [101], [103], implying that the minor fraction of this enzyme, carried by HDL, displays antiatherogenic properties [114], [115], [116]. Furthermore, PAF-AH is capable of hydrolyzing oxPLs within oxidized LDL; in this case, free fatty acid hydroperoxides are transferred to HDL for subsequent reduction to corresponding hydroxides by apo A-I [117]. It is worth noting that due to substrate overlapping, PAF-AH activity is difficult to separate from that of PON1 [118]. Nevertheless, as mentioned above, the majority of PAF-AH is carried by LDL, where its activity represents an independent risk factor for coronary heart disease [119]. Mechanisms underlying this association may involve release of lysophospholipids by LDL-associated PAF-AH in the subendothelial space where they become harmful to cells and membranes [119]. Interestingly, a rare example of HDL-mediated protection from two-electron oxidants consists in the inactivation by enzymes, such as PAF-AH and/or PON1 with a participation of apo A-I [120], of phospholipid core aldehydes generated upon HDL oxidation by peroxynitrite.
-
•
Glutathione selenoperoxidase 3 (GPX3), also known as glutathione peroxidase 3, is another antioxidant enzyme associated with HDL [121]. It is mainly secreted by the kidney [122] and, as all GPX enzymes, it protects biomolecules from oxidative damage by catalyzing the reduction of hydrogen peroxide, lipid peroxides and organic hydroperoxides, in a reaction which depends on the availability of reduced glutathione [123].
-
•
Lipid components also contribute to antioxidative activity of HDL (Table 3). As previously mentioned, such activity is negatively associated with free cholesterol and sphingomyelin content of HDL [42], both of which increase particle surface rigidity. In contrast, HDL ability to protect LDL oxidation correlates positively with sphingosine-1-phosphate [42] and phosphatidylserine [124] levels. Interestingly, earlier studies have shown an inverse association between phosphatidylcholine-based readily-oxidizable polyunsaturated fatty acid content [125] and antioxidative activity of HDL [126]. Furthermore, such activity is negatively associated with the content of proinflammatory PLs commonly present in these particles, such as phosphatidic acid [127], [128], ceramide [124], [126] and lysophosphatidylcholine [128]. Finally, both cholesteryl ester and triglyceride content have previously shown negative correlations with antioxidative activity of HDL [124].
Table 2.
Major lipoprotein-associated proteins harboring antioxidative activities (Ref. [43]).
| Protein | Main source | Preferential location in the circulation | Mechanism of antioxidative activity | Comments |
|---|---|---|---|---|
| ApoA-I | Liver, small intestine | HDL | Reduction of LOOH to redox-inactive LOH, ROS scavenging, PON1 activation | Largely determined by oxidative status of Met residues |
| ApoA-II | Liver | Large HDL | Reduction of LOOH to redox-inactive LOH | Largely determined by oxidative status of Met residues |
| ApoA-IV | Intestine | Chylomicrons, HDL | Removal of oxidized lipids from cells and lipoproteins, ROS scavenging | Primarily in a lipid-free form |
| ApoD | Brain, testes | HDL | Inhibition of lipid peroxidation by reduction of LOOH | Binds to LCAT, favors HDL to LDL association |
| ApoE | Liver | HDL3 | Inhibition of lipid peroxidation | Binds PON1, activity is allele-specific |
| ApoF | Liver | HDL, LDL | Modulator of CETP activity | |
| ApoJ | Brain, testes, ovary, liver, pancreas | HDL | Removal of oxidation products | |
| ApoL1 | Pancreas, lung, prostate, liver, placenta, spleen | HDL | Unknown | |
| ApoM | Liver, kidney | HDL | Unknown | |
| SAA | Liver | HDL | Binding of LOOH | |
| PON1 | Liver | HDL | Hydrolysis of short-chain oxidized PLs | Weak activity towards LOOH |
| LCAT | Liver | HDL | ||
| PAF-AH | Macrophages | LDL, Lp(a), HDL | Strong activity towards LOOH | |
| PON3 | Liver, kidney | HDL | Lactonase activity | |
| GPX3 | Kidney | HDL | Reduction of LOOH | Glutathione-dependent activity |
| CETP | Liver, adipose tissue | HDL | Enhanced transfer of oxidized lipids between LDL and HDL | Exchanges CE for TG between VLDL, LDL and HDL |
| PLTP | Placenta, pancreas, lung, kidney, heart, liver, muscle, brain | HDL | Unknown |
Apo, apolipoprotein; HDL, high density lipoprotein; LDL, low density lipoprotein; VLDL, very low density lipoprotein; SAA, serum amyloid A; LCAT, lecithin cholesterol acyltransferase; PON, paraoxonase; PAF-AH, platelet activating factor acetyl hydrolase; GPX3, glutathion selenoperoxidase 3; CETP, cholesteryl-ester transfer protein; PLTP, phospholipid transfer protein; ROS, recative oxygen species; LOOH, lipid hydroperoxide; LOH, lipid hydroxide; PL, phospholipids; CE, cholesteryl ester; TG, triglyceride.
Table 3.
Effects on antioxidative activity and preferential location of HDL-associated lipids.
| Effect on antioxidative activity | Preferential location | |
|---|---|---|
| SM | Inhibition | HDL2 |
| CE | Inhibition | HDL2 |
| FC | Inhibition | HDL2 |
| TG | Inhibition | HDL2 |
| Cer | Inhibition | HDL2 |
| LysoPC | Inhibition | HDL3 |
| PA | Inhibition | HDL3 |
| PS | Enhancement | HDL3 |
| S1P | Enhancement | HDL3 |
HDL, high density lipoprotein; SM, sphingomyelin; CE, cholesteryl ester; FC, free cholesterol; TG, triglyceride; Cer, ceramide; LysoPC, lysophophatidylcholine; PA, phopshatidic acid; PS, phosphatidyl serine; S1P, sphingosine-1 phosphate.
4. HDL and non-radical oxidants
There are two types of naturally occurring oxidant compounds, those that possess unpaired electrons, termed one-electron oxidants or free radicals (hydroxyl radicals, lipoxyl radicals, superoxide, etc.), and those which have all electrons paired, referred to as two-electron non-radical oxidants (HOCl, ONOO−, etc.) [129]. Traditionally, studies have focused on HDL ability to protect LDL from oxidation mediated by free radicals, while non-radical oxidants remain poorly studied in this regard. Ahmed et al. [89], [120] found that apo A-I and PON1 can act in tandem to neutralize the oxidative damage caused on HDL particles by ONOO−, both in isolated and reconstituted HDL. Introduction of His and Cys residues at the positions 2/3 and 12, respectively, of apo A-I remarkably enhanced its capacity to prevent LDL oxidation and to remove HOCl [130]. Nevertheless, evidence suggests that native HDL are rather ineffective against HOCl-mediated oxidation. HOCl can target and oxidize Tyr, Met and Trp residues in the apo A-I moiety leading to a loss of functionality [131], [132]. Furthermore, HOCl can induce impairment of PON1 activity [133]. The oxidative modification of apo A-I and PON1 produces defective HDL particles with diminished atheroprotective potential. Thus, HDL capacity to protect against non-radical induced oxidation seems to be dependent on the nature of the oxidant molecule.
5. Heterogeneity of antioxidative activities of HDL
HDL particles are highly heterogeneous in their structure, metabolism and biological functions [24], [50], [134] (Table 3). Among the major HDL subpopulations, small, dense, protein-rich HDL3 display the highest efficiency at protecting LDL from free radical-induced oxidative damage [35] and inhibiting oxLDL-induced apoptosis of endothelial cells [135]. HDL3 are characterized by distinct proteome and lipidome [41], [42], [124].
In particular, HDL3c showed a high potency to protect LDL from free-radical-induced oxidation; its proteome predominantly contained apo A-I, apo J, apo L-1, apo F, PON1/3, PLTP and PAF-AH, in addition to apo A-II, apo D, apo M, SAA 1, 2 and 4, apo C-I, apo C-II and apo E [41]. Furthermore, antioxidative activity of HDL subpopulations was positively correlated with the presence of apo J, apo M, SAA4, apo D, apo L-1 and PON1/3 [41]. It is relevant that the simultaneous presence of PON1 and apo A-IV on a subset of small HDLs endowed these particles with potent antioxidative activity in apo E-/-mice [136]. Moreover, PLTP is present on HDL together with apo J, apo L-I, apo D, apo A-I, apo A-II and apo E, suggesting direct functional interaction between them [137].
Furthermore, small, dense HDL3 is distinct in its lipidome which is deficient in sphingomyelin, a structural lipid with positive impact on surface rigidity and negative impact on LCAT activity, and in free cholesterol which is equally capable of increasing phospholipid monolayer rigidity [42]. Finally, the distinctly low lipid content of small, dense HDL3 induces conformational changes in apo A-I relative to large, light HDL2. This may result in enhanced exposure of the protein to the aqueous phase, thereby facilitating the redox reaction between Met residues and LOOH [138], [139].
6. Pathophysiological relevance of antioxidative activities of HDL
LDL-derived LOOH display multiple pro-oxidative and pro-inflammatory properties. Moreover, LOOH can decompose into short-chain oxPL and aldehydes, all of which are proatherogenic. These data highlight the relevance of HDL-mediated neutralization of diverse products of lipid peroxidation [3]. Such activity can be especially important in microenvironments depleted of other antioxidant defenses, such as in the arterial intima.
Different studies have reported that HDL particles display impaired antioxidative activity in atherogenic dyslipidemias characterized by low HDL-cholesterol (HDL-C) levels. In vitro studies reveal that the capacity of small, dense HDL3 subfractions to decrease LOOH accumulation in LDL particles was significantly reduced in patients with metabolic syndrome (MetS) [140]. Similar result was obtained for HDL3 particles isolated from patients with well-controlled type 2 diabetes (T2D), who also exhibited diminished PON1 and PAF-AH activities in HDL [141], but not when analyzing antioxidative activity of total HDL [142]. Regarding poorly-controlled T2D, one study showed that large, light, cholesteryl ester-rich HDL2 exhibited decreased capacity to protect LDL from macrophage-mediated oxidation [143], whilst other authors found decreased antioxidative activity in HDL3c [126]. Furthermore, all HDL subfractions from normoglycemic patients with isolated low HDL-C levels displayed reduced antioxidative activity relative to their counterparts from normolipidemic controls [144]. Interestingly, the intrinsic antioxidative activity of HDL particles was equally reduced in patients with hyperalphalipoproteinemia associated with low activity of hepatic lipase and high HDL content of triglyceride [145]. In all these studies, HDL particles presented similar alterations in their composition and/or activity of associated enzymes despite pronounced differences in plasma HDL-C levels, suggesting that intrinsic properties of HDL particles, rather than plasma HDL-C levels per se, accounted for the observed functional deficiency.
In a recent study, antioxidative capacity of apo B-depleted serum, measured as its ability to prevent LDL oxidation, was reported to be reduced in acute coronary syndrome but not in stable coronary artery disease when compared to healthy controls [146]. Using similar technique, apo B-depleted serum from patients with polycystic ovary syndrome, a condition characterized by insulin resistance, presented reduced capacity to protect LDL from oxidation [147]. It is essential to emphasize that only apo B-containing lipoproteins are removed from plasma by apoB-precipitation which leaves in the solution all other components, primarily albumin, a major antioxidant of human plasma, together with numerous low-molecular-weight hydrophilic antioxidants, such as ascorbate, urate and others [148]. Importantly, HDL only provides a minor contribution to the capacity of apo B-depleted plasma or serum to inhibit LDL oxidation [148], [149]. Results of the studies reporting altered antioxidative activity of HDL isolated by apo B precipitation cannot therefore be directly interpreted as alterations in HDL properties and may reflect other modifications of plasma or serum, such as decreased albumin levels under acute conditions.
Another study, which evaluated total antioxidative capacity of plasma, reported that this parameter was decreased in parallel to PON1 activity in polycystic ovary syndrome [150].
Alterations in antioxidative activity of HDL are not exclusive of diseases with a metabolic component. Several investigators reported that antioxidative activity of HDL was impaired in chronic inflammatory diseases. In a recent study, we found that antioxidative activity of HDL3 was reduced in patients with rheumatoid arthritis (RA) who presented high levels of inflammation (hsCRP > 10 mg/l) [127]. Consistent with these data, McMahon et al. [151] observed a decrease in antioxidative activity of total HDL in patients with systemic lupus erythematosus (SLE). This finding was corroborated by a recent study which also reported decreased PON1 activity in this condition [152].
In renal diseases, which are known causes of secondary dyslipidemia, Moradi et al. [153] found a decrease in antioxidative activity of total HDL in patients with end stage renal disease (ESRD). HDL particles from these patients also showed diminished PON1 and GPX activity, in addition to reduced apo A-I concentration. Furthermore, studies in patients undergoing hemodialysis documented impairment of HDL-mediated inhibition of LDL oxidation [154] and increase in oxidized HDL [155]. Remarkably, such increase in oxidized HDL was associated with elevated occurrence of cardiovascular disease and increased mortality.
Interestingly, patients with iron deficiency anemia (IDA) showed a decrease in antioxidative activity of total HDL associated with impairment in PON1 activity [156]. Similar results were observed in postmenopausal women but exclusively in the subgroup with PON1 QR phenotype [157].
In several studies mentioned above, HDL3 subfractions were triglyceride-enriched and cholesteryl ester-depleted [140], [141], [144], alterations that were correlated with the diminished antioxidative activity. Such modifications of lipid composition are in accordance with elevated CETP activity and/or reduced hepatic lipase activity. Different in vitro experiments have provided potential mechanisms to explain the impairment of HDL antioxidative capacity by triglyceride enrichment. Replacement of cholesteryl esters by triglycerides in the HDL lipid core considerably alters the conformation of the central and C-terminal domains of apo A-I [138], [139], thereby potentially modifying the accessibility of Met residues to LOOH. Moreover, replacement of cholesteryl esters by triglycerides in spherical rHDL decreases the conformational stability of apo A-I [138], resulting in unstable particles which readily lose apo A-I.
Other alterations at the level of the HDL lipidome which can impact antioxidative properties of HDL involve enrichment in sphingomyelin and depletion of polyunsaturated fatty acids, both resulting in elevated rigidity of the phospholipid surface monolayer of HDL. Such structural anomalies may cause a deficiency in the capacity of HDL3 to acquire LDL-derived LOOH and, consequently, to protect LDL from free radical-induced oxidative damage. In addition, we found that HDL from RA patients displayed increased content of phosphatidic acid, which was associated with decreased antioxidative activity [127].
In addition to alterations in the HDL lipidome, modifications of the proteome can affect HDL functionality in different diseases. Under conditions of acute inflammation, HDL particles are enriched in SAA [21]. Replacement of apo A-I by acute phase proteins, primarily SAA, in small, dense HDL particles under conditions of chronic inflammation [158] might lead to deficient inactivation of LOOH by HDL, increasing their accumulation in LDL. Indeed, apo A-I content in HDL tends to be reduced in metabolic [140], [141], [144] and inflammatory [127], [159] diseases. Interestingly, therapies targeting tumor necrosis factor α, a well-known activator of SAA synthesis [160], improved antioxidative activity of HDL in SLE patients [152]. However, it is important to note that recent evidence suggests potential antioxidant role for SAA-enriched HDL particles. Similarly to apo A-I, antioxidative activity of SAA depends on the presence of thiol groups, implying that this activity can partially compensate for the loss of apo A-I commonly observed during inflammation [85], [86].
Oxidative modifications of apo A-I represent another pathway potentially resulting in the decreased inactivation of LOOH by HDL. Indeed, apo A-I content of oxidized Met residues is elevated in type 1 diabetic patients at all three positions (Met86, Met112 and Met148) [161]. Finally, non-enzymatic glycation, a post-translational modification of apo A-I, can be equally involved in the impairment of antioxidative properties of HDL [162]. In addition to apo A-I, glycation may impair activities of HDL-associated enzymes. In this regard, high levels of apo A-I glycation and low PON1 activity were associated with the severity of coronary artery disease in T2D patients [163].
7. Lifestyle influence on antioxidative activity of HDL and therapeutic perspectives
HDL-targeting therapy has traditionally been directed to increase HDL-C levels. However, there is evidence pointing to HDL functionality as a better therapeutic target. In the case of antioxidative activity, evidence is still limited. Nevertheless, it is important to summarize all current findings on the subject.
Lifestyle differences are known to affect HDL composition and functionality. Several studies have explored the influence of dietary and exercise habits on antioxidative activity of HDL. Normocholesterolemic patients with metabolic syndrome presented significant improvement in antioxidative activity of HDL3c after 12 weeks of individually tailored diet and exercise intervention [164]. Moreover, dyslipidemic patients with metabolic syndrome exhibited an increase in antioxidative potential of HDL2a and 3b, in parallel to an increment in PON1 activity after 3 months of moderate intensity physical activity [165]. Likewise, triathletes with the PON1 QR phenotype exhibited an increase in antioxidative activity of total HDL [166], potentially reflecting adaptative increase in antioxidant defenses in response to oxidative stress resulting from increased oxygen consumption. Regarding smoking, Park et al. found that smokers presented impaired antioxidative activity of HDL and elevated LDL oxidation as compared to non-smoking controls [167].
In addition to lifestyle intervention several pharmacological alternatives have been developed which can potentially improve antioxidative activity of HDL:
7.1. Statins
Atorvastatin increased plasma total antioxidant status in parallel to PON1 activity in hypercholesterolemic patients [168], [169], [170], [171]. Other statins also showed positive effects on PON1 activity in different studies [172], [173], [174] and pitavastatin was able to increase both PON1 transcription [175], [176] and activity [177].
7.2. Fibrates
Fenofibrate therapy caused a redistribution of PAF-AH from apo B-containing liporpoteins to HDL, enhancing its anti-atherogenic potential, in dyslipidemic patients [115]. Moreover, administration of fenofibrate caused an increase in GSX activity together with a decrease in systemic oxidative stress in obese hyperlipidemic patients [178], [179]. Finally, several studies reported improvements in PON1 activity in patients with different types of dyslipidemia after treatment with fenofibrate [180], [181], [182]. By contrast, short-term administration of bezafibrate in combination with simvastatin failed to increase PON1 activity [183].
7.3. Niacin
Niacin intake correlated negatively with plasma levels of oxLDL in healthy adults [184]. Moreover, niacin treatment resulted in a decrease in systemic oxidative stress, in addition to an increase in PON1 activity in hypercholesterolemic patients [185]. However, it is worth noting that niacin/laropiprant treatment failed to increase antioxidative activity of HDL or PON1 activity in statin-resistant dyslipidemic patients [186]. Batuca et al. found that even though niacin administration raised HDL-C levels, it also led to an increase in anti-apo A-I antibodies and impairment of PON1 activity [187].
7.4. Apo A-I mimetics
Several Apo A-I mimetic peptides have been developed, with bihelical peptides being superior to their monohelical counterparts due to enhanced lipid affinity of the latter [188]. Navab et al. [59], [189] tested the ability of a series of apo A-I peptides to inhibit LDL oxidation, and found that peptides 4F and 5F were highly effective. Both 5F synthesized from l-amino acids (L-5F) and 4F synthesized from d-amino acids (D-4F) were tested in animal models and displayed the ability to protect from atherosclerosis [190]. Moreover, inserting antioxidant amino acid residues at different positions in the apo A-I moiety produced mimetic peptides with enhanced antioxidative activity [130]. Another apo A-I mimetic, L-6F, displayed antioxidant properties in several mouse models [191], [192]. In particular, feeding atherosclerotic mice a Western diet which included transgenic tomatoes expressing L-6F improved an array of biomarkers of inflammation and oxidative stress [191]. ETC-642 is a 22-amino acid peptide which has been shown to reduce proinflammatory oxidized LDL in rabbits [193]. 5A is another apo A-I mimetic which displays antioxidant properties both in vitro and in animal models [194]. D'Souza et al. [195] investigated the effect on cholesterol efflux and anti-inflammatory and antioxidant properties of 22 different bihelical apo A-I mimetic peptides and found that none of the compounds was superior in all the antiatherogenic functions studied, and that each of the examined biological activities was primarily affected by specific structural features. These results suggest that combining several apo A-I mimetic peptides, each mimicking different structural aspects of apo A-I, may prove to be a valuable strategy to mimic multiple anti-atherosclerotic properties of apo A-I.
7.5. rHDL
rHDL containing apo A-I mutant peptides V156 K and R173C displayed elevated antioxidative activity as compared to normal HDL in vitro [196]. Moreover, mice injected with V156K-rHDL and R173C-rHDL particles exhibited improvements in lipid parameters in addition to increases in HDL antioxidant activity [196]. A recent study [197] found that rHDL which carried growth hormone 2 showed enhanced antioxidative activity. Similar results were observed for growth hormone 1 [198]. Furthermore, apo A-I Milano-containing rHDL showed higher antioxidative activity than wild-type rHDL in rats, resulting in the protection from endotoxin-induced multiple organ injury [199]. CSL112, a formulation of rHDL comprised of apo A-I and phosphatidylcholne, currently under development for the prevention of atherothrombotic events, caused a remodeling of HDL particles in human test subjects [200]. Such remodeling resulted in the formation of three distinct HDL species, two of which displayed potent antioxidative and anti-inflammatory properties. Finally, rHDL which contained ω-3 fatty acids, showed greater capacity to protect LDL from oxidation than rHDL carryng ω-6 fatty acids both in cell culture and in zebra fish embryos [201].
7.6. Antibodies
Infliximab, an antibody against tumor necrosis factor α used in the treatment of rheumatoid arthritis, reportedly increased total HDL antioxidant function and PON1 activity six months after administration [202].
8. Conclusions
Circulating HDL particles, primarily small, dense, protein-rich HDL3, may provide potent protection to LDL from oxidative damage by free radicals in the arterial intima, resulting in the inhibition of the generation of pro-inflammatory oxidized lipids, primarily LOOH but also short-chain oxPL and reactive aldehydes. HDL-mediated inactivation of LOOH involves initial transfer of PLOOH from LDL to HDL, which is governed by the rigidity of the surface monolayer of HDL, and subsequent reduction of PLOOH by redox-active Met residues of apo A-I. PON1, PAF-AH, and LCAT are present at elevated concentrations in HDL3 relative to HDL2 and might be involved in the inactivation of short-chain oxPLs. Therefore, HDL represents a lipoprotein particle capable of acquiring potentially toxic proatherogenic lipids and restricting them to an environment where they can be safely inactivated and/or from which they can be delivered to the liver for excretion.
Different metabolic and inflammatory diseases present an impairment of HDL antioxidative function. Evidence suggests that structural and compositional anomalies in the proteome and lipidome of HDL underlie such functional deficiency. We therefore propose that concomitant normalization of the metabolism, circulating levels, composition and biological activities of HDL particles, primarily those of biologically potent small, dense HDL3, can constitute a therapeutic target of future interest.
Conflict of interest
There is no conflict of interest to declare.
Transparency document
Transparency document.
Acknowledgments
Acknowledgements
These studies were primarily supported by a collaborative grant from CONICET (Buenos Aires, Argentina) and National Institute of Health and Medical Research (INSERM, Paris, France). We gratefully acknowledge further support from University of Pierre and Marie Curie (UPMC), University of Buenos Aires (UBACyT CB23), Craveri Foundation and CONICET (PIP 516).
Footnotes
The Transparency document associated with this article can be found, in online version.
References
- 1.Hopkins P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013;93(3):1317–1542. doi: 10.1152/physrev.00004.2012. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg D., Witztum J.L. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010;30(12):2311–2316. doi: 10.1161/ATVBAHA.108.179697. [DOI] [PubMed] [Google Scholar]
- 3.Stocker R., Keaney J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004;84(4):1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 4.Simionescu M., Simionescu N. Proatherosclerotic events: pathobiochemical changes occurring in the arterial wall before monocyte migration. FASEB J. 1993;7(14):1359–1366. doi: 10.1096/fasebj.7.14.8224609. [DOI] [PubMed] [Google Scholar]
- 5.Camejo G., Fager G., Rosengren B., Hurt-Camejo E., Bondjers G. Binding of low density lipoproteins by proteoglycans synthesized by proliferating and quiescent human arterial smooth muscle cells. J. Biol. Chem. 1993;268(19):14131–14137. [PubMed] [Google Scholar]
- 6.Yla-Herttuala S., Solakivi T., Hirvonen J., Laaksonen H., Mottonen M., Pesonen E. Glycosaminoglycans and apolipoproteins B and A-I in human aortas. Chemical and immunological analysis of lesion-free aortas from children and adults. Arteriosclerosis. 1987;7(4):333–340. doi: 10.1161/01.atv.7.4.333. [DOI] [PubMed] [Google Scholar]
- 7.Williams K.J., Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995;15(5):551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabas I., Williams K.J., Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 9.Boren J., Olin K., Lee I., Chait A., Wight T.N., Innerarity T.L. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J. Clin. Invest. 1998;101(12):2658–2664. doi: 10.1172/JCI2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frei B., Stocker R., Ames B.N. Antioxidant defenses and lipid peroxidation in human blood plasma. Proc. Natl. Acad. Sci. U. S. A. 1988;85(24):9748–9752. doi: 10.1073/pnas.85.24.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J. Lipid Res. 2009;50(Suppl):S376–81. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itabe H. Oxidative modification of LDL: its pathological role in atherosclerosis. Clin Rev Allergy Immunol. 2009;37(1):4–11. doi: 10.1007/s12016-008-8095-9. [DOI] [PubMed] [Google Scholar]
- 13.Milanlioglu A., Aslan M., Ozkol H., Cilingir V., Nuri Aydin M., Karadas S. Serum antioxidant enzymes activities and oxidative stress levels in patients with acute ischemic stroke: influence on neurological status and outcome. Wien. Klin. Wochenschr. 2016;128(5–6):169–174. doi: 10.1007/s00508-015-0742-6. [DOI] [PubMed] [Google Scholar]
- 14.Traber M.G., Stevens J.F. Vitamins C and E: beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011;51(5):1000–1013. doi: 10.1016/j.freeradbiomed.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller Y.I., Choi S.H., Fang L., Tsimikas S. Lipoprotein modification and macrophage uptake: role of pathologic cholesterol transport in atherogenesis. Subcell. Biochem. 2010;51:229–251. doi: 10.1007/978-90-481-8622-8_8. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida H., Kisugi R. Mechanisms of LDL oxidation. Clin. Chim. Acta. 2010;411(23–24):1875–1882. doi: 10.1016/j.cca.2010.08.038. [DOI] [PubMed] [Google Scholar]
- 17.Fruebis J., Parthasarathy S., Steinberg D. Evidence for a concerted reaction between lipid hydroperoxides and polypeptides. Proc. Natl. Acad. Sci. U. S. A. 1992;89(22):10588–10592. doi: 10.1073/pnas.89.22.10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witztum J.L., Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc. Med. 2001;11(3–4):93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 19.Yla-Herttuala S., Palinski W., Rosenfeld M.E., Parthasarathy S., Carew T.E., Butler S. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 1989;84(4):1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holvoet P., Vanhaecke J., Janssens S., Van de Werf F., Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98(15):1487–1494. doi: 10.1161/01.cir.98.15.1487. [DOI] [PubMed] [Google Scholar]
- 21.Navab M., Ananthramaiah G.M., Reddy S.T., Van Lenten B.J., Ansell B.J., Fonarow G.C. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J. Lipid Res. 2004;45(6):993–1007. doi: 10.1194/jlr.R400001-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Rosenson R.S., Brewer H.B., Jr., Chapman M.J., Fazio S., Hussain M.M., Kontush A. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011;57(3):392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- 23.Pirillo A., Norata G.D., Catapano A.L. High-density lipoprotein subfractions—what the clinicians need to know. Cardiology. 2013;124(2):116–125. doi: 10.1159/000346463. [DOI] [PubMed] [Google Scholar]
- 24.Camont L., Chapman M.J., Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011;17(10):594–603. doi: 10.1016/j.molmed.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Kontush A., Chapman M.J. Antiatherogenic function of HDL particle subpopulations: focus on antioxidative activities. Curr. Opin. Lipidol. 2010;21(4):312–318. doi: 10.1097/MOL.0b013e32833bcdc1. [DOI] [PubMed] [Google Scholar]
- 26.Mackness M.I., Arrol S., Durrington P.N. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991;286(1–2):152–154. doi: 10.1016/0014-5793(91)80962-3. [DOI] [PubMed] [Google Scholar]
- 27.Durrington P.N., Mackness B., Mackness M.I. Paraoxonase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001;21(4):473–480. doi: 10.1161/01.atv.21.4.473. [DOI] [PubMed] [Google Scholar]
- 28.Mackness M.I., Arrol S., Abbott C., Durrington P.N. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis. 1993;104(1–2):129–135. doi: 10.1016/0021-9150(93)90183-u. [DOI] [PubMed] [Google Scholar]
- 29.Mackness M.I., Abbott C., Arrol S., Durrington P.N. The role of high-density lipoprotein and lipid-soluble antioxidant vitamins in inhibiting low-density lipoprotein oxidation. Biochem. J. 1993;294(Pt 3):829–834. doi: 10.1042/bj2940829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerrad-Saadi A., Therond P., Chantepie S., Couturier M., Rye K.A., Chapman M.J. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009;29(12):2169–2175. doi: 10.1161/ATVBAHA.109.194555. [DOI] [PubMed] [Google Scholar]
- 31.Ferretti G., Bacchetti T., Moroni C., Vignini A., Curatola G. Copper-induced oxidative damage on astrocytes: protective effect exerted by human high density lipoproteins. Biochim. Biophys. Acta. 2003;1635(1):48–54. doi: 10.1016/j.bbalip.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Ferretti G., Bacchetti T., Moroni C., Vignini A., Curatola G. Protective effect of human HDL against Cu(2 +)-induced oxidation of astrocytes. J. Trace Elem. Med. Biol. 2003;17(Suppl. 1):25–30. [PubMed] [Google Scholar]
- 33.Klimov A.N., Kozhevnikova K.A., Kuzmin A.A., Kuznetsov A.S., Belova E.V. On the ability of high density lipoproteins to remove phospholipid peroxidation products from erythrocyte membranes. Biochemistry (Mosc) 2001;66(3):300–304. doi: 10.1023/a:1010203930470. [DOI] [PubMed] [Google Scholar]
- 34.Vila A., Korytowski W., Girotti A.W. Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low-density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry. 2002;41(46):13705–13716. doi: 10.1021/bi026467z. [DOI] [PubMed] [Google Scholar]
- 35.Kontush A., Chantepie S., Chapman M.J. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2003;23(10):1881–1888. doi: 10.1161/01.ATV.0000091338.93223.E8. [DOI] [PubMed] [Google Scholar]
- 36.Yoshikawa M., Sakuma N., Hibino T., Sato T., Fujinami T. HDL3 exerts more powerful anti-oxidative, protective effects against copper-catalyzed LDL oxidation than HDL2. Clin. Biochem. 1997;30(3):221–225. doi: 10.1016/s0009-9120(97)00031-3. [DOI] [PubMed] [Google Scholar]
- 37.Sakuma N., Yoshikawa M., Hibino T., Ohte N., Kamiya T., Kunimatsu M. HDL3 exerts a more powerful antiperoxidative and protective effect against peroxidative modification of LDL than HDL2 does. J. Nutr. Sci. Vitaminol. (Tokyo) 2002;48(4):278–282. doi: 10.3177/jnsv.48.278. [DOI] [PubMed] [Google Scholar]
- 38.Kumpula L.S., Kumpula J.M., Taskinen M.R., Jauhiainen M., Kaski K., Ala-Korpela M. Reconsideration of hydrophobic lipid distributions in lipoprotein particles. Chem. Phys. Lipids. 2008;155(1):57–62. doi: 10.1016/j.chemphyslip.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Miyazaki A., Sakai M., Suginohara Y., Hakamata H., Sakamoto Y., Morikawa W. Acetylated low density lipoprotein reduces its ligand activity for the scavenger receptor after interaction with reconstituted high density lipoprotein. J. Biol. Chem. 1994;269(7):5264–5269. [PubMed] [Google Scholar]
- 40.Hsieh J.Y., Chang C.T., Huang M.T., Chang C.M., Chen C.Y., Shen M.Y. Biochemical and functional characterization of charge-defined subfractions of high-density lipoprotein from normal adults. Anal. Chem. 2013;85(23):11440–11448. doi: 10.1021/ac402516u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davidson W.S., Silva R.A., Chantepie S., Lagor W.R., Chapman M.J., Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009;29(6):870–876. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kontush A., Therond P., Zerrad A., Couturier M., Negre-Salvayre A., de Souza J.A. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler. Thromb. Vasc. Biol. 2007;27(8):1843–1849. doi: 10.1161/ATVBAHA.107.145672. [DOI] [PubMed] [Google Scholar]
- 43.Kontush A., Chapman M. John Wiley and Sons; 2012. High-density Lipoproteins: Structure, Metabolism, Function and Therapeutics. [Google Scholar]
- 44.Garner B., Waldeck A.R., Witting P.K., Rye K.A., Stocker R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J. Biol. Chem. 1998;273(11):6088–6095. doi: 10.1074/jbc.273.11.6088. [DOI] [PubMed] [Google Scholar]
- 45.Panzenbock U., Stocker R. Formation of methionine sulfoxide-containing specific forms of oxidized high-density lipoproteins. Biochim. Biophys. Acta. 2005;1703(2):171–181. doi: 10.1016/j.bbapap.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Boisfer E., Stengel D., Pastier D., Laplaud P.M., Dousset N., Ninio E. Antioxidant properties of HDL in transgenic mice overexpressing human apolipoprotein A-II. J. Lipid Res. 2002;43(5):732–741. [PubMed] [Google Scholar]
- 47.Ostos M.A., Conconi M., Vergnes L., Baroukh N., Ribalta J., Girona J. Antioxidative and antiatherosclerotic effects of human apolipoprotein A-IV in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001;21(6):1023–1028. doi: 10.1161/01.atv.21.6.1023. [DOI] [PubMed] [Google Scholar]
- 48.Gaidukov L., Viji R.I., Yacobson S., Rosenblat M., Aviram M., Tawfik D.S. ApoE induces serum paraoxonase PON1 activity and stability similar to ApoA-I. Biochemistry. 2010;49(3):532–538. doi: 10.1021/bi9013227. [DOI] [PubMed] [Google Scholar]
- 49.Elsoe S., Ahnstrom J., Christoffersen C., Hoofnagle A.N., Plomgaard P., Heinecke J.W. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221(1):91–97. doi: 10.1016/j.atherosclerosis.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 50.Kontush A., Chapman M.J. Antiatherogenic small, dense HDL—guardian angel of the arterial wall? Nat. Clin. Pract. Cardiovasc. Med. 2006;3(3):144–153. doi: 10.1038/ncpcardio0500. [DOI] [PubMed] [Google Scholar]
- 51.Kontush A., Chapman M.J. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006;58(3):342–374. doi: 10.1124/pr.58.3.1. [DOI] [PubMed] [Google Scholar]
- 52.Christison J.K., Rye K.A., Stocker R. Exchange of oxidized cholesteryl linoleate between LDL and HDL mediated by cholesteryl ester transfer protein. J. Lipid Res. 1995;36(9):2017–2026. [PubMed] [Google Scholar]
- 53.Girotti A.W. Translocation as a means of disseminating lipid hydroperoxide-induced oxidative damage and effector action. Free Radic. Biol. Med. 2008;44(6):956–968. doi: 10.1016/j.freeradbiomed.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goulinet S., Chapman M.J. Plasma LDL and HDL subspecies are heterogenous in particle content of tocopherols and oxygenated and hydrocarbon carotenoids. Relevance to oxidative resistance and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1997;17(4):786–796. doi: 10.1161/01.atv.17.4.786. [DOI] [PubMed] [Google Scholar]
- 55.Bowry V.W., Stanley K.K., Stocker R. High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors. Proc. Natl. Acad. Sci. U. S. A. 1992;89(21):10316–10320. doi: 10.1073/pnas.89.21.10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Proudfoot J.M., Barden A.E., Loke W.M., Croft K.D., Puddey I.B., Mori T.A. HDL is the major lipoprotein carrier of plasma F2-isoprostanes. J. Lipid Res. 2009;50(4):716–722. doi: 10.1194/jlr.M800607-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rye K.A., Barter P.J. Cardioprotective functions of HDLs. J. Lipid Res. 2014;55(2):168–179. doi: 10.1194/jlr.R039297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bashtovyy D., Jones M.K., Anantharamaiah G.M., Segrest J.P. Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J. Lipid Res. 2011;52(3):435–450. doi: 10.1194/jlr.R012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navab M., Hama S.Y., Cooke C.J., Anantharamaiah G.M., Chaddha M., Jin L. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J. Lipid Res. 2000;41(9):1481–1494. [PubMed] [Google Scholar]
- 60.Karlsson H., Kontush A., James R.W. Functionality of HDL: antioxidation and detoxifying effects. Handb. Exp. Pharmacol. 2015;224:207–228. doi: 10.1007/978-3-319-09665-0_5. [DOI] [PubMed] [Google Scholar]
- 61.Ribas V., Sanchez-Quesada J.L., Anton R., Camacho M., Julve J., Escola-Gil J.C. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: a new mechanism linking HDL protein composition and antiatherogenic potential. Circ. Res. 2004;95(8):789–797. doi: 10.1161/01.RES.0000146031.94850.5f. [DOI] [PubMed] [Google Scholar]
- 62.Gaidukov L., Tawfik D.S. High affinity, stability, and lactonase activity of serum paraoxonase PON1 anchored on HDL with ApoA-I. Biochemistry. 2005;44(35):11843–11854. doi: 10.1021/bi050862i. [DOI] [PubMed] [Google Scholar]
- 63.Segrest J.P., Garber D.W., Brouillette C.G., Harvey S.C., Anantharamaiah G.M. The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv. Protein Chem. 1994;45:303–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 64.Ghiselli G., Krishnan S., Beigel Y., Gotto A.M., Jr. Plasma metabolism of apolipoprotein A-IV in humans. J. Lipid Res. 1986;27(8):813–827. [PubMed] [Google Scholar]
- 65.Ohta T., Fidge N.H., Nestel P.J. Studies on the in vivo and in vitro distribution of apolipoprotein A-IV in human plasma and lymph. J. Clin. Invest. 1985;76(3):1252–1260. doi: 10.1172/JCI112081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo J.A., Lee E.Y., Park J.Y., Lee S.T., Ham S., Cho K.H. Different functional and structural characteristics between ApoA-I and ApoA-4 in lipid-free and reconstituted HDL state: ApoA-4 showed less anti-atherogenic activity. Mol. Cells. 2015;38(6):573–579. doi: 10.14348/molcells.2015.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyata M., Smith J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996;14(1):55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 68.Filou S., Lhomme M., Karavia E.A., Kalogeropoulou C., Theodoropoulos V., Zvintzou E. Distinct roles of apolipoproteins A1 and E in the modulation of high-density lipoprotein composition and function. Biochemistry. 2016;55(27):3752–3762. doi: 10.1021/acs.biochem.6b00389. [DOI] [PubMed] [Google Scholar]
- 69.Zhang X.Y., Dong X., Zheng L., Luo G.H., Liu Y.H., Ekstrom U. Specific tissue expression and cellular localization of human apolipoprotein M as determined by in situ hybridization. Acta Histochem. 2003;105(1):67–72. doi: 10.1078/0065-1281-00687. [DOI] [PubMed] [Google Scholar]
- 70.Xu N., Dahlback B. A novel human apolipoprotein (apoM) J. Biol. Chem. 1999;274(44):31286–31290. doi: 10.1074/jbc.274.44.31286. [DOI] [PubMed] [Google Scholar]
- 71.Mahley R.W., Innerarity T.L., Rall S.C., Jr., Weisgraber K.H. Plasma lipoproteins: apolipoprotein structure and function. J. Lipid Res. 1984;25(12):1277–1294. [PubMed] [Google Scholar]
- 72.Smith K.M., Lawn R.M., Wilcox J.N. Cellular localization of apolipoprotein D and lecithin:cholesterol acyltransferase mRNA in rhesus monkey tissues by in situ hybridization. J. Lipid Res. 1990;31(6):995–1004. [PubMed] [Google Scholar]
- 73.Braesch-Andersen S., Beckman L., Paulie S., Kumagai-Braesch M. ApoD mediates binding of HDL to LDL and to growing T24 carcinoma. PLoS One. 2014;9(12) doi: 10.1371/journal.pone.0115180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhatia S., Knoch B., Wong J., Kim W.S., Else P.L., Oakley A.J. Selective reduction of hydroperoxyeicosatetraenoic acids to their hydroxy derivatives by apolipoprotein D: implications for lipid antioxidant activity and Alzheimer's disease. Biochem. J. 2012;442(3):713–721. doi: 10.1042/BJ20111166. [DOI] [PubMed] [Google Scholar]
- 75.Morton R.E., Gnizak H.M., Greene D.J., Cho K.H., Paromov V.M. Lipid transfer inhibitor protein (apolipoprotein F) concentration in normolipidemic and hyperlipidemic subjects. J. Lipid Res. 2008;49(1):127–135. doi: 10.1194/jlr.M700258-JLR200. [DOI] [PubMed] [Google Scholar]
- 76.Park S., Mathis K.W., Lee I.K. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev. Endocr. Metab. Disord. 2014;15(1):45–53. doi: 10.1007/s11154-013-9275-3. [DOI] [PubMed] [Google Scholar]
- 77.Perez-Morga D., Vanhollebeke B., Paturiaux-Hanocq F., Nolan D.P., Lins L., Homble F. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309(5733):469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 78.Wan G., Zhaorigetu S., Liu Z., Kaini R., Jiang Z., Hu C.A. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem. 2008;283(31):21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chapman M.J., Le Goff W., Guerin M., Kontush A. Cholesteryl ester transfer protein: at the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur. Heart J. 2010;31(2):149–164. doi: 10.1093/eurheartj/ehp399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hine D., Mackness B., Mackness M. Cholesteryl-ester transfer protein enhances the ability of high-density lipoprotein to inhibit low-density lipoprotein oxidation. IUBMB Life. 2011;63(9):772–774. doi: 10.1002/iub.508. [DOI] [PubMed] [Google Scholar]
- 81.Albers J.J., Cheung M.C. Emerging roles for phospholipid transfer protein in lipid and lipoprotein metabolism. Curr. Opin. Lipidol. 2004;15(3):255–260. doi: 10.1097/00041433-200406000-00004. [DOI] [PubMed] [Google Scholar]
- 82.Uhlar C.M., Whitehead A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 1999;265(2):501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 83.Zhang N., Ahsan M.H., Purchio A.F., West D.B. Serum amyloid A-luciferase transgenic mice: response to sepsis, acute arthritis, and contact hypersensitivity and the effects of proteasome inhibition. J. Immunol. 2005;174(12):8125–8134. doi: 10.4049/jimmunol.174.12.8125. [DOI] [PubMed] [Google Scholar]
- 84.Steel D.M., Sellar G.C., Uhlar C.M., Simon S., DeBeer F.C., Whitehead A.S. A constitutively expressed serum amyloid A protein gene (SAA4) is closely linked to, and shares structural similarities with, an acute-phase serum amyloid A protein gene (SAA2) Genomics. 1993;16(2):447–454. doi: 10.1006/geno.1993.1209. [DOI] [PubMed] [Google Scholar]
- 85.Sato M., Ohkawa R., Yoshimoto A., Yano K., Ichimura N., Nishimori M. Effects of serum amyloid A on the structure and antioxidant ability of high-density lipoprotein. Biosci. Rep. 2016;36(4) doi: 10.1042/BSR20160075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jayaraman S., Haupt C., Gursky O. Paradoxical effects of SAA on lipoprotein oxidation suggest a new antioxidant function for SAA. J. Lipid Res. 2016;57(12):2138–2149. doi: 10.1194/jlr.M071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackness B., Durrington P.N., Mackness M.I. Human serum paraoxonase. Gen. Pharmacol. 1998;31(3):329–336. doi: 10.1016/s0306-3623(98)00028-7. [DOI] [PubMed] [Google Scholar]
- 88.Harel M., Aharoni A., Gaidukov L., Brumshtein B., Khersonsky O., Meged R. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004;11(5):412–419. doi: 10.1038/nsmb767. [DOI] [PubMed] [Google Scholar]
- 89.Ahmed Z., Ravandi A., Maguire G.F., Emili A., Draganov D., La Du B.N. Multiple substrates for paraoxonase-1 during oxidation of phosphatidylcholine by peroxynitrite. Biochem. Biophys. Res. Commun. 2002;290(1):391–396. doi: 10.1006/bbrc.2001.6150. [DOI] [PubMed] [Google Scholar]
- 90.Khersonsky O., Tawfik D.S. Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry. 2005;44(16):6371–6382. doi: 10.1021/bi047440d. [DOI] [PubMed] [Google Scholar]
- 91.Furlong C.E., Richter R.J., Seidel S.L., Costa L.G., Motulsky A.G. Spectrophotometric assays for the enzymatic hydrolysis of the active metabolites of chlorpyrifos and parathion by plasma paraoxonase/arylesterase. Anal. Biochem. 1989;180(2):242–247. doi: 10.1016/0003-2697(89)90424-7. [DOI] [PubMed] [Google Scholar]
- 92.Nevin D.N., Zambon A., Furlong C.E., Richter R.J., Humbert R., Hokanson J.E. Paraoxonase genotypes, lipoprotein lipase activity, and HDL. Arterioscler. Thromb. Vasc. Biol. 1996;16(10):1243–1249. doi: 10.1161/01.atv.16.10.1243. [DOI] [PubMed] [Google Scholar]
- 93.Shih D.M., Xia Y.R., Wang X.P., Miller E., Castellani L.W., Subbanagounder G. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J. Biol. Chem. 2000;275(23):17527–17535. doi: 10.1074/jbc.M910376199. [DOI] [PubMed] [Google Scholar]
- 94.Ng D.S., Chu T., Esposito B., Hui P., Connelly P.W., Gross P.L. Paraoxonase-1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovasc. Pathol. 2008;17(4):226–232. doi: 10.1016/j.carpath.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 95.She Z.G., Zheng W., Wei Y.S., Chen H.Z., Wang A.B., Li H.L. Human paraoxonase gene cluster transgenic overexpression represses atherogenesis and promotes atherosclerotic plaque stability in ApoE-null mice. Circ. Res. 2009;104(10):1160–1168. doi: 10.1161/CIRCRESAHA.108.192229. [DOI] [PubMed] [Google Scholar]
- 96.Mackness B., Durrington P., McElduff P., Yarnell J., Azam N., Watt M. Low paraoxonase activity predicts coronary events in the Caerphilly Prospective Study. Circulation. 2003;107(22):2775–2779. doi: 10.1161/01.CIR.0000070954.00271.13. [DOI] [PubMed] [Google Scholar]
- 97.Wang M., Lang X., Cui S., Zou L., Cao J., Wang S. Quantitative assessment of the influence of paraoxonase 1 activity and coronary heart disease risk. DNA Cell Biol. 2012;31(6):975–982. doi: 10.1089/dna.2011.1478. [DOI] [PubMed] [Google Scholar]
- 98.Tang W.H., Hartiala J., Fan Y., Wu Y., Stewart A.F., Erdmann J. Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2012;32(11):2803–2812. doi: 10.1161/ATVBAHA.112.253930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhattacharyya T., Nicholls S.J., Topol E.J., Zhang R., Yang X., Schmitt D. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA. 2008;299(11):1265–1276. doi: 10.1001/jama.299.11.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mackness M.I., Arrol S., Mackness B., Durrington P.N. Alloenzymes of paraoxonase and effectiveness of high-density lipoproteins in protecting low-density lipoprotein against lipid peroxidation. Lancet. 1997;349(9055):851–852. doi: 10.1016/S0140-6736(05)61755-2. [DOI] [PubMed] [Google Scholar]
- 101.Connelly P.W., Draganov D., Maguire G.F. Paraoxonase-1 does not reduce or modify oxidation of phospholipids by peroxynitrite. Free Radic. Biol. Med. 2005;38(2):164–174. doi: 10.1016/j.freeradbiomed.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 102.Draganov D.I., Teiber J.F., Speelman A., Osawa Y., Sunahara R., La Du B.N. Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res. 2005;46(6):1239–1247. doi: 10.1194/jlr.M400511-JLR200. [DOI] [PubMed] [Google Scholar]
- 103.Marathe G.K., Zimmerman G.A., McIntyre T.M. Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J. Biol. Chem. 2003;278(6):3937–3947. doi: 10.1074/jbc.M211126200. [DOI] [PubMed] [Google Scholar]
- 104.Teiber J.F., Draganov D.I., La Du B.N. Purified human serum PON1 does not protect LDL against oxidation in the in vitro assays initiated with copper or AAPH. J. Lipid Res. 2004;45(12):2260–2268. doi: 10.1194/jlr.M400213-JLR200. [DOI] [PubMed] [Google Scholar]
- 105.Huang Y., Wu Z., Riwanto M., Gao S., Levison B.S., Gu X. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J. Clin. Invest. 2013;123(9):3815–3828. doi: 10.1172/JCI67478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang C., Peng W., Wang M., Zhu J., Zang Y., Shi W. Studies on protective effects of human paraoxonases 1 and 3 on atherosclerosis in apolipoprotein E knockout mice. Gene Ther. 2010;17(5):626–633. doi: 10.1038/gt.2010.11. [DOI] [PubMed] [Google Scholar]
- 107.Dobiasova M., Frohlich J.J. Advances in understanding of the role of lecithin cholesterol acyltransferase (LCAT) in cholesterol transport. Clin. Chim. Acta. 1999;286(1–2):257–271. doi: 10.1016/s0009-8981(99)00106-0. [DOI] [PubMed] [Google Scholar]
- 108.Goyal J., Wang K., Liu M., Subbaiah P.V. Novel function of lecithin-cholesterol acyltransferase. Hydrolysis of oxidized polar phospholipids generated during lipoprotein oxidation. J. Biol. Chem. 1997;272(26):16231–16239. doi: 10.1074/jbc.272.26.16231. [DOI] [PubMed] [Google Scholar]
- 109.McPherson P.A., Young I.S., McEneny J. A dual role for lecithin:cholesterol acyltransferase (EC 2.3.1.43) in lipoprotein oxidation. Free Radic. Biol. Med. 2007;43(11):1484–1493. doi: 10.1016/j.freeradbiomed.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 110.Holleboom A.G., Daniil G., Fu X., Zhang R., Hovingh G.K., Schimmel A.W. Lipid oxidation in carriers of lecithin:cholesterol acyltransferase gene mutations. Arterioscler. Thromb. Vasc. Biol. 2012;32(12):3066–3075. doi: 10.1161/ATVBAHA.112.255711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mallat Z., Lambeau G., Tedgui A. Lipoprotein-associated and secreted phospholipases A(2) in cardiovascular disease: roles as biological effectors and biomarkers. Circulation. 2010;122(21):2183–2200. doi: 10.1161/CIRCULATIONAHA.110.936393. [DOI] [PubMed] [Google Scholar]
- 112.McIntyre T.M., Prescott S.M., Stafforini D.M. The emerging roles of PAF acetylhydrolase. J. Lipid Res. 2009;50(Suppl):S255–9. doi: 10.1194/jlr.R800024-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tselepis A.D., Dentan C., Karabina S.A., Chapman M.J., Ninio E. PAF-degrading acetylhydrolase is preferentially associated with dense LDL and VHDL-1 in human plasma. Catalytic characteristics and relation to the monocyte-derived enzyme. Arterioscler. Thromb. Vasc. Biol. 1995;15(10):1764–1773. doi: 10.1161/01.atv.15.10.1764. [DOI] [PubMed] [Google Scholar]
- 114.Quarck R., De Geest B., Stengel D., Mertens A., Lox M., Theilmeier G. Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;103(20):2495–2500. doi: 10.1161/01.cir.103.20.2495. [DOI] [PubMed] [Google Scholar]
- 115.Tsimihodimos V., Kakafika A., Tambaki A.P., Bairaktari E., Chapman M.J., Elisaf M. Fenofibrate induces HDL-associated PAF-AH but attenuates enzyme activity associated with apoB-containing lipoproteins. J. Lipid Res. 2003;44(5):927–934. doi: 10.1194/jlr.M200452-JLR200. [DOI] [PubMed] [Google Scholar]
- 116.Zalewski A., Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler. Thromb. Vasc. Biol. 2005;25(5):923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 117.Kotosai M., Shimada S., Kanda M., Matsuda N., Sekido K., Shimizu Y. Plasma HDL reduces nonesterified fatty acid hydroperoxides originating from oxidized LDL: a mechanism for its antioxidant ability. Lipids. 2013;48(6):569–578. doi: 10.1007/s11745-013-3779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rodrigo L., Mackness B., Durrington P.N., Hernandez A., Mackness M.I. Hydrolysis of platelet-activating factor by human serum paraoxonase. Biochem. J. 2001;354(Pt 1):1–7. doi: 10.1042/0264-6021:3540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rosenson R.S., Hurt-Camejo E. Phospholipase A2 enzymes and the risk of atherosclerosis. Eur. Heart J. 2012;33(23):2899–2909. doi: 10.1093/eurheartj/ehs148. [DOI] [PubMed] [Google Scholar]
- 120.Ahmed Z., Ravandi A., Maguire G.F., Emili A., Draganov D., La Du B.N. Apolipoprotein A-I promotes the formation of phosphatidylcholine core aldehydes that are hydrolyzed by paraoxonase (PON-1) during high density lipoprotein oxidation with a peroxynitrite donor. J. Biol. Chem. 2001;276(27):24473–24481. doi: 10.1074/jbc.M010459200. [DOI] [PubMed] [Google Scholar]
- 121.Maddipati K.R., Marnett L.J. Characterization of the major hydroperoxide-reducing activity of human plasma. Purification and properties of a selenium-dependent glutathione peroxidase. J. Biol. Chem. 1987;262(36):17398–17403. [PubMed] [Google Scholar]
- 122.Avissar N., Ornt D.B., Yagil Y., Horowitz S., Watkins R.H., Kerl E.A. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am. J. Phys. 1994;266(2 Pt 1):C367–75. doi: 10.1152/ajpcell.1994.266.2.C367. [DOI] [PubMed] [Google Scholar]
- 123.Chen N., Liu Y., Greiner C.D., Holtzman J.L. Physiologic concentrations of homocysteine inhibit the human plasma GSH peroxidase that reduces organic hydroperoxides. J. Lab. Clin. Med. 2000;136(1):58–65. doi: 10.1067/mlc.2000.107692. [DOI] [PubMed] [Google Scholar]
- 124.Camont L., Lhomme M., Rached F., Le Goff W., Negre-Salvayre A., Salvayre R. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013;33(12):2715–2723. doi: 10.1161/ATVBAHA.113.301468. [DOI] [PubMed] [Google Scholar]
- 125.Parthasarathy S., Khoo J.C., Miller E., Barnett J., Witztum J.L., Steinberg D. Low density lipoprotein rich in oleic acid is protected against oxidative modification: implications for dietary prevention of atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 1990;87(10):3894–3898. doi: 10.1073/pnas.87.10.3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gomez Rosso L., Lhomme M., Merono T., Dellepiane A., Sorroche P., Hedjazi L. Poor glycemic control in type 2 diabetes enhances functional and compositional alterations of small, dense HDL3c. Biochim. Biophys. Acta. 2017;1862(2):188–195. doi: 10.1016/j.bbalip.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 127.Gomez Rosso L., Lhomme M., Merono T., Sorroche P., Catoggio L., Soriano E. Altered lipidome and antioxidative activity of small, dense HDL in normolipidemic rheumatoid arthritis: relevance of inflammation. Atherosclerosis. 2014;237(2):652–660. doi: 10.1016/j.atherosclerosis.2014.09.034. [DOI] [PubMed] [Google Scholar]
- 128.Rached F., Lhomme M., Camont L., Gomes F., Dauteuille C., Robillard P. Defective functionality of small, dense HDL3 subpopulations in ST segment elevation myocardial infarction: Relevance of enrichment in lysophosphatidylcholine, phosphatidic acid and serum amyloid A. Biochim. Biophys. Acta. 2015;1851(9):1254–1261. doi: 10.1016/j.bbalip.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 129.Ohara A., Sayuri M. 2011. Oxygen and Its Metabolites Imprinted the Evolution of Life. [Google Scholar]
- 130.Nguyen S.D., Jeong T.S., Sok D.E. Apolipoprotein A-I-mimetic peptides with antioxidant actions. Arch. Biochem. Biophys. 2006;451(1):34–42. doi: 10.1016/j.abb.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 131.Bergt C., Marsche G., Panzenboeck U., Heinecke J.W., Malle E., Sattler W. Human neutrophils employ the myeloperoxidase/hydrogen peroxide/chloride system to oxidatively damage apolipoprotein A-I. Eur. J. Biochem. 2001;268(12):3523–3531. doi: 10.1046/j.1432-1327.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- 132.Hadfield K.A., Pattison D.I., Brown B.E., Hou L., Rye K.A., Davies M.J. Myeloperoxidase-derived oxidants modify apolipoprotein A-I and generate dysfunctional high-density lipoproteins: comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCl) Biochem. J. 2013;449(2):531–542. doi: 10.1042/BJ20121210. [DOI] [PubMed] [Google Scholar]
- 133.Sutherland W.H., de Jong S.A., Walker R.J. Hypochlorous acid and low serum paraoxonase activity in haemodialysis patients: an in vitro study. Nephrol. Dial. Transplant. 2004;19(1):75–82. doi: 10.1093/ndt/gfg484. [DOI] [PubMed] [Google Scholar]
- 134.Rosenson R.S., Brewer H.B., Jr., Chapman M.J., Fazio S., Hussain M.M., Kontush A. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011;57(3):392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- 135.de Souza J.A., Vindis C., Negre-Salvayre A., Rye K.A., Couturier M., Therond P. Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J. Cell. Mol. Med. 2010;14(3):608–620. doi: 10.1111/j.1582-4934.2009.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Arbones-Mainar J.M., Navarro M.A., Carnicer R., Guillen N., Surra J.C., Acin S. Accelerated atherosclerosis in apolipoprotein E-deficient mice fed Western diets containing palm oil compared with extra virgin olive oils: a role for small, dense high-density lipoproteins. Atherosclerosis. 2007;194(2):372–382. doi: 10.1016/j.atherosclerosis.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 137.Heinecke J. The many transport functions of HDL. Circulation. 2009 [Google Scholar]
- 138.Sparks D.L., Davidson W.S., Lund-Katz S., Phillips M.C. Effects of the neutral lipid content of high density lipoprotein on apolipoprotein A-I structure and particle stability. J. Biol. Chem. 1995;270(45):26910–26917. doi: 10.1074/jbc.270.45.26910. [DOI] [PubMed] [Google Scholar]
- 139.Curtiss L.K., Bonnet D.J., Rye K.A. The conformation of apolipoprotein A-I in high-density lipoproteins is influenced by core lipid composition and particle size: a surface plasmon resonance study. Biochemistry. 2000;39(19):5712–5721. doi: 10.1021/bi992902m. [DOI] [PubMed] [Google Scholar]
- 140.Hansel B., Giral P., Nobecourt E., Chantepie S., Bruckert E., Chapman M.J. Metabolic syndrome is associated with elevated oxidative stress and dysfunctional dense high-density lipoprotein particles displaying impaired antioxidative activity. J. Clin. Endocrinol. Metab. 2004;89(10):4963–4971. doi: 10.1210/jc.2004-0305. [DOI] [PubMed] [Google Scholar]
- 141.Nobecourt E., Jacqueminet S., Hansel B., Chantepie S., Grimaldi A., Chapman M.J. Defective antioxidative activity of small dense HDL3 particles in type 2 diabetes: relationship to elevated oxidative stress and hyperglycaemia. Diabetologia. 2005;48(3):529–538. doi: 10.1007/s00125-004-1655-5. [DOI] [PubMed] [Google Scholar]
- 142.Sanguinetti S.M., Brites F.D., Fasulo V., Verona J., Elbert A., Wikinski R.L. HDL oxidability and its protective effect against LDL oxidation in Type 2 diabetic patients. Diabetes Nutr. Metab. 2001;14(1):27–36. [PubMed] [Google Scholar]
- 143.Gowri M.S., Van der Westhuyzen D.R., Bridges S.R., Anderson J.W. Decreased protection by HDL from poorly controlled type 2 diabetic subjects against LDL oxidation may be due to the abnormal composition of HDL. Arterioscler. Thromb. Vasc. Biol. 1999;19(9):2226–2233. doi: 10.1161/01.atv.19.9.2226. [DOI] [PubMed] [Google Scholar]
- 144.Kontush A., de Faria E.C., Chantepie S., Chapman M.J. A normotriglyceridemic, low HDL-cholesterol phenotype is characterised by elevated oxidative stress and HDL particles with attenuated antioxidative activity. Atherosclerosis. 2005;182(2):277–285. doi: 10.1016/j.atherosclerosis.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 145.Kontush A., de Faria E.C., Chantepie S., Chapman M.J. Antioxidative activity of HDL particle subspecies is impaired in hyperalphalipoproteinemia: relevance of enzymatic and physicochemical properties. Arterioscler. Thromb. Vasc. Biol. 2004;24(3):526–533. doi: 10.1161/01.ATV.0000118276.87061.00. [DOI] [PubMed] [Google Scholar]
- 146.Patel P.J., Khera A.V., Jafri K., Wilensky R.L., Rader D.J. The anti-oxidative capacity of high-density lipoprotein is reduced in acute coronary syndrome but not in stable coronary artery disease. J. Am. Coll. Cardiol. 2011;58(20):2068–2075. doi: 10.1016/j.jacc.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 147.Zhang J., Zhang Y., Liu H., Bai H., Wang Y., Jiang C. Antioxidant properties of high-density lipoproteins are impaired in women with polycystic ovary syndrome. Fertil. Steril. 2015;103(5):1346–1354. doi: 10.1016/j.fertnstert.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 148.Wayner D.D., Burton G.W., Ingold K.U., Barclay L.R., Locke S.J. The relative contributions of vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity of human blood plasma. Biochim. Biophys. Acta. 1987;924(3):408–419. doi: 10.1016/0304-4165(87)90155-3. [DOI] [PubMed] [Google Scholar]
- 149.Nyyssonen K., Porkkala-Sarataho E., Kaikkonen J., Salonen J.T. Ascorbate and urate are the strongest determinants of plasma antioxidative capacity and serum lipid resistance to oxidation in Finnish men. Atherosclerosis. 1997;130(1–2):223–233. doi: 10.1016/s0021-9150(96)06064-9. [DOI] [PubMed] [Google Scholar]
- 150.Mohamadin A.M., Habib F.A., Elahi T.F. Serum paraoxonase 1 activity and oxidant/antioxidant status in Saudi women with polycystic ovary syndrome. Pathophysiology. 2010;17(3):189–196. doi: 10.1016/j.pathophys.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 151.McMahon M., Grossman J., FitzGerald J., Dahlin-Lee E., Wallace D.J., Thong B.Y. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006;54(8):2541–2549. doi: 10.1002/art.21976. [DOI] [PubMed] [Google Scholar]
- 152.Gaal K., Tarr T., Lorincz H., Borbas V., Seres I., Harangi M. High-density lipopoprotein antioxidant capacity, subpopulation distribution and paraoxonase-1 activity in patients with systemic lupus erythematosus. Lipids Health Dis. 2016;15:60. doi: 10.1186/s12944-016-0229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moradi H., Pahl M.V., Elahimehr R., Vaziri N.D. Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl. Res. 2009;153(2):77–85. doi: 10.1016/j.trsl.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 154.Morena M., Cristol J.P., Dantoine T., Carbonneau M.A., Descomps B., Canaud B. Protective effects of high-density lipoprotein against oxidative stress are impaired in haemodialysis patients. Nephrol. Dial. Transplant. 2000;15(3):389–395. doi: 10.1093/ndt/15.3.389. [DOI] [PubMed] [Google Scholar]
- 155.Honda H., Ueda M., Kojima S., Mashiba S., Michihata T., Takahashi K. Oxidized high-density lipoprotein as a risk factor for cardiovascular events in prevalent hemodialysis patients. Atherosclerosis. 2012;220(2):493–501. doi: 10.1016/j.atherosclerosis.2011.10.038. [DOI] [PubMed] [Google Scholar]
- 156.Merono T., Dauteuille C., Tetzlaff W., Martin M., Botta E., Lhomme M. Oxidative stress, HDL functionality and effects of intravenous iron administration in women with iron deficiency anemia. Clin. Nutr. (Edinburgh, Scotland) 2016 doi: 10.1016/j.clnu.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 157.Zago V., Sanguinetti S., Brites F., Berg G., Verona J., Basilio F. Impaired high density lipoprotein antioxidant activity in healthy postmenopausal women. Atherosclerosis. 2004;177(1):203–210. doi: 10.1016/j.atherosclerosis.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 158.Van Lenten B.J., Navab M., Shih D., Fogelman A.M., Lusis A.J. The role of high-density lipoproteins in oxidation and inflammation. Trends Cardiovasc. Med. 2001;11(3–4):155–161. doi: 10.1016/s1050-1738(01)00095-0. [DOI] [PubMed] [Google Scholar]
- 159.Charles-Schoeman C., Fleischmann R., Davignon J., Schwartz H., Turner S.M., Beysen C. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. 2015;67(3):616–625. doi: 10.1002/art.38974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ghezzi P., Sipe J.D. Dexamethasone modulation of LPS, IL-1, and TNF stimulated serum amyloid A synthesis in mice. Lymphokine Res. 1988;7(2):157–166. [PubMed] [Google Scholar]
- 161.Brock J.W., Jenkins A.J., Lyons T.J., Klein R.L., Yim E., Lopes-Virella M. Increased methionine sulfoxide content of apoA-I in type 1 diabetes. J. Lipid Res. 2008;49(4):847–855. doi: 10.1194/jlr.M800015-JLR200. [DOI] [PubMed] [Google Scholar]
- 162.Zhou H., Tan K.C., Shiu S.W., Wong Y. Increased serum advanced glycation end products are associated with impairment in HDL antioxidative capacity in diabetic nephropathy. Nephrol. Dial. Transplant. 2008;23(3):927–933. doi: 10.1093/ndt/gfm631. [DOI] [PubMed] [Google Scholar]
- 163.Shen Y., Ding F.H., Sun J.T., Pu L.J., Zhang R.Y., Zhang Q. Association of elevated apoA-I glycation and reduced HDL-associated paraoxonase1, 3 activity, and their interaction with angiographic severity of coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015;14:52. doi: 10.1186/s12933-015-0221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hansel B., Bonnefont-Rousselot D., Orsoni A., Bittar R., Giral P., Roussel R. Lifestyle intervention enhances high-density lipoprotein function among patients with metabolic syndrome only at normal low-density lipoprotein cholesterol plasma levels. J. Clin. Lipidol. 2016;10(5):1172–1181. doi: 10.1016/j.jacl.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 165.Casella-Filho A., Chagas A.C., Maranhao R.C., Trombetta I.C., Cesena F.H., Silva V.M. Effect of exercise training on plasma levels and functional properties of high-density lipoprotein cholesterol in the metabolic syndrome. Am. J. Cardiol. 2011;107(8):1168–1172. doi: 10.1016/j.amjcard.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 166.Brites F., Zago V., Verona J., Muzzio M.L., Wikinski R., Schreier L. HDL capacity to inhibit LDL oxidation in well-trained triathletes. Life Sci. 2006;78(26):3074–3081. doi: 10.1016/j.lfs.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 167.Park K.H., Shin D.G., Cho K.H. Dysfunctional lipoproteins from young smokers exacerbate cellular senescence and atherogenesis with smaller particle size and severe oxidation and glycation. Toxicol. Sci. 2014;140(1):16–25. doi: 10.1093/toxsci/kfu076. [DOI] [PubMed] [Google Scholar]
- 168.Fuhrman B., Koren L., Volkova N., Keidar S., Hayek T., Aviram M. Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis. 2002;164(1):179–185. doi: 10.1016/s0021-9150(02)00063-1. [DOI] [PubMed] [Google Scholar]
- 169.Harangi M., Seres I., Varga Z., Emri G., Szilvassy Z., Paragh G. Atorvastatin effect on high-density lipoprotein-associated paraoxonase activity and oxidative DNA damage. Eur. J. Clin. Pharmacol. 2004;60(10):685–691. doi: 10.1007/s00228-004-0820-6. [DOI] [PubMed] [Google Scholar]
- 170.Harangi M., Mirdamadi H.Z., Seres I., Sztanek F., Molnar M., Kassai A. Atorvastatin effect on the distribution of high-density lipoprotein subfractions and human paraoxonase activity. Transl. Res. 2009;153(4):190–198. doi: 10.1016/j.trsl.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 171.Nagila A., Permpongpaiboon T., Tantrarongroj S., Porapakkham P., Chinwattana K., Deakin S. Effect of atorvastatin on paraoxonase1 (PON1) and oxidative status. Pharmacol. Rep. 2009;61(5):892–898. doi: 10.1016/s1734-1140(09)70146-x. [DOI] [PubMed] [Google Scholar]
- 172.Bergheanu S.C., Van Tol A., Dallinga-Thie G.M., Liem A., Dunselman P.H., Van der Bom J.G. Effect of rosuvastatin versus atorvastatin treatment on paraoxonase-1 activity in men with established cardiovascular disease and a low HDL-cholesterol. Curr. Med. Res. Opin. 2007;23(9):2235–2240. doi: 10.1185/030079907X226104. [DOI] [PubMed] [Google Scholar]
- 173.Mirdamadi H.Z., Sztanek F., Derdak Z., Seres I., Harangi M., Paragh G. The human paraoxonase-1 phenotype modifies the effect of statins on paraoxonase activity and lipid parameters. Br. J. Clin. Pharmacol. 2008;66(3):366–374. doi: 10.1111/j.1365-2125.2008.03213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Muacevic-Katanec D., Bradamante V., Poljicanin T., Reiner Z., Babic Z., Simeon-Rudolf V. Clinical study on the effect of simvastatin on paraoxonase activity. Arzneimittelforschung. 2007;57(10):647–653. doi: 10.1055/s-0031-1296663. [DOI] [PubMed] [Google Scholar]
- 175.Yamashita S., Tsubakio-Yamamoto K., Ohama T., Nakagawa-Toyama Y., Nishida M. Molecular mechanisms of HDL-cholesterol elevation by statins and its effects on HDL functions. J. Atheroscler. Thromb. 2010;17(5):436–451. doi: 10.5551/jat.5405. [DOI] [PubMed] [Google Scholar]
- 176.Arii K., Suehiro T., Ota K., Ikeda Y., Kumon Y., Osaki F. Pitavastatin induces PON1 expression through p44/42 mitogen-activated protein kinase signaling cascade in Huh7 cells. Atherosclerosis. 2009;202(2):439–445. doi: 10.1016/j.atherosclerosis.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 177.Miyamoto-Sasaki M., Yasuda T., Monguchi T., Nakajima H., Mori K., Toh R. Pitavastatin increases HDL particles functionally preserved with cholesterol efflux capacity and antioxidative actions in dyslipidemic patients. J. Atheroscler. Thromb. 2013;20(9):708–716. doi: 10.5551/jat.17210. [DOI] [PubMed] [Google Scholar]
- 178.Tkac I., Molcanyiova A., Javorsky M., Kozarova M. Fenofibrate treatment reduces circulating conjugated diene level and increases glutathione peroxidase activity. Pharmacol. Res. 2006;53(3):261–264. doi: 10.1016/j.phrs.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 179.Broncel M., Cieslak D., Koter-Michalak M., Duchnowicz P., Mackiewicz K., Chojnowska-Jezierska J. The anti-inflammatory and antioxidants effects of micronized fenofibrate in patients with visceral obesity and dyslipidemia. Pol. Merkur. Lekarski. 2006;20(119):547–550. [PubMed] [Google Scholar]
- 180.Paragh G., Seres I., Harangi M., Balogh Z., Illyes L., Boda J. The effect of micronised fenofibrate on paraoxonase activity in patients with coronary heart disease. Diabetes Metab. 2003;29(6):613–618. doi: 10.1016/s1262-3636(07)70077-0. [DOI] [PubMed] [Google Scholar]
- 181.Phuntuwate W., Suthisisang C., Koanantakul B., Chaloeiphap P., Mackness B., Mackness M. Effect of fenofibrate therapy on paraoxonase1 status in patients with low HDL-C levels. Atherosclerosis. 2008;196(1):122–128. doi: 10.1016/j.atherosclerosis.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 182.Yesilbursa D., Serdar A., Saltan Y., Serdar Z., Heper Y., Guclu S. The effect of fenofibrate on serum paraoxonase activity and inflammatory markers in patients with combined hyperlipidemia. Kardiol. Pol. 2005;62(6):526–530. [PubMed] [Google Scholar]
- 183.Dullaart R.P., de Vries R., Voorbij H.A., Sluiter W.J., van Tol A. Serum paraoxonase-I activity is unaffected by short-term administration of simvastatin, bezafibrate, and their combination in type 2 diabetes mellitus. Eur. J. Clin. Investig. 2009;39(3):200–203. doi: 10.1111/j.1365-2362.2009.02090.x. [DOI] [PubMed] [Google Scholar]
- 184.Kaplon R.E., Gano L.B., Seals D.R. Vascular endothelial function and oxidative stress are related to dietary niacin intake among healthy middle-aged and older adults. J. Appl. Physiol. (1985) 2014;116(2):156–163. doi: 10.1152/japplphysiol.00969.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hamoud S., Kaplan M., Meilin E., Hassan A., Torgovicky R., Cohen R. Niacin administration significantly reduces oxidative stress in patients with hypercholesterolemia and low levels of high-density lipoprotein cholesterol. Am J Med Sci. 2013;345(3):195–199. doi: 10.1097/MAJ.0b013e3182548c28. [DOI] [PubMed] [Google Scholar]
- 186.Yadav R., Liu Y., Kwok S., Hama S., France M., Eatough R. Effect of extended-release niacin on high-density lipoprotein (HDL) functionality, lipoprotein metabolism, and mediators of vascular inflammation in statin-treated patients. J. Am. Heart Assoc. 2015;4(9) doi: 10.1161/JAHA.114.001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Batuca J.R., Amaral M.C., Favas C., Paula F.S., Ames P.R., Papoila A.L. Extended-release niacin increases anti-apolipoprotein A-I antibodies that block the antioxidant effect of high-density lipoprotein-cholesterol: the EXPLORE clinical trial. Br. J. Clin. Pharmacol. 2016 doi: 10.1111/bcp.13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wool G.D., Reardon C.A., Getz G.S. Apolipoprotein A-I mimetic peptide helix number and helix linker influence potentially anti-atherogenic properties. J. Lipid Res. 2008;49(6):1268–1283. doi: 10.1194/jlr.M700552-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Navab M., Hama S.Y., Anantharamaiah G.M., Hassan K., Hough G.P., Watson A.D. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J. Lipid Res. 2000;41(9):1495–1508. [PubMed] [Google Scholar]
- 190.Navab M., Shechter I., Anantharamaiah G.M., Reddy S.T., Van Lenten B.J., Fogelman A.M. Structure and function of HDL mimetics. Arterioscler. Thromb. Vasc. Biol. 2010;30(2):164–168. doi: 10.1161/ATVBAHA.109.187518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chattopadhyay A., Navab M., Hough G., Gao F., Meriwether D., Grijalva V. A novel approach to oral apoA-I mimetic therapy. J. Lipid Res. 2013;54(4):995–1010. doi: 10.1194/jlr.M033555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Navab M., Hough G., Buga G.M., Su F., Wagner A.C., Meriwether D. Transgenic 6F tomatoes act on the small intestine to prevent systemic inflammation and dyslipidemia caused by Western diet and intestinally derived lysophosphatidic acid. J. Lipid Res. 2013;54(12):3403–3418. doi: 10.1194/jlr.M042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Di Bartolo B.A., Vanags L.Z., Tan J.T., Bao S., Rye K.A., Barter P.J. The apolipoprotein A-I mimetic peptide, ETC-642, reduces chronic vascular inflammation in the rabbit. Lipids Health Dis. 2011;10:224. doi: 10.1186/1476-511X-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tabet F., Remaley A.T., Segaliny A.I., Millet J., Yan L., Nakhla S. The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler. Thromb. Vasc. Biol. 2010;30(2):246–252. doi: 10.1161/ATVBAHA.109.200196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.D'Souza W., Stonik J.A., Murphy A., Demosky S.J., Sethi A.A., Moore X.L. Structure/function relationships of apolipoprotein A-I mimetic peptides: implications for antiatherogenic activities of high-density lipoprotein. Circ. Res. 2010;107(2):217–227. doi: 10.1161/CIRCRESAHA.110.216507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cho K.H., Park S.H., Han J.M., Kim H.C., Choi Y.K., Choi I. ApoA-I mutants V156K and R173C promote anti-inflammatory function and antioxidant activities. Eur. J. Clin. Investig. 2006;36(12):875–882. doi: 10.1111/j.1365-2362.2006.01737.x. [DOI] [PubMed] [Google Scholar]
- 197.Kim S.H., Lee E.Y., Cho K.H. Incorporation of human growth hormone-2 into proteoliposome enhances tissue regeneration with anti-oxidant and anti-senescence activities. Rejuvenation Res. 2015;18(1):20–29. doi: 10.1089/rej.2014.1594. [DOI] [PubMed] [Google Scholar]
- 198.Lee E.Y., Kim S.H., Cho K.H. Reconstituted high-density lipoprotein containing human growth hormone-1 shows potent tissue regeneration activity with enhancement of anti-oxidant and anti-atherosclerotic activities. Rejuvenation Res. 2015;18(3):245–256. doi: 10.1089/rej.2014.1644. [DOI] [PubMed] [Google Scholar]
- 199.Zhang X., Wang L., Chen B. Recombinant HDL (Milano) protects endotoxin-challenged rats from multiple organ injury and dysfunction. Biol. Chem. 2015;396(1):53–60. doi: 10.1515/hsz-2014-0188. [DOI] [PubMed] [Google Scholar]
- 200.Didichenko S.A., Navdaev A.V., Cukier A.M., Gille A., Schuetz P., Spycher M.O. Enhanced HDL functionality in small HDL species produced upon remodeling of HDL by reconstituted HDL, CSL112: effects on cholesterol efflux, anti-inflammatory and antioxidative activity. Circ. Res. 2016;119(6):751–763. doi: 10.1161/CIRCRESAHA.116.308685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Park K.H., Kim J.Y., Choi I., Kim J.R., Cho K.H. omega-6 (18:2) and omega-3 (18:3) fatty acids in reconstituted high-density lipoproteins show different functionality of anti-atherosclerotic properties and embryo toxicity. J. Nutr. Biochem. 2015;26(12):1613–1621. doi: 10.1016/j.jnutbio.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 202.Popa C., van Tits L.J., Barrera P., Lemmers H.L., van den Hoogen F.H., van Riel P.L. Anti-inflammatory therapy with tumour necrosis factor alpha inhibitors improves high-density lipoprotein cholesterol antioxidative capacity in rheumatoid arthritis patients. Ann. Rheum. Dis. 2009;68(6):868–872. doi: 10.1136/ard.2008.092171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.