

Abstract
This review focuses on the biogenesis and composition of the eukaryotic DNA replication fork, with an emphasis on the enzymes that synthesize DNA and repair discontinuities on the lagging strand of the replication fork. Physical and genetic methodologies aimed at understanding these processes are discussed. The preponderance of evidence supports a model in which DNA polymerase ε (Pol ε) carries out the bulk of leading strand DNA synthesis at an undisturbed replication fork. DNA polymerases α and β carry out the initiation of Okazaki fragment synthesis and its elongation and maturation, respectively. This review also discusses alternative proposals, including cellular processes during which alternative forks may be utilized, and new biochemical studies with purified proteins that are aimed at reconstituting leading and lagging strand DNA synthesis separately and as an integrated replication fork.
Keywords: DNA polymerase, DNA primase, CMG helicase, Okazaki fragment, replisome coordination
1. INTRODUCTION
Cellular DNA replication mechanisms are highly conserved. All organisms in the three kingdoms of life carry out semiconservative DNA replication, as originally hypothesized by Watson and Crick (1). All organisms also have solved the mode of replication of antiparallel double-stranded DNA (dsDNA) in a similar fashion. The strand that is synthesized in the same direction as that of the moving replication fork is replicated continuously, whereas the strand synthesized in the opposite direction is replicated discontinuously. The small fragments laid down on the lagging strand are termed Okazaki fragments, in honor of Reji Okazaki & Tuneko Okazaki (2), who first proposed the model for their synthesis in 1968. The term semidiscontinuous should not be interpreted too literally. It is generally assumed that the leading strand is replicated continuously, although the incorporation of noncanonical nucleotides by the DNA polymerases, particularly uracil and ribonucleotides, followed by their subsequent excision repair, has given the appearance that the leading strand may also be replicated somewhat discontinuously (3, 4). This was in fact one of the models originally proposed by the Okazakis (2).
Beyond this simple replication model, the details vary considerably among kingdoms and even within kingdoms, in particular within the bacterial and archaeal kingdoms. The inherent asymmetry of the fork imposes an iterative priming mechanism on the lagging strand, which in eukaryotes is coupled to a polymerization machinery that is distinct from that on the leading strand. To understand these machineries, how they are assembled during the initiation of DNA replication at origins is important. We briefly describe this process and then discuss methodologies and experimental approaches that have resulted in the proposal of current models of the eukaryotic replication fork.
Ideally one would like to take a snapshot of the replication fork using a biophysical analysis of unperturbed cells. Recently advented biochemical studies of the initiation and elongation of DNA replication using purified yeast replication factors promise to become powerful tools for in-depth studies of the replication fork (5). However, much of the current information we have still derives from genetic analyses based on the use of informative replication mutants. Unfortunately, this genetic analysis can be complicated by the fact that the very alterations introduced to assess the structure of the replication fork through measuring perturbations in mutants may also alter the structure of the replication fork itself (6).1 This may occur through recruitment of alternative factors that suppress growth defects resulting from our alterations and also through an altered deployment of those factors already present. Therefore, ideally, the perturbations introduced by such an analysis should be as minimal as possible. In this review, we discuss the methodologies and experiments that have led to our current proposal of an unperturbed fork and briefly indicate situations in the cell and cellular responses to stress that may alter the fork structure.
2. REPLICATION FORK ASSEMBLY
A recent review in this series focused on the selection of replication initiation sites in eukaryotes and their control (7). A second review in this series discussed both the commonalities and the critical differences in replisome assembly and activity in the three kingdoms of life (8). Additional reviews focus on the binding and activities of the many replication initiation proteins in eukaryotes and the step-by-step assembly of distinct replication initiation intermediates and their progression into a functional replisome (9, 10). Here, we briefly summarize replisome assembly with a focus on those factors that end up moving with the replication fork. We divide initiation into four broad stages (Figure 1): (a) loading of the minichromosome maintenance (MCM) helicase as a double hexamer at sites marked by the origin recognition complex (ORC); (b) loading of other accessory factors to form a preinitiation complex; (c) rearrangement of the MCM complex from an inactive to an active helicase; and (d ) priming of DNA replication.
Figure 1.
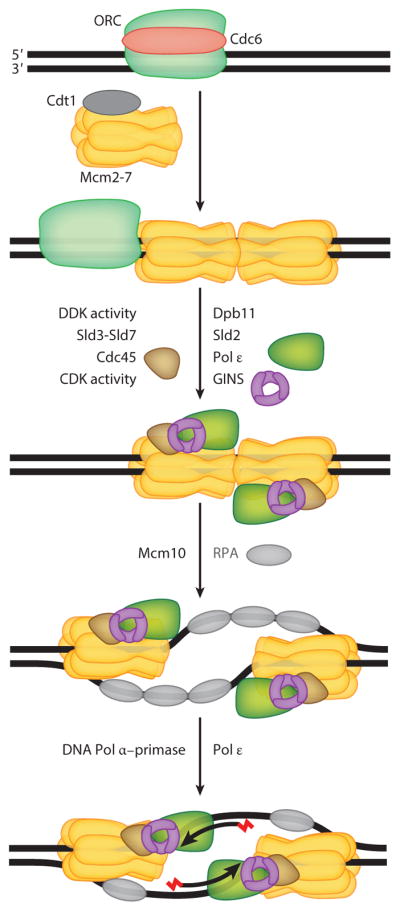
Assembly of the eukaryotic replisome. The origin-bound ORC–Cdc6 complex initially recruits one Cdt1–Mcm2-7 complex, followed by a second complex, to form a double Mcm2-7 hexamer. Further assembly requires Dpb11, Sld2, Sld3, and Sld7, which are not thought to be associated with the mature replisome, and Cdc45, GINS, and Pol ε, which are associated with it, as well as DDK and CDK kinase activity to complete assembly and prime the complex for helicase activation that is accomplished by Mcm10 and RPA. Abbreviations: CDK, cyclin-dependent kinase; DDK, Cdc7/Dbf4 kinase; GINS, Sld5, Psf1, Psf2, and Psf3 complex; Mcm2-7, helicase complex; ORC, origin recognition complex; Pol, DNA polymerase; RPA, replication protein A.
The initial loading of the heterohexameric MCM helicase to form a preinitiation complex involves Cdc6 and Cdt1 as loading factors, and it proceeds in two distinct steps. Initially, the ORC–Cdc6 complex recruits a single MCM hexamer in a complex with Cdt1. Subsequently, the second MCM–Cdt1 complex is recruited and both MCM hexamers, in the form of a symmetrical double hexamer, surround the dsDNA (reviewed in 9–11). During the second stage of replisome assembly, many additional factors are recruited to this inactive double hexamer, and the assembly is driven by both DDK (Cdc7/Dbf4 kinase) and CDK (cyclin-dependent kinase) protein kinase activity. During this assembly, the heterotetrameric ring-like complex GINS, consisting of the Sld5, Psf1, Psf2, and Psf3 subunits, is loaded as a tightly interacting complex with DNA polymerase ε (Pol ε), the first DNA polymerase to be incorporated into the ever-growing conglomerate of initiation factors (12–14).
The next stage of replication initiation, the transformation of the MCM double hexamer surrounding the dsDNA into that of a single MCM hexamer encircling the leading strand at each of the two divergent forks, is currently not well understood. It is known to depend on the preceding phosphorylation of initiation factors by the CDK and DDK protein kinases. This stage appears to be catalyzed by Mcm10 and the single-stranded binding protein RPA (replication protein A) (15–19). At this point, the active DNA helicase consists of three factors—Cdc45, Mcm2-7, and GINS—i.e., the CMG complex (20, 21). Movement of the CMG helicase along each of the two leading strands of the nascent replication forks generates single-stranded DNA (ssDNA) coated with RPA, which provides sites for priming by the DNA polymerase α (Pol α)–DNA primase complex for leading strand DNA synthesis.
3. METHODOLOGIES TO LOCALIZE REPLICATION FACTORS TO LEADING AND LAGGING STRANDS IN CELLS
The techniques described below have been primarily developed to address a central question in eukaryotic DNA replication: Which DNA polymerase replicates which strand at the replication fork? In addition, these techniques have also been useful in addressing other replication fork–related questions, such as that of chromatin dynamics behind the fork.
3.1. Biochemical Techniques
Ideally, one would like to take a snapshot of replication forks in unperturbed cells during the course of DNA replication and identify individual proteins that are associated with either the leading or the lagging strand. Only then can one proceed in determining how these associations change when cells are subjected to replication inhibitors or genotoxic agents. Methodologies have been developed to probe the association of proteins with replication forks inside living cells (22, 23). For example, replicating DNA can be pulse labeled by incorporation of a nucleotide analog that allows purification of the labeled DNA together with proteins associated with this DNA by a cross-linking step. This step is followed by the detection of the cross-linked proteins, specific DNA sequences, or both. The success of this technique depends critically on the sharpness and effectiveness of the pulse, i.e., a rapid on phase when the analog is added followed by a rapid and effective chase phase. For example, in one pulse-labeling study of DNA replication in mammalian cells, a 2.5-min pulse yielded PCNA (proliferating cell nuclear antigen) and the chromatin assembly factor CAF-1 as the most abundant cross-linked species, whereas a 5-min pulse yielded core histones as the more abundant species (22). Therefore, depending on pulse labeling conditions, post-fork processes may actually predominate the analysis.
Pulse labeling of nascent DNA in yeast has been combined with strand-specific sequencing of the labeled DNA that was cross-linked to replication proteins (23). Given that origins in yeast are known with high accuracy, this technique allows mapping of the nascent labeled strands, and by inference the proteins that were associated with it, to either the leading or lagging strand of replication forks (Figure 2a). The technique was used to map Pol ε, Cdc45, and the Mcm6 subunit of CMG predominantly to the leading strand and RPA, Pol α, and Pol δ mainly to the lagging strand (23). There are two potential concerns with the interpretation of these data. First, a preferential association of a replication protein with one strand does not imply a lack of association with the opposite strand. As an example, RPA is preferentially associated with the lagging strand where it binds to the exposed ssDNA of unreplicated Okazaki fragments. However, a more limited association of RPA with the leading strand would also be consistent with the data. Second, the specific association of an enzyme with a certain strand does not necessarily imply that the enzyme carries out its catalytic function on that strand. This unusual explanation has been put forward to argue that Pol ε, although associated with the leading strand, does not carry out DNA synthesis of the leading strand (24).
Figure 2.

Strand-specific mapping techniques. (a) Mapping of strand-specific protein binding. Replicating cells are pulse labeled with bromodeoxyuridine (BrdU), followed by chromatin-immunoprecipitation (ChIP) of a lagging strand–associated protein. Protein-associated nascent single-stranded DNA (ssDNA) is enriched by immunoprecipitation (IP) with antibodies against BrdU, and the isolated ssDNA is subjected to strand-specific sequencing. The sequence reads are mapped to both the Watson (W) and Crick (C) strands and plotted as a ratio of W/C reads. The opposite result is expected when the experiment is carried out with a protein associated with the nascent leading strand. (b) Mapping of ribonucleotide monophosphate (rNMP) incorporation by the rNMP-prone lagging strand polymerase variant. The frequent rNMP incorporation by DNA polymerase δ (L612M) was detected in an RNH201 strain that eliminates ribonucleotide excision repair. After cleavage of ribonucleotides in the isolated DNA with alkali or ribonuclease H2 (RNase H2), various technologies have been used to target these ends (either the ribose-2′- or 3′-phosphate end or the 5′-phosphate end) for strand-specific sequencing. The sequence reads are mapped to both the Watson (W) and Crick (C) strands and plotted as a ratio of W/C reads. The opposite result is expected when the experiment is carried out with the rNMP-prone leading strand polymerase variant.
3.2. Genetic Techniques: Strand-Specific Ribonucleotide Incorporation
The roles of Pol α, Pol δ, and Pol ε in replicating the two strands of the nuclear genome have been estimated from genetic studies of their abilities to incorporate noncanonical nucleotides during replication of DNA that has not been exposed to external environmental stress. The most abundant noncanonical nucleotide precursors present in eukaryotic cells are the ribonucleotide triphosphates (rNTPs). Most DNA polymerases discriminate well against inserting rNTPs during DNA synthesis in vitro, by factors ranging from 1,000-fold to >1,000,000-fold (25–27). However, the four rNTPs are present in eukaryotic cells in 10-fold to >100-fold excess over the four deoxynucleotide triphosphates (dNTPs) normally used to synthesize DNA (28, 29). Thus, when the four rNTPs and the four dNTPs are all present in polymerization reactions at concentrations estimated to be present in vivo (4), yeast Pol α, Pol δ, and Pol εincorporate one rNTP for every 625, 5,000, or 1,250 dNTPs incorporated in vitro, respectively, and their mammalian equivalents were subsequently found to behave similarly (30, 31).
Such frequent incorporation of ribonucleotides into DNA in vitro predicts that ribonucleotides should be readily incorporated into DNA during replication of the nuclear genome in vivo. If so, ribonucleotide incorporation could be used to estimate the roles of Pol α, Pol δ, and Pol ε in replicating the leading and lagging strands of undamaged DNA in vivo, using variant polymerases with amino acid substitutions in the polymerase active site that enhance ribonucleotide incorporation. The presence of the 2′-hydroxyl group makes RNA exquisitely sensitive to alkaline degradation compared with DNA, and this chemical property has been utilized to cleave genomic DNA specifically at ribonucleotide positions. Moreover, repair of single genomic ribonucleotides is initiated by ribonuclease H2 (RNase H2) (32–34). Therefore, ribonucleotide mapping experiments have been carried out in an RNH201-defective mutant lacking RNase H2 to prevent the excision of ribonucleotides.
The Pol ε (M644G) variant shows an 11-fold increase in the incorporation of ribonucleotides into DNA in vitro (35). The sharp decrease in the size of fragments resulting from alkaline hydrolysis of chromosomal DNA isolated from an RNase H2–defective strain containing the Pol ε (M644G) variant compared with wild-type Pol ε has been taken as evidence that M644G Pol ε also more readily incorporates ribonucleotides during DNA replication in vivo. Importantly, these small fragments mapped predominantly to the leading strand from the well-behaved early replication origin autonomously replicating sequence 301 (ARS301), suggesting that the variant Pol ε, and by implication also the wild-type Pol ε, is primarily responsible for leading strand DNA replication (35). A similar pattern of strand-specific ribonucleotide incorporation was observed at a well-defined replication origin using a similar variant of Schizosaccharomyces pombe Pol ε (36). In comparison, alkaline hydrolysis of genomic DNA from RNase H2–defective yeast containing ribonucleotide-promiscuous variants of Pol α and Pol δ showed preferential incorporation of ribonucleotides into the nascent lagging strand near ARS301 (37).
More recently, several studies have measured ribonucleotide incorporation by variant yeast nuclear replicases across the whole genome. Three independent studies used different approaches for mapping ribonucleotides in Saccharomyces cerevisiae (Figure 2b). In one study, isolated ribonucleotide-containing genomic DNA was cleaved by RNase H2, and the product DNA with a free 5′-hydroxyl group was targeted for strand-specific sequencing (38). In two other studies, the genomic DNA was treated with alkali and either the product DNA with a free 5′-hydroxyl group (39) or the product DNA with a 2′-3′-cyclic phosphate-terminated ribonucleotide (40) and was targeted for further isolation and for strand-specific sequencing. These three S. cerevisiae studies show remarkable agreement. Ribonucleotides incorporated during replication in untreated yeast cells by the M644G variant of Pol ε are primarily present in the nascent leading strand, whereas ribonucleotides incorporated by ribonucleotide-promiscuous variants of Pol α and Pol δ are primarily present in the nascent lagging strand. Similar results have been reported in an analogous study in S. pombe (41). Collectively, these results strongly suggest that in undamaged yeast cells, the leading strand is primarily replicated by Pol ε, whereas the lagging strand is primarily replicated by Pol α and Pol δ. These data of course do not exclude that Pol α (and possibly Pol δ) initiates replication of both strands at origins and that Pol δ can replicate the leading strand under special circumstances, e.g., upon replication restart after blockage by natural but difficult-to-replicate sequences (e.g., see 42), or following bypass of lesions resulting from endogenous or exogenous environmental stress.
Ribonucleotides that are incorporated into DNA can have both beneficial and detrimental consequences. On the beneficial side, two studies suggest that ribonucleotides incorporated into DNA by Pol ε, but not those incorporated by Pol α or Pol δ, can act as strand discrimination signals for repairing mismatches (37, 43). Other evidence indicates that two ribonucleotides in DNA may act as an imprint for mating type switching in fission yeast (44). On the detrimental side, a subset of ribonucleotides incorporated into DNA can be mutagenic. For example, DNA topoisomerase 1 can cleave the DNA backbone at the site of a ribonucleotide, which can result in a nick bounded by a ribonucleotide 2′-3′-cyclic phosphate. If this occurs in a repetitive DNA element, short deletions can occur. This pathway was discovered in a RNase H2–defective yeast that accumulates ribonucleotides (35, 45). Interestingly, these effects are observed for ribonucleotides incorporated into DNA by a variant of Pol ε, but not for variants of Pol α and Pol δ, revealing asymmetric consequences of the three replicases on genome stability (46). Ribonucleotides in DNA also lead to large forms of chromosomal rearrangements (47–49), including gross chromosomal rearrangements, loss of heterozygosity, and nonallelic homologous recombination. Readers interested in further details are encouraged to read recent reviews on the causes and consequences of ribonucleotides incorporated into DNA by replicases (50–53).
3.3. Genetic Techniques: Strand-Specific Replication Errors
Pol δ and Pol ε were originally suggested to operate on opposite DNA strands in eukaryotic cells (54). However, budding and fission yeast strains lacking Pol ε catalytic and exonuclease activities ( pol2-16) were subsequently demonstrated to be viable, although multiple deleterious phenotypes are associated with such domain deletions (55–58). These studies clearly show that in the absence of catalysis by Pol ε, other DNA polymerases can synthesize both the leading and the lagging DNA strands. This type of replication is consistent with a recent study suggesting that Pol δ is the major replicase for both the leading and lagging strands (24; reviewed in 6). In this model, the polymerase activity of Pol ε is suggested to not be important for the bulk of DNA replication, but its 3′-exonuclease activity is important for editing some of the errors made by Pol δ during leading strand replication. Interestingly, although Pol ε polymerase domain deletion mutants are viable, polymerase catalytic site mutants of Pol ε are not, suggesting that when Pol ε is properly engaged at the leading strand, it is required to fulfill its polymerization function (56, 59).
The variant DNA polymerases that turned out to be so useful for ribonucleotide incorporation mapping were originally designed for asymmetric mutation mapping. For example, during DNA synthesis in vitro, the L612M variant of yeast Pol δ generates a template dG-dTTP mismatch at a 28-fold higher rate than it generates a dC-dATP mismatch, the other misincorporation event that could explain the origin of a G-C to A-T mutation arising during replication of dsDNA (60). Accordingly, the base substitution specificity of a pol3-L612M strain deficient in mismatch repair was measured using the URA3 gene placed in each of the two orientations adjacent to origin ARS306 on S. cerevisiae chromosome III. The observed pattern of G-C to A-T mutations, and other types of point mutations and 1-nt deletions that exhibited asymmetry, was consistent with the primary participation of Pol δ in lagging strand replication (61). Similar experiments were also performed with the homologous L868M Pol α variant (62) and with the M644G Pol ε variant (63). Mutation rates for various types of replication errors were consistent with the primary participation of Pol α in nascent lagging strand replication, whereas a different pattern of mutations was consistent with the participation of Pol ε in leading strand replication. In addition, and consistent with the evolutionary conservation of Pol δ and Pol ε in all eukaryotes, the base substitution specificity and ribonucleotide incorporation patterns observed in S. pombe strains with the analogous Pol δ and Pol ε variants led to the same conclusion (36).
Because the studies just mentioned monitored mutations in reporter genes near one strong, early firing origin, which surveyed only ~0.01% of the yeast genome, more recent studies of strand-specific mutagenesis have also been performed across the whole yeast genome (37, 64). As illustrated by the strand specificity of mutagenesis in those studies, the results again suggest that Pol α and Pol δ primarily perform lagging strand replication, whereas Pol ε primarily performs leading strand replication. This model is consistent with the mutational specificity observed in Pol ε exonuclease-defective human tumors, which have strand-specific mutational patterns near origins that are similar to those in cell extracts, but only if Pol ε is assumed to primarily synthesize the leading strand (65).
The asymmetric mutator approach to polymerase mapping has two drawbacks that make this method less reliable than the ribonucleotide incorporation approach discussed above. First, the density of mutations generated along the genome is 100- to 1,000-fold lower than the density of ribonucleotides inserted by the variant DNA polymerases, lending superior statistical strength to the latter method. Second, to obtain interpretable spectra, the analysis needs to be carried out with strong polymerase mutators and preferably in a mismatch repair–defective background, so that polymerase misinsertions remain detectable (61). These highly mutable backgrounds are deleterious for growth and could potentially alter the results, through further mutations and/or by altering the fork itself. In a comment on the Johnson et al. (24) study, which concluded that Pol δ carries out replication of both the leading and the lagging strands in S. cerevisiae, we have argued that this may have occurred in their strains but not in ours (61). The reader is invited to peruse the Johnson et al. article (24), our commentary on the article (66), the response on our commentary by Johnson et al. (67), and a recent review by Stillman (6). In the sections below, we continue our discussion on the model that leading strand replication is predominantly carried out by Pol ε, with the reservation that alternatives are possible under some conditions.
4. LEADING STRAND REPLICATION
4.1. The CMG DNA Helicase
In the last few years, biochemical studies of DNA replication have shown great progress. Indeed, in 2015, the complete reconstitution of DNA replication initiation and elongation has been reported with purified proteins from yeast (5). In this section, we focus on the function of the leading strand replicase consisting of the CMG helicase and Pol ε. Initiation of DNA replication is associated with the rearrangement of the Mcm2-7 core helicase from an inactive form encircling dsDNA to that of an active helicase, which encircles ssDNA and unwinds parental dsDNA using the energy of adenosine triphosphate (ATP) hydrolysis (Figure 1). Several lines of evidence support the localization of the MCM core around the leading strand. The CMG complex is associated with the leading strand Pol ε, making its association with the leading strand plausible (13, 14). CMG is a 3′-5′ helicase on model DNA substrates (20), as is the homologous archaeal MCM helicase (68, 69). This directionality would place the complex on the leading strand when moving in the direction of the fork. Furthermore, lagging strand roadblocks, but not leading strand roadblocks, are bypassed by the CMG helicase, suggesting a tight association with the leading strand (70).
Although X-ray and cryo–electron microscopy (cryo-EM) structures have been available for each of the three subassemblies of the CMG for some time (see references in 71, 72), two exciting new studies have yielded high-resolution cryo-EM structures of the CMG complex. Both studies reveal a similar overall arrangement of the three subassemblies—Mcm2-7, Cdc45, and GINS—providing new information on the organization of the leading strand replicase. The structure of yeast CMG was obtained without DNA at 3.7–4.8 Å resolution (73). These cryo-EM data are consistent with the presence of two different conformers, one compact form and one extended form. The conversion from the extended to the compact form upon ATP binding is proposed to be associated with movement of the six AAA+ domains of the Mcm2-7 core, although hydrolysis of ATP reverses this motion, allowing the CMG helicase to move along ssDNA in an inchworm motion. A second cryo-EM structure, of the Drosophila CMG complex, was obtained in the presence of a partially dsDNA molecule mimicking a stable replication fork (74). Again, two main conformers were obtained, a compact form at 7.4 Å resolution and a relaxed form at 9.8 Å resolution. In the compact form, the ssDNA portion of the DNA can be visualized to thread through the Mcm2-7 ring, supporting the conclusions from previous studies that CMG encircles the leading strand. The second, relaxed structure lacks DNA density in the core and shows a gap between the Mcm2 and Mcm5 subunits. Interestingly, both the initial loading of the MCM complex around dsDNA and the subsequent conversion to that of an active helicase surrounding ssDNA are proposed to be mediated through an opening between the Mcm2 and Mcm5 subunits (75, 76). The compact form of CMG predominates in the presence of nonhydrolyzable adenosine 5′-γ-thio-triphosphate (ATPγS), whereas the relaxed form predominates in the presence of ATP. The latter form likely represents a structure in which the ATP has been hydrolyzed, supporting a model in which ATP hydrolysis would convert the compact form into a relaxed form. Interestingly, this conformational change is associated with only minimal stretching movement of CMG, which was observed with yeast CMG and formed the basis for an inchworm model for helicase action (73). These new structures of the CMG complex are just the beginning of a new direction for the biophysics of DNA replication studies aided by cryo-EM studies.
4.2. DNA Polymerase ε and Leading Strand DNA Replication
The three replicative DNA polymerases, α, δ, and ε, are members of the B family of DNA poly-merases. A thorough review of these enzymes falls outside the scope of this review, and the reader is referred to recent comprehensive reviews discussing both the structure and function of Pol α, Pol δ, and Pol ε and their evolutionary relationships (77, 78). Another recent review describes the consequence of replicative polymerase mutations on cancer disposition in humans (79). In this section, we briefly consider the properties that predispose these polymerases to replicate the two strands of the nuclear genome (Figure 3).
Figure 3.

Eukaryotic DNA replicases. DNA polymerase α (Pol α, red ) and Pol ε ( green) contain four subunits, and Saccharomyces cerevisiae Pol δ (blue) contains three subunits, whereas human Pol δand Schizosaccharomyces pombe Pol δhave an additional small fourth subunit (not shown). Demonstrated [4Fe–4S] iron–sulfur clusters are indicated with large orange balls, and bound zinc atoms with small gray balls. Catalytic properties and protein–protein interactions are listed. Note that Pol δhas a high fidelity for base–base mismatches but lower fidelity for single-nucleotide deletions in repetitive sequences. Abbreviations: GINS, Sld5, Psf1, Psf2, and Psf3 complex; n.d., not determined; PCNA, proliferating cell nuclear antigen.
The properties of Pol ε are distinguished from those of Pol α and Pol δ in several ways. Pol ε has a high-molecular-weight subunit whose N-terminal domain encodes DNA polymerization and 3′-exonucleolytic activity and a homologous but catalytically inactive C-terminal domain (CTD) that is required for replisome assembly and checkpoint activation (55–59, 80, 81). The holoenzyme form of Pol ε also contains a noncatalytic Dpb2 subunit that is essential and Dpb3 and Dpb4 subunits that are nonessential (82). The presence of a small domain in the catalytic subunit allows Pol ε to encircle the nascent dsDNA (83), which is likely responsible for the high intrinsic processivity of this enzyme. This high processivity is increased even further by the nonessential Dpb3 and Dpb4 subunits and through interactions with the replication clamp PCNA. In contrast, the very low intrinsic processivity of DNA synthesis by Pol δ is vastly enhanced by PCNA, such that in the presence of PCNA both Pol ε and Pol δ have comparable processivities (84).
The capacity for strand displacement synthesis by the lagging strand DNA polymerase is essential for the efficient maturation of Okazaki fragments on the lagging strand. Compared with Pol δ, Pol ε does not perform efficient strand displacement synthesis (85). To a large degree, this is a consequence of its very active 3′-exonuclease activity, as strand displacement synthesis is observable in the exonuclease-defective enzyme (86). The intrinsic 3′-exonuclease activity of Pol ε proofreads its own replication errors (87), and it does this so efficiently that Pol ε synthesizes DNA more accurately than proofreading-proficient Pol δ and much more accurately than the proofreading-deficient Pol α. Fortunately, this lower fidelity on the lagging strand is counterbalanced by more active mismatch repair on that strand (64). Thus, the lack of strand displacement capacity makes Pol ε less suitable to serve as the lagging strand replicase. In contrast, its interactions with several components of the CMG complex cause its unique targeting to the leading strand of the replication fork (Figure 3). Previous biochemical and genetic studies established an essential interaction between the Dpb2 subunit of Pol ε and the Psf1 subunit of GINS (14). A low-resolution cryo-EM study of the CMG helicase in a complex with Pol ε is consistent with these interactions, and this structure revealed additional interactions between Mcm5 and the C-terminal half of Pol2 (88). The functional significance of these interactions with Pol ε is supported by recent biochemical studies by O’Donnell and coworkers (89, 90). The CMG helicase, preloaded on the leading strand of a model replication fork, recruited Pol ε to the leading strand in preference to Pol δ and in a manner that was dependent on Pol ε’s Dpb2 subunit. Furthermore, even when Pol δ was prebound to the leading strand, Pol ε readily displaced it if CMG complexes were present.
The cryo-EM structure of the CMG–Pol ε complex not only revealed its overall architecture and protein interactions but also suggested a path for threading the leading ssDNA through the CMG complex and Pol ε (88) (Figure 4). In previous biochemical studies in the Xenopus egg extract–based DNA replication system, the replication fork was allowed to run into a precisely positioned interstrand cross-link (91). Transient stalling of leading strand replication was observed ~40 nt prior to the cross-link, followed by a more pronounced stall at a position ~20 nt before the cross-link. The ~20-nt stall is consistent with the length of DNA occluded by the Mcm2-7 hexamer (74), whereas the ~40-nt stall is proposed to represent additional threading of the leading strand through Cdc45 and GINS prior to entering the Pol ε active site (88). These biochemical and structural considerations would suggest that the leading strand preferentially threads through the entire CMG complex before binding Pol ε, but it may have the flexibility to release readily from Cdc45–GINS, perhaps to allow for a more flexible response to challenges in leading strand DNA replication (Figure 4).
Figure 4.

Replisome structure and interactions. Two models for the pathway taken by the leading strand prior to entry into the DNA polymerase ε (Pol ε) catalytic site. Either (a) ~40-nt or (b) ~20-nt lengths of single-stranded DNA are occluded. The proposal has been made that these two forms can also interconvert. The lagging strand is shown looped such that both Pol α and Pol ε move in the same direction while held in a complex by Ctf4. Abbreviations: GINS, Sld5, Psf1, Psf2, and Psf3 complex; Mcm2-7, helicase complex; PCNA, proliferating cell nuclear antigen; RPA, replication protein A.
5. LAGGING STRAND REPLICATION
5.1. Priming of Okazaki Fragments
Both leading and lagging strand DNA syntheses are initiated by the Pol α –RNA primase complex. This highly conserved heterotetrameric complex contains two catalytic activities, the RNA primase activity in the smallest p48 subunit (Pri1) and the polymerase activity in the largest p180 subunit (Pol1), and two regulatory subunits (92) (Figure 3). The catalytic subunit comprises a conserved polymerase core and a separate CTD connected to the core by a flexible linker (93, 94). The CTD is unique to the eukaryotic members of the B-family DNA polymerases, Pol α, Pol δ, Pol ε, and the mutagenic DNA polymerase ζ (Pol ζ), and it forms the structural basis for their multisubunit nature. Currently, structural information is available only for the CTD of Pol α (93, 94). The CTD has a bilobal shape that is stabilized by the binding of two metal ions to each of a set of four cysteine residues. The structure of the CTD of Pol α displays a zinc atom bound in both positions. However, biochemical studies of the CTDs of Pol δ and Pol ζ show that one of the 4-cysteine motifs contains an iron–sulfur cluster of the [4Fe–4S] type (95, 96), and the suggestion has been made that Pol α and Pol ε may also contain iron–sulfur clusters in their CTDs. The presence of iron–sulfur clusters as part of eukaryotic DNA polymerases remains enigmatic. Additional iron–sulfur clusters have been found in the primase accessory subunit and in the catalytic domain of Pol ε (97–99). Whether they are merely structural building blocks or are subject to oxidation and reduction, perhaps as part of a signaling pathway in response to the changing redox environments in the cell, remains to be established (100).
Attached to the catalytic polymerase domain by a flexible linker, the structure of the CTD of Pol α makes the majority of interactions with the other subunits (94, 101). The primase-accessory subunit also contains two domains connected by a flexible linker (97, 102, 103). These flexible linkers allow the complex to go through several large-scale motions to synthesize the RNA primers that start the millions of Okazaki fragments required for replication of eukaryotic chromosomes (103, 104). Priming is initiated at the interface of the primase and the primase-accessory subunit. Primer elongation by the primase subunit is aided by binding of the 5′-end of the nascent primer to the primase-accessory subunit. Growing steric clashes as the RNA primer increases in length limit the primer length to ~10 nt. The primase-to-polymerase switch is proposed to be mediated by a large rotation of the C-terminus of the primase accessory subunit with bound RNA to deliver this primer to the Pol α active site for DNA synthesis (103).
On the lagging strand, Pol α –mediated DNA synthesis is terminated after ~20–30 nt of DNA synthesis to allow initiation of Pol δ –dependent replication. These estimates are based on classical studies of SV40 viral DNA replication (105, 106), which also uses the Pol α –RNA primase complex for primer synthesis. However, in contrast to chromosomal DNA replication, SV40 uses solely Pol δ for primer elongation on both the leading and the lagging strands of the replication fork (107, 108). How the length of the DNA portion of the primer synthesized by Pol α is regulated is still uncertain, and several mechanisms have been proposed. In one model, Pol α –mediated DNA synthesis is abrogated by the loading of PCNA by replication factor C (RFC) (109, 110). A second model is based on the observation that RNA–DNA duplexes often assume an A helix conformation, to which the Pol α polymerase domain binds with very high affinity. Elongation of the primer and subsequent assumption of the B DNA form would reduce the affinity of binding by Pol α and lead to its dissociation (111). Unfortunately, these studies were carried out on poly(dT) templates that are prone to triple-strand helix formation when partially replicated, which causes polymerase dissociation (112, 113). Therefore, although a combination of factors likely contribute to abrogation of DNA synthesis by the Pol α polymerase subunit, the relative contributions of these factors still await determination.
5.2. Elongation and Maturation of Okazaki Fragments
Pol α –synthesized primers are extended by Pol δ. The replication clamp PCNA enhances not only the processivity of Pol δ but also its actual rate of catalysis, such that at saturating dNTP concentrations, Pol δ replicates at a rate of ~250 nt/sec (114). Pol ε displays a similarly high rate of DNA synthesis (115). However, dNTP levels in the cell are far below the Km values for these enzymes (4, 116). When measured at physiological dNTP levels, and in the presence of competing rNTPs, DNA synthesis proceeds at ~50 nt/sec, which is commensurate with rates of fork movement in the cell (117).
When Pol δ reaches the 5′-end of the preceding Okazaki fragment, it initiates strand displacement synthesis. Several biochemical mechanisms are in place to ensure that the 5′-flaps generated by strand displacement synthesis are kept to a minimal size, i.e., generally not more than a single nucleotide (Figure 5). First, the rates of DNA synthesis decrease sharply with each consecutive nucleotide being displaced by Pol δ. In part, this progressive molecular brake applied by the growing 5′-flap is due to a sharp decrease in the macroscopic rate of polymerization. But, in addition, kinetic modeling has shown that the polymerase-DNA complex equilibrates between an elongation-competent form and an elongation-incompetent form, and longer flaps show an increased partitioning to the elongation-incompetent form (114). In part, the elongation-incompetent form signifies a switch to the 3′-exonuclease domain of Pol δ, which degrades the primer terminus back to that of the nick position. This continuous elongation and degradation by Pol δ are in essence a futile cycle, termed idling, and constitute a second mechanism to restrain the formation of long flaps (reviewed in 118). However, the elongation-incompetent form can also be a structure in which the primer terminus has been released by Pol δ to provide access to flap endonuclease 1 (FEN1) for 5′-flap cutting. This initiates a degradation process termed nick translation (Figure 5).
Figure 5.

Okazaki fragment maturation. Primers on the lagging strand are elongated by Pol δ (DNA polymerase δ) until the downstream Okazaki fragment is reached. Subsequent strand displacement synthesis by Pol δ is counteracted by its 3′-exonuclease activity (idling). In the presence of FEN1, the nascent flap is cut and strand displacement synthesis restarts. This iterative process (nick translation) predominantly releases mononucleotides. Occasional excess strand displacement synthesis yields very long 5′-flaps that are processed to short flaps by the nuclease activity of Dna2. After degradation of all primer RNA, ligation of the DNA–DNA nick is performed by DNA ligase 1. Abbreviations: FEN1, 5′-flap endonuclease 1; PCNA, proliferating cell nuclear antigen; RPA, replication protein A.
Unlike the image often depicted in textbooks, the mechanism of FEN1 catalysis actually does not involve simple cutting at the base of the 5′-flap (reviewed in 119, 120). Rather, the nascent 5′-flap generated by strand displacement synthesis re-equilibrates to form a single-nucleotide 3′-flap. This 3′-flap binds FEN1 with high specificity, directing precise cutting by the enzyme one nucleotide into the dsDNA, which has been unraveled inside the active site. Single-nucleotide 5′-flaps form inefficient substrates for FEN1, because they lack a distinct 5′-flap after re-equilibration (119). Yet, the single-nucleotide 5′-flap is the predominant substrate in the course of primer RNA degradation during nick translation (114). Increased strand displacement synthesis to form longer 5′-flaps does occur, particularly if the DNA has decreased duplex stability, e.g., in AT-rich regions. The catalytic activity of FEN1 increases on these longer flaps, and this more avid activity of FEN1 on longer flaps can be thought of as a third mechanism to keep 5′-flaps short.
Occasionally, strand displacement can become decoupled from FEN1 activity, and the long flaps that are generated are resistant to FEN1, because of either secondary structure formation or coating of the 5′-flap by RPA. In yeast, the processing of long flaps is carried out by the 5′-endonuclease activity of Dna2, an essential multifunctional nuclease/helicase (reviewed in 121, 122). Cutting of 5′-flaps by Dna2 occurs with lower precision than cutting by FEN1, with the remaining flap size varying from 0 to 5 nt in several studies (discussed in 121, 123). Some ends produced by Dna2 are ligatable by DNA ligase 1. In addition, when Dna2 activity is coupled to strand displacement synthesis by Pol δ, ligation is more efficient because the 3′-exonuclease of Pol δ can trim the imprecise ends left by Dna2 into a ligatable nick (123, 124). However, the percentage of imprecise ends remaining would be too high for successful completion of the many Okazaki fragments produced even in a yeast cell with its small, compact chromosomes. Therefore, long flaps trimmed by Dna2 are generally thought to proceed through a pathway that requires further cutting by FEN1 (Figure 5). Given the importance of FEN1 in Okazaki fragment maturation, it is somewhat surprising that yeast FEN1 deletions are viable. However, it is very likely that other, related nucleases, e.g., Exo1, can substitute for FEN1 albeit with reduced efficiency and fidelity (reviewed in 118, 125).
Each of the three enzymes that make up the core Okazaki fragment maturation machinery—Pol δ, FEN1, and ligase—has one or more PCNA-interaction motifs. Given that PCNA is a homotrimer, it is possible that each of these enzymes could occupy one monomer of PCNA and thereby carry out processive Okazaki fragment maturation without enzyme dissociation. This has been termed the toolbelt model (126). In archaea, strong evidence exists for this toolbelt model in Okazaki fragment maturation (127). In yeast, a toolbelt mechanism involving just Pol δ and FEN1 has been demonstrated (114). Although processive maturation is likely more efficient, it is not essential. This follows from a biochemical study of PCNA heterotrimers in which only one monomer has the capacity to bind either Pol δ or FEN1, thereby enforcing a distributive mechanism (128), and from the very mild phenotype displayed by a PCNA interaction–defective mutant of FEN1, compared with that of the FEN1 deletion (129).
After degradation of primer RNA, nick translation is terminated by the action of DNA ligase. DNA ligase also has a PCNA-binding motif that can stabilize ligase onto DNA substrate (130). However, in one biochemical study of Okazaki fragment maturation, DNA ligase acted distributively in this process (131) and the position after the RNA–DNA junction where ligation occurred was determined largely by the concentration of DNA ligase, rather than by the ability to make a complex with PCNA on the DNA. How closely behind the RNA–DNA junction this ligation step occurs is of some importance, because it determines to what extent the primer DNA synthesized by the lower-fidelity Pol α survives. Nick translation through that region would serve to substitute lower-fidelity DNA with higher-fidelity DNA synthesized by Pol δ (132). Despite this possible fidelity mechanism, remnants of Pol α –synthesized DNA with lower fidelity remain (38, 62, 133). That the nick translation machinery has the capacity to carry out extensive nick translation inside the cell follows from an experiment in which DNA ligase was shut off in yeast cells (134). Okazaki fragments grew beyond their customary length through continuous nick translation until termination after collision with downstream chromatin.
6. REPLISOME COORDINATION
Given the differences in the machineries and mechanisms for carrying out leading and lagging strand DNA replication, it is obvious that additional factors and/or mechanisms must exist to enforce coordinated replication of both strands. On the basis of their studies of the bacteriophage T4 DNA replication system, Alberts and coworkers (135) proposed a novel mechanism, termed the trombone model, in which the two polymerases on both strands could coordinately replicate DNA by bending the lagging strand back upon itself (Figure 4). This model has been supported both by electron microscopy (136) and by measuring trombone loop dynamics in single-molecule studies (137). In addition, these two polymerization machineries require a physical linkage between the two sides. In T4, this linkage is mediated by the polymerase itself (138), whereas in Escherichia coli this linkage is mediated by two τ subunits of the clamp loader that bind the DNA polymerase III replicases at either strand (139). Recent studies of the yeast Ctf4 protein suggest that it may be the sought-after replisome coordinator in eukaryotes (140, 141). Ctf4 forms a homotrimer and exhibits protein–protein interactions with both Pol α on the lagging strand and GINS and Pol ε on the leading strand, thereby linking the two machineries (Figure 4). The human Ctf4 homolog AND-1 shows additional interactions with Pol δ (142). Other replication proteins, such as Dna2 and the sister chromatin cohesion protein Chl1, also bind Ctf4, marking this factor as an important interaction hub within the replisome (141, 143). Surprisingly, a deletion of CTF4, or of the S. pombe homolog mcl1+, is viable. However, the deletion shows various defects in genome stability (144–146). Possibly, other factor(s) contribute to coordinating the leading and lagging strands of the replication fork. Among these could be the Tof1–Csm3–Mrc1 replication pausing complex that also is physically associated with multiple factors in the replisome (147).
7. FUTURE PERSPECTIVES
Over the years, many investigators have performed structural, biophysical, biochemical, and genetic studies to inform us about the properties of partial replication machines. These efforts can now be expanded to the more complete systems that are being developed to study eukaryotic DNA replication. In the last few years, major new insights into eukaryotic replication fork structure, fidelity, and dynamics have been gained through striking advances in the development of several key technologies. Improvements in cryo-EM have made it possible to study ever-larger complexes, such as those of the CMG helicase complex, and with resolution approaching that of X-ray crystallography. Single-molecule approaches have been developed that allow fork movement in simple replication systems to be visualized and are now making it possible to study the kinetics and dynamic properties of more complex eukaryotic replicases and the eukaryotic replisome. The advancement of genome-mapping technologies based on next-generation sequencing has also made it possible to obtain exquisite coverage of DNA alterations that inform us about the behavior of the replication fork inside the cell. These approaches offer a bright future for understanding how normal eukaryotic DNA replication occurs as well as how perturbations in normal replication influence evolution and disease.
Acknowledgments
The authors thank Joseph Stodola and Scott Lujan for advice with the writing of this review. The research in the Burgers laboratory is supported by grants from the National Institutes of Health (NIH) (GM032431, GM083970, and GM118129 to P.M.J.B.). Research in the Kunkel laboratory is supported by project numbers ES065070 and ES065089 from the Division of Intramural Research of the National Institute of Environmental Health Sciences at the NIH.
Footnotes
In this review, we largely cite recent publications. Readers who are further interested in any topic mentioned here are encouraged to read other outstanding articles cited in the reviews that we mention.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Watson JD, Crick FH. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–67. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 2.Okazaki R, Okazaki T, Sakabe K, Sugimoto K, Kainuma R, et al. In vivo mechanism of DNA chain growth. Cold Spring Harb Symp Quant Biol. 1968;33:129–43. [Google Scholar]
- 3.Tye BK, Nyman PO, Lehman IR, Hochhauser S, Weiss B. Transient accumulation of Okazaki fragments as a result of uracil incorporation into nascent DNA. PNAS. 1977;74:154–57. doi: 10.1073/pnas.74.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, et al. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. PNAS. 2010;107:4949–54. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015;519:431–35. doi: 10.1038/nature14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stillman B. Reconsidering DNA polymerases at the replication fork in eukaryotes. Mol Cell. 2015;59:139–41. doi: 10.1016/j.molcel.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- 8.Costa A, Hood IV, Berger JM. Mechanisms for initiating cellular DNA replication. Annu Rev Biochem. 2013;82:25–54. doi: 10.1146/annurev-biochem-052610-094414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell SP, Labib K. Chromosome duplication in Saccharomyces cerevisiae. Genetics. 2016;203:1027–67. doi: 10.1534/genetics.115.186452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deegan TD, Diffley JF. MCM: one ring to rule them all. Curr Opin Struct Biol. 2016;37:145–51. doi: 10.1016/j.sbi.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Ticau S, Friedman LJ, Ivica NA, Gelles J, Bell SP. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell. 2015;161:513–25. doi: 10.1016/j.cell.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takayama Y, Kamimura Y, Okawa M, Muramatsu S, Sugino A, Araki H. GINS, a novel multi-protein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 2003;17:1153–65. doi: 10.1101/gad.1065903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muramatsu S, Hirai K, Tak YS, Kamimura Y, Araki H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes Dev. 2010;24:602–12. doi: 10.1101/gad.1883410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengupta S, van Deursen F, de Piccoli G, Labib K. Dpb2 integrates the leading-strand DNA polymerase into the eukaryotic replisome. Curr Biol. 2013;23:543–52. doi: 10.1016/j.cub.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell. 2011;146:80–91. doi: 10.1016/j.cell.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanke M, Kodama Y, Takahashi TS, Nakagawa T, Masukata H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 2012;31:2182–94. doi: 10.1038/emboj.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Deursen F, Sengupta S, De Piccoli G, Sanchez-Diaz A, Labib K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 2012;31:2195–206. doi: 10.1038/emboj.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quan Y, Xia Y, Liu L, Cui J, Li Z, et al. Cell-cycle-regulated interaction between Mcm10 and double hexameric Mcm2-7 is required for helicase splitting and activation during S phase. Cell Rep. 2015;13:2576–86. doi: 10.1016/j.celrep.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Arnaiz P, Bruck I, Kaplan DL. Mcm10 coordinates the timely assembly and activation of the replication fork helicase. Nucleic Acids Res. 2016;44:315–29. doi: 10.1093/nar/gkv1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. PNAS. 2006;103:10236–41. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol Cell. 2006;21:581–87. doi: 10.1016/j.molcel.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 22.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–27. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu C, Gan H, Han J, Zhou ZX, Jia S, et al. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell. 2014;56:551–63. doi: 10.1016/j.molcel.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson RE, Klassen R, Prakash L, Prakash S. A major role of DNA polymerase δ in replication of both the leading and lagging DNA strands. Mol Cell. 2015;59:163–75. doi: 10.1016/j.molcel.2015.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joyce CM. Choosing the right sugar: how polymerases select a nucleotide substrate. PNAS. 1997;94:1619–22. doi: 10.1073/pnas.94.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roettger MP, Fiala KA, Sompalli S, Dong Y, Suo Z. Pre-steady-state kinetic studies of the fidelity of human DNA polymerase μ. Biochemistry. 2004;43:13827–38. doi: 10.1021/bi048782m. [DOI] [PubMed] [Google Scholar]
- 27.Brown JA, Suo Z. Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry. 2011;50:1135–42. doi: 10.1021/bi101915z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 29.Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 30.Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair. 2012;12:121–27. doi: 10.1016/j.dnarep.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goksenin AY, Zahurancik W, LeCompte KG, Taggart DJ, Suo Z, Pursell ZF. Human DNA polymerase εis able to efficiently extend from multiple consecutive ribonucleotides. J Biol Chem. 2012;287:42675–84. doi: 10.1074/jbc.M112.422733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eder PS, Walder JA. Ribonuclease H from K562 human erythroleukemia cells. Purification, characterization, and substrate specificity. J Biol Chem. 1991;266:6472–79. [PubMed] [Google Scholar]
- 33.Rydberg B, Game J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. PNAS. 2002;99:16654–59. doi: 10.1073/pnas.262591699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, et al. RNase H2-initiated ribonucleotide excision repair. Mol Cell. 2012;47:980–86. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, et al. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol. 2010;6:774–81. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases ε and δ at the eukaryotic replication fork are evolutionarily conserved. PLOS Genet. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol Cell. 2014;50:437–43. doi: 10.1016/j.molcel.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reijns MAM, Kemp H, Ding J, de Procé SM, Jackson AP, Taylor MS. Lagging-strand replication shapes the mutational landscape of the genome. Nature. 2015;518:502–6. doi: 10.1038/nature14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, et al. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat Struct Mol Biol. 2015;22:185–91. doi: 10.1038/nsmb.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koh KD, Balachander S, Hesselberth JR, Storici F. Ribose-seq: global mapping of ribonucleotides embedded in genomic DNA. Nat Methods. 2015;12:251–57. doi: 10.1038/nmeth.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daigaku Y, Keszthelyi A, Muller CA, Miyabe I, Brooks T, et al. A global profile of replicative polymerase usage. Nat Struct Mol Biol. 2015;22:192–98. doi: 10.1038/nsmb.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyabe I, Mizuno K, Keszthelyi A, Daigaku Y, Skouteri M, et al. Polymerase δ replicates both strands after homologous recombination-dependent fork restart. Nat Struct Mol Biol. 2015;22:932–38. doi: 10.1038/nsmb.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Boran M, Cejka P, et al. Ribonu-cleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol Cell. 2014;50:323–32. doi: 10.1016/j.molcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vengrova S, Dalgaard JZ. The wild-type Schizosaccharomyces pombe mat1 imprint consists of two ribonucleotides. EMBO Rep. 2006;7:59–65. doi: 10.1038/sj.embor.7400576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim N, Huang SN, Williams JS, Li YC, Clark AB, et al. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science. 2011;332:1561–64. doi: 10.1126/science.1205016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams JS, Clausen AR, Lujan SA, Marjavaara L, Clark AB, et al. Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading-strand specific. Nat Struct Mol Biol. 2015;22:291–97. doi: 10.1038/nsmb.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–22. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen-Soltero S, Martinez SL, Putnam CD, Kolodner RD. A Saccharomyces cerevisiae RNase H2 interaction network functions to suppress genome instability. Mol Cell Biol. 2014;34:1521–34. doi: 10.1128/MCB.00960-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conover HN, Lujan SA, Chapman MJ, Cornelio DA, Sharif R, et al. Stimulation of chromosomal rearrangements by ribonucleotides. Genetics. 2015;201:951–61. doi: 10.1534/genetics.115.181149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams JS, Kunkel TA. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair. 2014;19:27–37. doi: 10.1016/j.dnarep.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Potenski CJ, Klein HL. How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res. 2014;42:10226–34. doi: 10.1093/nar/gku773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerritelli SM, Crouch RJ. The balancing act of ribonucleotides in DNA. Trends Biochem Sci. 2016;41:434–45. doi: 10.1016/j.tibs.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams JS, Lujan SA, Kunkel TA. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat Rev Mol Cell Biol. 2016;17:350–63. doi: 10.1038/nrm.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison A, Araki H, Clark AB, Hamatake RK, Sugino A. A third essential DNA polymerase in S. cerevisiae. Cell. 1990;62:1143–51. doi: 10.1016/0092-8674(90)90391-q. [DOI] [PubMed] [Google Scholar]
- 55.Kesti T, Flick K, Keranen S, Syvaoja JE, Wittenberg C. DNA polymerase ε catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol Cell. 1999;3:679–85. doi: 10.1016/s1097-2765(00)80361-5. [DOI] [PubMed] [Google Scholar]
- 56.Dua R, Levy DL, Campbell JL. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol ε and its unexpected ability to support growth in the absence of the DNA polymerase domain. J Biol Chem. 1999;274:22283–88. doi: 10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- 57.Feng W, D’Urso G. Schizosaccharomyces pombe cells lacking the amino-terminal catalytic domains of DNA polymerase ε are viable but require the DNA damage checkpoint control. Mol Cell Biol. 2001;21:4495–504. doi: 10.1128/MCB.21.14.4495-4504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohya T, Kawasaki Y, Hiraga S, Kanbara S, Nakajo K, et al. The DNA polymerase domain of Pol ε is required for rapid, efficient, and highly accurate chromosomal DNA replication, telomere length maintenance, and normal cell senescence in Saccharomyces cerevisiae. J Biol Chem. 2002;277:28099–108. doi: 10.1074/jbc.M111573200. [DOI] [PubMed] [Google Scholar]
- 59.Pavlov YI, Shcherbakova PV, Kunkel TA. In vivo consequences of putative active site mutations in yeast DNA polymerases α, ε, δ , and ζ. Genetics. 2001;159:47–64. doi: 10.1093/genetics/159.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McElhinny SA, Stith CM, Burgers PM, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase δ. J Biol Chem. 2007;282:2324–32. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–44. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nick McElhinny SA, Kissling GE, Kunkel TA. Differential correction of lagging-strand replication errors made by DNA polymerases α and δ. PNAS. 2010;107:21070–75. doi: 10.1073/pnas.1013048107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase ε participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lujan SA, Williams JS, Pursell ZF, Abdulovic-Cui AA, Clark AB, et al. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLOS Genet. 2012;8:e1003016. doi: 10.1371/journal.pgen.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shinbrot E, Henninger EE, Weinhold N, Covington KR, Goksenin AY, et al. Exonuclease mutations in DNA polymerase ε reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24:1740–50. doi: 10.1101/gr.174789.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burgers PM, Gordenin D, Kunkel TA. Who is leading the replication fork, Pol ε or Pol δ ? Mol Cell. 2016;61:492–93. doi: 10.1016/j.molcel.2016.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson RE, Klassen R, Prakash L, Prakash S. Response to Burgers et al. Mol Cell. 2016;61:494–95. doi: 10.1016/j.molcel.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 68.Kelman Z, Lee JK, Hurwitz J. The single minichromosome maintenance protein of Methanobac-terium thermoautotrophicum ΔH contains DNA helicase activity. PNAS. 1999;96:14783–88. doi: 10.1073/pnas.96.26.14783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shechter DF, Ying CY, Gautier J. The intrinsic DNA helicase activity of Methanobacterium thermoautotrophicum ΔH minichromosome maintenance protein. J Biol Chem. 2000;275:15049–59. doi: 10.1074/jbc.M000398200. [DOI] [PubMed] [Google Scholar]
- 70.Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, et al. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–41. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li N, Zhai Y, Zhang Y, Li W, Yang M, et al. Structure of the eukaryotic MCM complex at 3.8 Å. Nature. 2015;524:186–91. doi: 10.1038/nature14685. [DOI] [PubMed] [Google Scholar]
- 72.Simon AC, Sannino V, Costanzo V, Pellegrini L. Structure of human Cdc45 and implications for CMG helicase function. Nat Commun. 2016;7:11638. doi: 10.1038/ncomms11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yuan Z, Bai L, Sun J, Georgescu R, Liu J, et al. Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat Struct Mol Biol. 2016;23:217–24. doi: 10.1038/nsmb.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abid Ali F, Renault L, Gannon J, Gahlon HL, Kotecha A, et al. Cryo-EM structures of the eukaryotic replicative helicase bound to a translocation substrate. Nat Commun. 2016;7:10708. doi: 10.1038/ncomms10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Samel SA, Fernandez-Cid A, Sun J, Riera A, Tognetti S, et al. A unique DNA entry gate serves for regulated loading of the eukaryotic replicative helicase MCM2-7 onto DNA. Genes Dev. 2014;28:1653–66. doi: 10.1101/gad.242404.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bruck I, Kaplan DL. The Dbf4-Cdc7 kinase promotes Mcm2-7 ring opening to allow for single-stranded DNA extrusion and helicase assembly. J Biol Chem. 2015;290:1210–21. doi: 10.1074/jbc.M114.608232. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Johansson E, Dixon N. Replicative DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5:1–14. doi: 10.1101/cshperspect.a012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Makarova KS, Krupovic M, Koonin EV. Evolution of replicative DNA polymerases in archaea and their contributions to the eukaryotic replication machinery. Front Microbiol. 2014;5:354. doi: 10.3389/fmicb.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16:71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 80.Navas TA, Zhou Z, Elledge SJ. DNA polymerase ε links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- 81.Lou H, Komata M, Katou Y, Guan Z, Reis CC, et al. Mrc1 and DNA polymerase ε function together in linking DNA replication and the S phase checkpoint. Mol Cell. 2008;32:106–17. doi: 10.1016/j.molcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hogg M, Johansson E. DNA polymerase ε. Subcell Biochem. 2012;62:237–57. doi: 10.1007/978-94-007-4572-8_13. [DOI] [PubMed] [Google Scholar]
- 83.Hogg M, Osterman P, Bylund GO, Ganai RA, Lundström EB, et al. Structural basis for processive DNA synthesis by yeast DNA polymerase ε. Nat Struct Mol Biol. 2014;21:49–55. doi: 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]
- 84.Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, et al. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res. 2007;35:6588–97. doi: 10.1093/nar/gkm741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM. Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004;18:2764–73. doi: 10.1101/gad.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ganai RA, Zhang XP, Heyer WD, Johansson E. Strand displacement synthesis by yeast DNA polymerase ε. Nucleic Acids Res. 2016;44:8229–40. doi: 10.1093/nar/gkw556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flood CL, Rodriguez GP, Bao G, Shockley AH, Kow YW, Crouse GF. Replicative DNA poly-merase δ but not ε proofreads errors in cis and in trans. PLOS Genet. 2015;11:e1005049. doi: 10.1371/journal.pgen.1005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sun J, Shi Y, Georgescu RE, Yuan Z, Chait BT, et al. The architecture of a eukaryotic replisome. Nat Struct Mol Biol. 2015;22:976–82. doi: 10.1038/nsmb.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, et al. CMG helicase and DNA polymerase ε form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. PNAS. 2014;111:15390–95. doi: 10.1073/pnas.1418334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, et al. Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat Struct Mol Biol. 2014;21:664–70. doi: 10.1038/nsmb.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–80. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pellegrini L. The Pol α-primase complex. Subcell Biochem. 2012;62:157–69. doi: 10.1007/978-94-007-4572-8_9. [DOI] [PubMed] [Google Scholar]
- 93.Klinge S, Nunez-Ramirez R, Llorca O, Pellegrini L. 3D architecture of DNA Pol α reveals the functional core of multi-subunit replicative polymerases. EMBO J. 2009;28:1978–87. doi: 10.1038/emboj.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suwa Y, Gu J, Baranovskiy AG, Babayeva ND, Pavlov YI, Tahirov TH. Crystal structure of the human Pol α B subunit in complex with the C-terminal domain of the catalytic subunit. J Biol Chem. 2015;290:14328–37. doi: 10.1074/jbc.M115.649954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Netz DJ, Stith CM, Stumpfig M, Kopf G, Vogel D, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2011;8:125–32. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Makarova AV, Stodola JL, Burgers PM. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012;40:11618–26. doi: 10.1093/nar/gks948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klinge S, Hirst J, Maman JD, Krude T, Pellegrini L. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nat Struct Mol Biol. 2007;14:875–77. doi: 10.1038/nsmb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weiner BE, Huang H, Dattilo BM, Nilges MJ, Fanning E, Chazin WJ. An iron-sulfur cluster in the C-terminal domain of the p58 subunit of human DNA primase. J Biol Chem. 2007;282:33444–51. doi: 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- 99.Jain R, Vanamee ES, Dzikovski BG, Buku A, Johnson RE, et al. An iron-sulfur cluster in the polymerase domain of yeast DNA polymerase ε. J Mol Biol. 2014;426:301–8. doi: 10.1016/j.jmb.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arnold AR, Grodick MA, Barton JK. DNA charge transport: from chemical principles to the cell. Cell Chem Biol. 2016;23:183–97. doi: 10.1016/j.chembiol.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kilkenny ML, De Piccoli G, Perera RL, Labib K, Pellegrini L. A conserved motif in the C-terminal tail of DNA polymerase α tethers primase to the eukaryotic replisome. J Biol Chem. 2012;287:23740–47. doi: 10.1074/jbc.M112.368951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kilkenny ML, Longo MA, Perera RL, Pellegrini L. Structures of human primase reveal design of nucleotide elongation site and mode of Pol α tethering. PNAS. 2013;110:15961–66. doi: 10.1073/pnas.1311185110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baranovskiy AG, Babayeva ND, Zhang Y, Gu J, Suwa Y, et al. Mechanism of concerted RNA-DNA primer synthesis by the human primosome. J Biol Chem. 2016;291:10006–20. doi: 10.1074/jbc.M116.717405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nunez-Ramirez R, Klinge S, Sauguet L, Melero R, Recuero-Checa MA, et al. Flexible tethering of primase and DNA Pol α in the eukaryotic primosome. Nucleic Acids Res. 2011;39:8187–99. doi: 10.1093/nar/gkr534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nethanel T, Reisfeld S, Dinter-Gottlieb G, Kaufmann G. An Okazaki piece of simian virus 40 may be synthesized by ligation of shorter precursor chains. J Virol. 1988;62:2867–73. doi: 10.1128/jvi.62.8.2867-2873.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bullock PA, Seo YS, Hurwitz J. Initiation of simian virus 40 DNA synthesis in vitro. Mol Cell Biol. 1991;11:2350–61. doi: 10.1128/mcb.11.5.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Waga S, Stillman B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature. 1994;369:207–12. doi: 10.1038/369207a0. [DOI] [PubMed] [Google Scholar]
- 108.Zlotkin T, Kaufmann G, Jiang Y, Lee MY, Uitto L, et al. DNA polymerase ε may be dispensable for SV40—but not cellular—DNA replication. EMBO J. 1996;15:2298–305. [PMC free article] [PubMed] [Google Scholar]
- 109.Tsurimoto T, Stillman B. Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase α and δ during initiation of leading and lagging strand synthesis. J Biol Chem. 1991;266:1961–68. [PubMed] [Google Scholar]
- 110.Yuzhakov A, Kelman Z, Hurwitz J, O’Donnell M. Multiple competition reactions for RPA order the assembly of the DNA polymerase δ holoenzyme. EMBO J. 1999;18:6189–99. doi: 10.1093/emboj/18.21.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perera RL, Torella R, Klinge S, Kilkenny ML, Maman JD, Pellegrini L. Mechanism for priming DNA synthesis by yeast DNA polymerase α. eLife. 2013;2:e00482. doi: 10.7554/eLife.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mikhailov VS, Bogenhagen DF. Termination within oligo(dT) tracts in template DNA by DNA polymerase γ occurs with formation of a DNA triplex structure and is relieved by mitochondrial single-stranded DNA-binding protein. J Biol Chem. 1996;271:30774–80. doi: 10.1074/jbc.271.48.30774. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Y, Baranovskiy AG, Tahirov ET, Tahirov TH, Pavlov YI. Divalent ions attenuate DNA synthesis by human DNA polymerase α by changing the structure of the template/primer or by perturbing the polymerase reaction. DNA Repair. 2016;43:24–33. doi: 10.1016/j.dnarep.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stodola JL, Burgers PM. Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat Struct Mol Biol. 2016;23:402–8. doi: 10.1038/nsmb.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ganai RA, Osterman P, Johansson E. Yeast DNA polymerase catalytic core and holoenzyme have comparable catalytic rates. J Biol Chem. 2015;290:3825–35. doi: 10.1074/jbc.M114.615278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dieckman LM, Johnson RE, Prakash S, Washington MT. Pre-steady state kinetic studies of the fidelity of nucleotide incorporation by yeast DNA polymerase δ. Biochemistry. 2010;49:7344–50. doi: 10.1021/bi100556m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, et al. Replication dynamics of the yeast genome. Science. 2001;294:115–21. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- 118.Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–45. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem. 2013;82:119–38. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tsutakawa SE, Lafrance-Vanasse J, Tainer JA. The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair. 2014;19:95–107. doi: 10.1016/j.dnarep.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kang YH, Lee CH, Seo YS. Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit Rev Biochem Mol Biol. 2010;45:71–96. doi: 10.3109/10409230903578593. [DOI] [PubMed] [Google Scholar]
- 122.Wanrooij PH, Burgers PM. Yet another job for Dna2: checkpoint activation. DNA Repair. 2015;32:17–23. doi: 10.1016/j.dnarep.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Levikova M, Cejka P. The Saccharomyces cerevisiae Dna2 can function as a sole nuclease in the processing of Okazaki fragments in DNA replication. Nucleic Acids Res. 2015;43:7888–97. doi: 10.1093/nar/gkv710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′-5′-exonuclease activities of Pol α in the creation of a ligatable nick. J Biol Chem. 2003;278:1626–33. doi: 10.1074/jbc.M209803200. [DOI] [PubMed] [Google Scholar]
- 125.Balakrishnan L, Bambara RA. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. J Biol Chem. 2010;286:6865–70. doi: 10.1074/jbc.R110.209502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Indiani C, McInerney P, Georgescu R, Goodman MF, O’Donnell M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol Cell. 2005;19:805–15. doi: 10.1016/j.molcel.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 127.Beattie TR, Bell SD. Coordination of multiple enzyme activities by a single PCNA in archaeal Okazaki fragment maturation. EMBO J. 2012;31:1556–67. doi: 10.1038/emboj.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dovrat D, Stodola JL, Burgers PM, Aharoni A. Sequential switching of binding partners on PCNA during in vitro Okazaki fragment maturation. PNAS. 2014;111:14118–23. doi: 10.1073/pnas.1321349111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gary R, Park MS, Nolan JP, Cornelius HL, Kozyreva OG, et al. A novel role in DNA metabolism for the binding of Fen1/Rad27 to PCNA and implications for genetic risk. Mol Cell Biol. 1999;19:5373–82. doi: 10.1128/mcb.19.8.5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vijayakumar S, Chapados BR, Schmidt KH, Kolodner RD, Tainer JA, Tomkinson AE. The C-terminal domain of yeast PCNA is required for physical and functional interactions with Cdc9 DNA ligase. Nucleic Acids Res. 2007;35:1624–37. doi: 10.1093/nar/gkm006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ayyagari R, Gomes XV, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 and DNA2. J Biol Chem. 2003;278:1618–25. doi: 10.1074/jbc.M209801200. [DOI] [PubMed] [Google Scholar]
- 132.Kadyrov FA, Genschel J, Fang Y, Penland E, Edelmann W, Modrich P. A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. PNAS. 2009;106:8495–500. doi: 10.1073/pnas.0903654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lujan SA, Clausen AR, Clark AB, MacAlpine HK, MacAlpine DM, et al. Heterogeneous poly-merase fidelity and mismatch repair bias genome variation and composition. Genome Res. 2014;24:1751–64. doi: 10.1101/gr.178335.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–38. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sinha NK, Morris CF, Alberts BM. Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J Biol Chem. 1980;255:4290–93. [PubMed] [Google Scholar]
- 136.Chastain PD, II, Makhov AM, Nossal NG, Griffith J. Architecture of the replication complex and DNA loops at the fork generated by the bacteriophage T4 proteins. J Biol Chem. 2003;278:21276–85. doi: 10.1074/jbc.M301573200. [DOI] [PubMed] [Google Scholar]
- 137.Hamdan SM, Loparo JJ, Takahashi M, Richardson CC, van Oijen AM. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature. 2009;457:336–39. doi: 10.1038/nature07512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ishmael FT, Trakselis MA, Benkovic SJ. Protein-protein interactions in the bacteriophage T4 replisome. The leading strand holoenzyme is physically linked to the lagging strand holoenzyme and the primosome. J Biol Chem. 2003;278:3145–52. doi: 10.1074/jbc.M209858200. [DOI] [PubMed] [Google Scholar]
- 139.McHenry CS. DNA replicases from a bacterial perspective. Annu Rev Biochem. 2011;80:403–36. doi: 10.1146/annurev-biochem-061208-091655. [DOI] [PubMed] [Google Scholar]
- 140.Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, et al. A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature. 2014;510:293–97. doi: 10.1038/nature13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Villa F, Simon AC, Ortiz Bazan MA, Kilkenny ML, Wirthensohn D, et al. Ctf4 is a hub in the eukaryotic replisome that links multiple CIP-box proteins to the CMG helicase. Mol Cell. 2016;63:385–96. doi: 10.1016/j.molcel.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bermudez VP, Farina A, Tappin I, Hurwitz J. Influence of the human cohesion establishment factor Ctf4/AND-1 on DNA replication. J Biol Chem. 2010;285:9493–505. doi: 10.1074/jbc.M109.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, et al. Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol Cell. 2016;63:371–84. doi: 10.1016/j.molcel.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hanna JS, Kroll ES, Lundblad V, Spencer FA. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 2001;21:3144–58. doi: 10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα /Primase/Ctf4 complex. Mol Cell. 2015;57:812–23. doi: 10.1016/j.molcel.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Williams DR, McIntosh JR. mcl1+, the Schizosaccharomyces pombe homologue of CTF4, is important for chromosome replication, cohesion, and segregation. Eukaryot Cell. 2002;1:758–73. doi: 10.1128/EC.1.5.758-773.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–19. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]