

Abstract
Huntington's disease (HD) is an autosomal-dominant neurodegenerative disorder caused by the expansion of a CAG triplet in the gene encoding for huntingtin (Htt). The resulting mutant protein (mHtt) with extended polyglutamine (polyQ) sequence at the N terminus leads to neuronal degeneration both in a cell-autonomous and a non-cell-autonomous manner. Recent studies identified mHtt in the extracellular environment and suggested that its spreading contributes to toxicity, but the mechanism of mHtt release from the cell of origin remains unknown. In this study, we performed a comprehensive, unbiased analysis of secretory pathways and identified an unconventional lysosomal pathway as an important mechanism for mHtt secretion in mouse neuroblastoma and striatal cell lines, as well as in primary neurons. mHtt secretion was dependent on synaptotagmin 7, a regulator of lysosomal secretion, and inhibited by chemical ablation of late endosomes/lysosomes, suggesting a lysosomal secretory pattern. mHtt was targeted preferentially to the late endosomes/lysosomes compared with wild-type Htt. Importantly, we found that late endosomal/lysosomal targeting and secretion of mHtt could be inhibited efficiently by the phosphatidylinositol 3-kinase and neutral sphingomyelinase chemical inhibitors, Ly294002 and GW4869, respectively. Together, our data suggest a lysosomal mechanism of mHtt secretion and offer potential strategies for pharmacological modulation of its neuronal secretion.
SIGNIFICANCE STATEMENT This is the first study examining the mechanism of mutant huntingtin (mHTT) secretion in an unbiased manner. We found that the protein is secreted via a late endosomal/lysosomal unconventional secretory pathway. Moreover, mHtt secretion can be reduced significantly by phosphatidylinositol 3-kinase and neutral sphingomyelinase inhibitors. Understanding and manipulating the secretion of mHtt is important because of its potentially harmful propagation in the brain.
Keywords: Huntington's disease, late endosome, lysosome, mutant huntingtin, secretion
Introduction
Huntington's disease (HD) is a neurodegenerative disorder characterized by motor, cognitive, and psychiatric symptoms that occur due to the progressive degeneration of striatal neurons and, ultimately, other brain regions. HD is an autosomal-dominant disorder caused by the expansion of CAG triplets in the gene encoding for huntingtin (Htt) and the resulting production of the mutant Htt (mHtt) protein with an extended polyglutamine sequence at the N terminus. The disease pathophysiology develops primarily due to a toxic gain-of-function of mHtt (Bates et al., 2015). mHtt and its proteolytic fragments display a propensity to aggregate and form toxic inclusions, although a number of studies suggest that soluble forms of mHtt may represent the harmful species (Kim et al., 1999; Dunah et al., 2002; Arrasate et al., 2004; Cui et al., 2006).
Although most of the early studies revealed that mHtt acts in a cell-autonomous manner (Ross and Tabrizi, 2011), a growing body of evidence has identified non-cell-autonomous mechanisms as an additional driving force for disease progression. mHtt has been found in the CSF (Wild et al., 2015) and in the neuronal allografts transplanted into the brains of HD patients (Cicchetti et al., 2014), suggesting that the protein can be exported from cells of origin. To study the effects of released mHtt, various experimental models expressing wild-type Htt (wtHtt) and mHtt have been developed (Pecho-Vrieseling et al., 2014; Babcock and Ganetzky, 2015; Jeon et al., 2016). These studies showed that, once released, mHtt gives rise to neurodegeneration in normal cells and tissues. The toxicity of secreted mHtt aggregates has been associated with their propensity to seed aggregation of the soluble protein in recipient cells (Chen et al., 2002; Ren et al., 2009; Tan et al., 2015). In light of the potential role of propagation in both HD and other neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) (Soto, 2012; Jucker and Walker, 2013), further characterizing the cellular mechanisms of protein secretion is critical for better understanding disease pathogenesis. Furthermore, designing preventative strategies in HD may require targeted approaches against the cellular secretion of mHtt.
Proteins can exit the cell via multiple mechanisms categorized as either conventional or unconventional pathways. The conventional pathway is used by the signal-peptide-containing secreted proteins that are translocated to the ER lumen and then to the Golgi and Golgi-derived vesicles, which ultimately fuse with the plasma membrane, thereby releasing their content to the extracellular environment (Lee et al., 2004). Alternatively, secreted cargo can follow one of the many unconventional pathways that include transfer of proteins via extracellular vesicles (ectosomes or exosomes) or in a free, nonvesicular form (Zhang and Schekman, 2013). Ectosomes are formed by the outward budding of the plasma membrane, whereas exosomes originate from the multivesicular bodies (MVBs) and are released upon MVB fusion with the plasma membrane. Free-form proteins can be secreted through translocation across plasma membrane, as in the case of FGF2 (Nickel, 2011), endolysosomal exocytosis (Andrews, 2000; Laulagnier et al., 2011), or secretory autophagy (Ponpuak et al., 2015). Neurons also use a specific, synaptic-vesicle-mediated secretory pathway for their neurotransmitters. Finally, many cell types can form tunneling nanotubes for intercellular exchange of various cargos (Abounit and Zurzolo, 2012).
Although the precise mechanism of mHtt secretion is not completely understood, several possibilities have been suggested, such as synaptic transmission (Pecho-Vrieseling et al., 2014), vesicular transport (Babcock and Ganetzky, 2015), exosomes/extracellular vesicles (Jeon et al., 2016; Zhang et al., 2016), and tunneling nanotubes (Costanzo et al., 2013). In this study, we analyzed in detail the mechanism of mHtt release and found that the extracellular mHtt mostly exists in a free form and follows the late endosomal/lysosomal (LE/Lys) secretory pattern. We also identified two potent inhibitors of mHtt secretion, Ly294002 and GW4869, which could prove useful for modulating mHtt secretion in the brain.
Materials and Methods
Cell culture, cDNAs, shRNAs, lentiviruses, antibodies, and reagents.
Neuro2A cells (ATCC catalog #CCL-131, RRID: CVCL_0470) were cultured in DMEM supplemented with 10% FCS (Invitrogen). Neuro2A cell lines stably expressing Htt 571 aa/72Q or 571 aa/25Q were generated by transducing Neuro2A cells with the lentiviral vectors encoding the first 571 aa of human Htt with 25 or 72 CAG repeats (Régulier et al., 2003). Cell cloning was performed by limiting dilution to obtain monoclonal cell population with high Htt expression. Striatal cells STHdh+/Hdh+, STHdhQ111/Hdh+, and STHdhQ111/HdhQ111 were a kind gift from Marcy MacDonald (Trettel et al., 2000). They were grown in DMEM with 10% FCS, penicillin/streptomycin, and 250 μg/ml geneticin (Thermo Fisher Scientific). Primary cortical neurons were isolated from Sprague Dawley rats at embryonic day 18 (E18), seeded in six-well plates for the secretion assay (700,000 neurons per well) or on 13 mm coverslips for immunofluorescence (100,000 neurons per coverslip), and grown in Neurobasal medium with glutamine, penicillin/streptomycin, and 2% B27 supplement (Thermo Fisher Scientific).
Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Medium was replaced 6 h after transfection. Lentiviral transductions of primary neurons were performed at a multiplicity of infection of 5 for mHtt lentiviruses, untagged Htt at day in vitro 7 (DIV7) for immunofluorescence and Flag-tagged Htt at DIV3 for the secretion analysis. Virus titer was determined using HIV type 1 p24 ELISA (ZeptoMetrix). One nanogram p24 was considered equal to 5000 infectious particles.
Mammalian expression plasmid encoding full-length Htt with N-terminal Flag tag was generated by subcloning Htt ORF of HD73 (Trottier et al., 1995) into pCGFlag vector (Mahajan et al., 2002). PolyQ length was engineered to have 97Q. mHtt571/72Q-GFP was generated by subcloning Htt ORF of Htt571/72Q into pEGFP-N1 vector. Other plasmids used in the study were as follows: Htt571/72Q and 25Q (Jeong et al., 2009); Htt 590/97Q and 25Q (Jeong et al., 2009); dsRed-2-ER-5 (calreticulin) and dsRed2-Mito-7 (COX8A) (Addgene catalog #55836 and #55838, respectively); and Vps34 shRNA (GE Healthcare/Dharmacon). pEGFP-2xFYVE was a kind gift from Harald Stenmark (Oslo University Hospital, Norway), Arf1-HA and Arf1T31N-HA (wild-type and dominant-negative mutant) from Julie Donaldson (National Institutes of Health, Bethesda), and synaptotagmin 7 shRNA (KD607) from Thomas Südhof (Stanford University Medical School) (Bacaj et al., 2013). Htt571/72Q-Flag plasmid for lentiviral production was generated by inserting a Flag tag at the C terminus of 571/72Q Htt using Q5 Site-Directed Mutagenesis Kit (New England Biolabs). Synaptotagmin 7- and scrambled shRNA-carrying lentiviruses were generated using the above-mentioned synaptotagmin 7 shRNA and scrambled shRNA control. Lamp2A antibody was a kind gift from Judith Blanz (Rothaug et al., 2015). Other antibodies were purchased from the following producers: monoclonal anti-synaptotagmin 7, cat #MABN665 anti-Htt MAB5490 and anti-Htt MAP2166 from Millipore (catalog #MAB5490 RRID: AB_2233522 and catalog #MAB2166 RRID: AB_2123255); anti-PolyQ from Millipore (catalog #MAB1574 RRID: AB_11211899); polyclonal anti-synaptotagmin 7 from Synaptic Systems (catalog #105 173 RRID: AB_887838); anti-Vps34 from Cell Signaling Technology; monoclonal anti-tubulin; anti-HA from Sigma-Aldrich (catalog #H9658 RRID: AB_260092); anti-Flag from Sigma-Aldrich (catalog #F3165 RRID: AB_439685 and catalog #F7425 RRID: AB_439687); anti-GFP from Sigma-Aldrich (catalog #G1544 RRID: AB_439690); anti-Lamp1 from Abcam (catalog #ab24170 RRID: AB_775978); anti-EEA1 from Abcam (catalog #ab2900 RRID: AB_2262056); HRP-conjugated secondary antibodies from Jackson ImmunoResearch; and fluorescently labeled secondary antibodies from Invitrogen. The following reagents were used: brefeldin A, Triton X-100, sodium taurocholate, EDTA, BSA, and glycine from Sigma-Aldrich; 4-methylumbelliferyl b-glucophyranoside (4-MU) from Chem-Impex; LDH cytotoxicity assay kit from Pierce; BAPTA AM and Alexa Fluor 488-conjugated transferrin from human serum from Life Technologies; cycloheximide, LY294002, 3-methyladenine (3-MA), GW4869, and bafilomycin A1 from Cayman Chemical; and CellLight Golgi-GFP and BacMam 2.0 from Thermo Fisher Scientific.
Secretion assay.
To collect secreted proteins, Neuro2A and striatal cells were grown to 90–100% confluency in 12-well plates (∼1,000,000 cells for Htt571 aa) or 6 cm dishes (∼5,500,000 cells for FL Htt) in OptiMEM medium (Invitrogen) for the indicated periods of time. When indicated, the media were preconditioned for 1 h on naive Neuro2A cells to enhance secretion. Upon collecting, media were centrifuged for 20 min at 2000 × g to eliminate cell debris and then concentrated using Amicon ultracentrifugation filter units NMWL 10. In media fractionation experiments, cell-debris-free medium was sequentially centrifuged for 30 min at 10,000 × g and for 1 h at 100,000 × g to sediment ectosomes and exosomes, respectively. The final volume of all pellets and supernatants was 50 μl and 15 μl of each was analyzed by Western blotting. For the secretion assay in neurons, 1 ml of Neurobasal medium with glutamine, penicillin/streptomycin, and 0.2% of B27 supplement was used to collect secreted proteins.
Cell fractionation.
For fractionation experiments, cells were grown to confluency (∼14,500,000 cells) in 10 cm dishes. Cell fractionation on sucrose density gradients was described previously (Aniento and Gruenberg, 2003). Briefly, cells were washed and scraped in cold PBS using a custom-made rubber cell scraper, followed by centrifugation for 5 min at 150 × g. Cell pellets were then resuspended in 8.5% sucrose/3 mm imidazole and centrifuged for 10 min at 700 × g. The resulting pellets were resuspended in 8.5% sucrose/3 mm imidazole containing protease inhibitor complex, homogenized by passing three times through a 22Gx1 ½ needle, and centrifuged for 10 min at 700 × g. Postnuclear supernatants were collected, diluted by 62% sucrose/3 mm imidazole to a final sucrose concentration of 40.6% (as determined by refractometer), and transferred to 11 × 60 mm centrifuge tubes. Sucrose step gradients were generated by loading 1.5 ml of 35% sucrose/3 mm imidazole and 3 ml of 8.5% sucrose/3 mm imidazole, respectively, and centrifuged for 1 h at 35,000 rpm in a Beckman SW60 rotor. Light membranes were collected from the interphase between 8.5% and 35% sucrose, heavy membranes from the interphase between 35% and 40.6% sucrose, and the remaining bottom layer (40.6% sucrose) represented cytosol. Protein concentration of the fractions was determined using the bicinchoninic acid assay (Sigma-Aldrich).
Endosome ablation.
The HRP-diaminobenzidine (DAB) cross-linking procedure was used for endolysosomal ablation (Stoorvogel, 1998; Laulagnier et al., 2011). Briefly, cells were incubated for 15 min with 0.5 g/L HRP diluted in OptiMEM at 37°C; washed 3 times in 5% BSA/PBS; followed by a 0, 30, or 90 min chase in OptiMEM to label consecutive stages of the endocytic pathway. The internalized HRP was then cross-linked for 30 min on ice in the presence of 100 μg/ml DAB and 0.003% H2O2 diluted in PBS. Cells were then washed extensively by PBS and incubated in preconditioned OptiMEM for an additional 30 min to collect secreted proteins.
β-Glucocerebrosidase (GCase) activity assay.
The GCase activity assay was described previously (Marshall et al., 2002; Mazzulli et al., 2011). Briefly, 15 μl of concentrated medium was incubated with 65 μl of activity assay buffer containing 0.25% (v/v) Triton X-100, 0.25% (w/v) Na-taurocholate, 1 mm EDTA, 1% BSA in citrate/phosphate buffer, pH 5.4, and 20 μl of 4-MU for 30 min at 37°C. The reaction was stopped by adding 100 μl of 1 m glycine, pH 12.5, and fluorescence (ex = 365 nm, em = 445) was determined in a SpectraMax i3x plate reader from Molecular Devices.
Western blot analysis.
Cells were lysed directly in 2× Laemmli buffer containing β-mercaptoethanol (200 μl of for 12-well plates and 1 ml for 6 cm dishes). Then, 8 μl of each lysate was analyzed by SDS-PAGE and Western blotting. In time course experiments with GW4869 treatment, cells were lysed in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, and protease inhibitor complex (Complete EDTA-free; Roche). Cell debris and nuclei were eliminated by centrifugation for 5 min at 5000 × g. Protein concentration was determined using the bicinchoninic acid assay. Immunolabeled proteins were revealed by chemiluminescence using the ChemiDoc System from Bio-Rad. Quantitative analysis of protein bands was performed using ImageJ software.
LDH cytotoxicity assay.
LDH cytotoxicity assay was performed according to the manufacturer's instructions using cell-debris-free medium before concentrating.
Immunofluorescence, confocal microscopy, and image analysis.
Cytosol washout was performed by washing 60–80% confluent living cells with cytosol buffer containing 25 mm HEPES-KOH, pH 7.4, 25 mm KCl, 2.5 mm Mg acetate, 5 mm EGTA, pH 8.0, and pre-permeabilization in the cytosol buffer with 0.05% saponin for 30 s at room temperature, followed by another wash in the cytosol buffer and fixation for 20 min in 3% formaldehyde/PBS. The immunofluorescence procedure was described previously (Song et al., 2016). The coverslips were mounted in DAPI Fluoromount-G from Southern Biotech. Images were obtained on a Leica DMI4000B inverted microscope and analyzed using ImageJ software. Colocalization was determined using the colocalization analysis module of the software (Mander's coefficients). A total of 80–200 cells per experiment were imaged and analyzed quantitatively.
Super-resolution microscopy was performed using a Nikon Structured Illumination Super-resolution Microscope (N-SIM).
Experimental design and statistical analysis.
All experiments were done at least three times (biological replicates). Mean values and SDs were obtained using Microsoft Excel. Significance was determined using unpaired Student's t test.
Results
mHtt is secreted preferentially compared with wtHtt
It has been shown previously both in vivo and in vitro that mHtt can be released from the cell of origin (Cicchetti et al., 2014; Pecho-Vrieseling et al., 2014; Babcock and Ganetzky, 2015; Wild et al., 2015; Jeon et al., 2016), but the specificity and mechanism of export have been largely unknown. To begin addressing these questions, we created Neuro2A cell lines stably expressing a biologically relevant 571 aa N-terminal Htt fragment with either 72 (mutant/m) or 25 (wild-type/wt) glutamines (Q). Our data obtained in these lines show that the ratio of secreted (SN) to intracellular (CL) mHtt is ∼three times higher compared with that of wtHtt (Fig. 1A). mHtt-expressing cells did not exhibit signs of toxicity-induced permeabilization, as shown by the LDH cytotoxicity assay (Fig. 1A). To further determine whether mHtt is secreted preferentially, we coexpressed mHtt and wtHtt transiently in naive Neuro2A cells. Consistent with the data obtained using Htt-expressing cell lines, we observed increased secretion of the mHtt compared with wtHtt (Fig. 1B). We next tested the possibility that some of the secreted protein was depleted from the medium through reinternalization into the cells. The media collected from mHtt or wtHtt-expressing stable Neuro2A cell lines were transferred to either cell-free dishes or dishes containing naive Neuro2A cells and analyzed for Htt content after a 4 h incubation period. No depletion of Htt was observed in the media incubated with cells compared with the media incubated without cells (Fig. 1C). Moreover, when we transferred the media from mHtt-GFP-transfected donor Neuro2A cells to untransfected Neuro2A cells, we failed to detect any GFP signal in the acceptor cells after 4 h of incubation (Fig. 1C). Together, these data suggest that there was no detectable loss of Htt in the media due to transmission of the protein to neighboring cells under our experimental conditions. To further characterize mHtt release into the media and confirm that this was not due to passive leakage of cellular proteins from dying cells, we took advantage of the fact that facilitated transport is generally temperature dependent. As shown in Figure 1D, the amount of mHtt in the extracellular medium was ∼60% lower at 25°C compared with 37°C, suggesting the involvement of active transport in mHtt secretion. To eliminate the possibility that diminished mHtt secretion at 25°C was due to reduced protein synthesis, mHtt secretion was assessed upon cycloheximide-mediated inhibition of protein synthesis. Using this approach, we found no difference in secretion between cells treated with cycloheximide or vehicle (Fig. 1D), suggesting that decreased secretion of mHtt at 25°C is not due to reduced protein synthesis.
Figure 1.
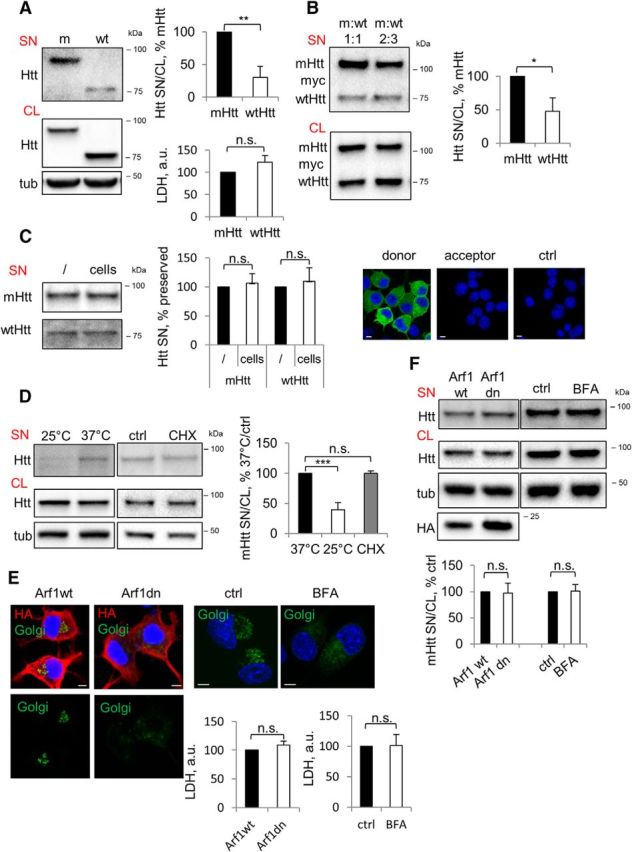
Preferential secretion of mHtt via unconventional secretory pathway. A, mHtt is secreted preferentially compared with wtHtt. Left, Neuro2A-mHtt cells were incubated for the final 16 h in OptiMEM of total 48 h after plating. Equal volumes of the concentrated media (SN) and cell lysates (CL) were analyzed by Western blotting using anti-Htt antibody. Tubulin was used as a loading control. Top right, Ratio of Htt in the media and cell lysates expressed as a percentage of mHtt. n = 3, p = 0.0021. Bottom right, LDH assay was performed on the media before concentrating. n = 3, p = 0.1601. B, mHtt is secreted preferentially from cells coexpressing mHtt and wtHtt. Left, Naive Neuro2A cells were cotransfected with 590aa Htt/myc/His/97Q (mHtt) and 25Q (wtHtt) at the ratios 1:1 (left lanes) and 2:3 (right lanes). Cells were incubated for the final 16 h of total 48 h after transfection in OptiMEM. Concentrated media (SN) and cell lysates (CL) were analyzed by Western blotting using anti-myc antibody. Right, Ratio of Htt in the media and cell lysates expressed as a percentage of mHtt. n = 3, p = 0.0114. C, Secreted Htt is not depleted from the media through endocytosis. Left, Media conditioned overnight on Neuro2A stably expressing mHtt or wtHtt was collected and further incubated in empty wells (/) or with naive Neuro2A cells (cells) for 4 h. Media was then concentrated and analyzed by Western blotting using anti-Htt antibody. Center, Htt in the media incubated with cells expressed as a percentage of Htt in the media incubated in empty wells. n = 3; p = 0.5230 and 0.5183. Right, Media conditioned on Neuro2A cells transiently expressing mHtt-GFP (donor cells) was transferred on naive Neuro2A (acceptor cells) and incubated for 4 h in the presence of 200 nm bafilomycin A1 to prevent degradation of putatively endocytosed Htt. Naive Neuro2A not exposed to Htt-conditioned media was used as a negative control (ctrl). Cells were then fixed, mounted, and analyzed by confocal microscopy. Nuclei were visualized using DAPI staining. n = 3. D, mHtt secretion is temperature dependent. Neuro2A-mHtt cells were incubated at 25°C or 37°C on the benchtop for 4 h in OptiMEM (left) and at 37°C with 10 μg/ml cycloheximide (CHX) or DMSO (ctrl) (center). Concentrated media and cell lysates were analyzed by Western blotting using anti-Htt antibody. Tubulin was used as a loading control. Right, Ratio of mHtt in the media and cell lysates expressed as a percentage of 37°C control. n = 3, p = 0.0009 and 0.957. E, Genetic and pharmacological disruption of the constitutive secretory pathway leads to collapse of the Golgi complex and is nontoxic. Left, Neuro2A-mHtt cells were transfected with HA-tagged Arf1wt or Arf1T31N (Arf1dn) and incubated for the last 16 of total 24 h after transfection in OptiMEM. Right, Cells were treated with DMSO (ctrl) or 5 μg/ml brefeldin A (BFA) in preconditioned OptiMEM for 1 h. Cells were then transduced with CellLight Golgi-GFP for the last 16 h of incubation and analyzed by confocal microscopy upon immunolabeling with anti-HA antibody (left) or without additional staining (right). Nuclei were visualized using DAPI staining. Scale bar, 10 μm. Bottom, LDH assay was performed on 30 μl of the media before concentrating. n = 3, p = 0.2189 and 0.9424. F, mHtt secretion does not follow a constitutive secretion pathway. Top, Cells were treated as in E. Concentrated media and the cell lysates were analyzed by Western blotting using anti-Htt antibody. Tubulin was used as a loading control. Bottom, Ratio of mHtt in the media and cell lysates expressed as a percentage of control. n = 3, p = 0.91 and 0.89. Error bars indicate SD. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., Not significant.
mHtt is secreted in an unconventional manner
Our results so far suggested that both wtHtt and mHtt can be secreted into the extracellular medium, but with preferential secretion of the mutant protein. To examine the mechanism of Htt secretion, we performed a comprehensive assessment of various secretion mechanisms. First, to determine whether mHtt is secreted via constitutive secretion, brefeldin A treatment or overexpressed Arf1 dominant-negative mutant was used to arrest the secretion (Klausner et al., 1992; Donaldson and Jackson, 2011), and Golgi marker N-acetylgalactosaminyltransferase–emGFP packaged in baculovirus to assess the dispersal of the Golgi apparatus. Although the Golgi complex was clearly disrupted by either of the treatments without accompanying cytotoxicity (Fig. 1E), the secretion of mHtt was unaffected under these conditions (Fig. 1F), suggesting that mHtt may be secreted in an unconventional manner.
Extracellular mHtt is predominantly in a free form, whereas the intracellular vesicular protein localizes to a Lamp1-positive compartment
Next, we investigated whether mHtt is secreted in association with extracellular vesicles or in a free form. Using sequential centrifugation of the extracellular medium, we found that ∼92% of the released mHtt is in a free form, 7% in the 100,000 × g exosome-containing pellet, and only 1% in the 10,000 × g ectosome-containing pellet (Fig. 2A). Free-form proteins can exit the cell either through direct translocation across the plasma membrane or through fusion of intracellular vesicular intermediates with the plasma membrane (Rabouille et al., 2012). Using cell fractionation experiments to assess the presence of mHtt in vesicles, we found ∼15% of the protein in the vesicle-enriched light membrane fraction (Fig. 2B). To visualize intravesicular mHtt, we used a cytosol-washout procedure that allows for permeabilization of the plasma membrane and leakage of the cytosolic proteins, whereas intracellular membranes remain intact with their contents preserved in the lumen. This approach revealed punctate staining pattern of mHtt, confirming that a portion of the protein is encapsulated within vesicles. To characterize mHtt-containing vesicles, we performed a colocalization analysis with various vesicular markers and found no obvious colocalization with major organelles, including the ER, Golgi, mitochondria, and recycling and early endosomes (Fig. 2C and data not shown). Instead, the bulk of vesicular mHtt colocalized with Lamp1 and Lamp2, LE/Lys markers (Fig. 2C). The colocalization with Lamp1 was confirmed in rat primary cortical neurons (Fig. 2D). Using N-SIM, we visualized mHtt within the lumen of Lamp1-positive compartments observed as large rings (Fig. 2E), raising the possibility that these Lamp1-positive compartments are the source of secreted mHtt.
Figure 2.
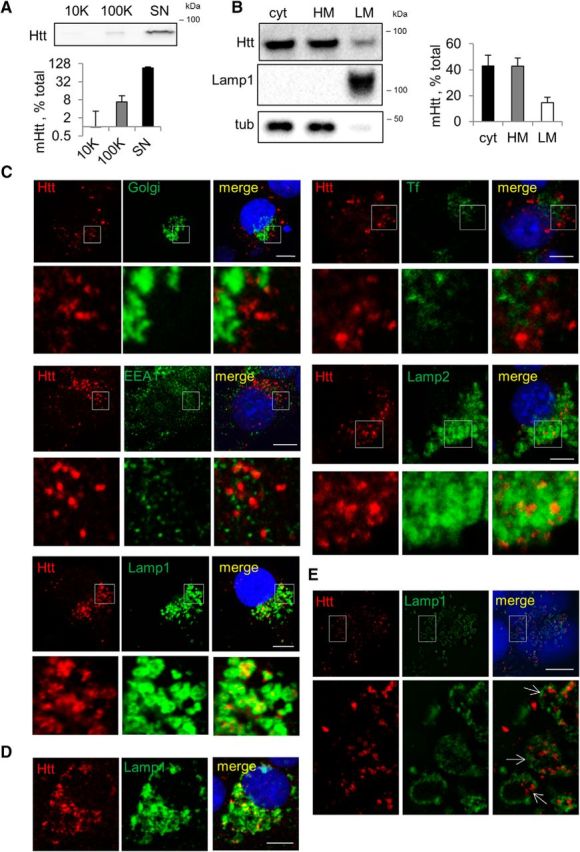
Intracellular mHtt localizes to LE/Lys vesicles, whereas secreted mHtt exists in a free form. A, Extracellular mHtt is predominantly in a free form. Top, OptiMEM conditioned for 24 h on Neuro2A-mHtt was collected and sequentially centrifuged on 2000 × g to eliminate cell debris, 10,000 × g to pellet ectosomes, and 100,000 × g to pellet exosomes. The remaining supernatant was concentrated. The 10,000 × g pellet, the 100,000 × g pellet, and the concentrated supernatant were analyzed by Western blotting using anti-Htt antibody. Bottom, Relative amount of mHtt in each fraction expressed as a percentage of total mHtt. n = 3. B, Portion of intracellular mHtt is localized in the light membrane fraction. Left, Postnuclear supernatant (PNS) obtained from Neuro2A-mHtt cells was subjected to floatation in the sucrose density gradient and 2 μg of cytosolic (cyt), heavy membrane (HM), and light membrane (LM) fractions were analyzed by Western blotting using anti-Htt antibody. Lamp1 and tubulin were used as light membrane and cytosolic markers, respectively. Right, Relative amount of mHtt in each fraction expressed as a percentage of total mHtt. n = 3. C, mHtt-containing vesicles colocalize with Lamp1-positive compartments. Neuro2A-mHtt cells were transduced with CellLight Golgi-GFP (top left), treated 20 min with 20 μg/ml transferrin-Alexa Fluor 488 (top right), or left untreated (remaining panels). Cells were then pre-permeabilized, fixed, immunolabeled with anti-Htt (all), anti-EEA1 (second raw left), anti-Lamp2 (second row right), or anti-Lamp1 (third row) antibodies; and analyzed by confocal microscopy. D, Primary cortical neurons derived from rat embryos and transduced with Htt571/72Q lentivirus were pre-permeabilized and fixed 7 d after transduction, immunolabeled with anti-Htt and anti-Lamp1 antibodies, and analyzed by confocal microscopy. E, Vesicular mHtt is found within the lumen of Lamp1-positive vesicles. Untreated cells were pre-permeabilized, fixed, immunolabeled with anti-Htt and anti-Lamp1 antibodies, and analyzed by N-SIM. Nuclei were visualized using DAPI staining. Scale bar, 10 μm. Error bars indicate SD.
LE/Lys secretion of mHtt
LE/Lys are acidic organelles containing lysosomal enzymes that are crucial for protein degradation. However, they have the ability to fuse with the plasma membrane and release their content into the extracellular environment in a Ca2+-dependent manner (Andrews, 2000). This process is regulated by synaptotagmin 7, a lysosome-specific calcium sensor (Martinez et al., 2000). To test the hypothesis that mHtt is released from the Lamp1 compartment, we analyzed mHtt secretion upon inhibition of lysosomal exocytosis through silencing of synaptotagmin 7 in mHtt-expressing Neuro2A (Fig. 3A). Interestingly, this led to a significant decrease in secretion of both 571 aa fragment and Flag-tagged full-length mHtt (Fig. 3A). To validate these results in more HD-relevant experimental systems, we developed secretion assays for the striatal cell line STHdh+/Hdh+ and primary cortical neurons (see Materials and Methods) and observed a significant decrease in mHtt secretion upon silencing of synaptotagmin 7 in both cell types (Fig. 3A). Moreover, short treatment of the Neuro2A-mHtt cells with BAPTA AM, a membrane-permeant calcium chelator, led to the intracellular accumulation of mHtt-positive punctae (Fig. 3B), consistent with synaptotagmin 7-dependent secretion of mHtt. To confirm that mHtt is secreted from LE/Lys, we performed an organelle ablation assay based on the selective loading of a cellular compartment with HRP and concomitant DAB/H2O2-mediated cross-linking, which renders the given compartment inactive (Stoorvogel, 1998; Laulagnier et al., 2011). To label LE/Lys, Neuro2A-mHtt cells were pulsed with the HRP-containing medium for 15 min to allow for fluid-phase endocytosis of the enzyme, which was then chased along the endocytic pathway for an additional 90 min. During this period of time, the enzyme transits through the consecutive stages of the endocytic pathway, early endosomes and intermediate compartments, and finally reaches the LE/Lys. Cells were then incubated with DAB in the presence of low concentrations of H2O2 to induce HRP cross-linking and concomitant chemical ablation of the LE/Lys and secretion of mHtt into the fresh medium was allowed to proceed for 30 min. This experiment revealed that the ablation of LE/Lys reduced mHtt secretion to ∼50% of the control, corroborating our finding that mHtt is secreted from LE/Lys (Fig. 3C). To confirm the validity of the assay, we measured the activity of a lysosomal enzyme GCase in the media upon LE/Lys ablation and found a similar decrease (Fig. 3C). Conversely, when we ablated early and intermediate endocytic compartments by performing cross-linking at earlier points of the chase period, after 0 and 30 min, respectively, we found the secretion of mHtt unaffected (Fig. 3C). Together, these results further suggested that at least a portion of mHtt is secreted from late endosomes and/or lysosomes.
Figure 3.

LE/Lys secretion of mHtt. A, Disruption of lysosomal exocytosis leads to decreased secretion of mHtt. Left, Neuro2A-mHtt cells were transfected with scrambled shRNA or shRNA against synaptotagmin 7; naive Neuro2A cells with Flag-tagged full-length mHtt (FL Htt) and scrambled or synaptotagmin 7 shRNAs at 3:1 ratio; primary cortical neurons were transduced with Htt571/72Q-Flag and scrambled or synaptotagmin 7 shRNA lentiviruses at 5:3 ratio; and STHdh+/Hdh+ cells were transfected with GFP-tagged 571 aa mHtt and scrambled or synaptotagmin 7 shRNAs. Silencing was allowed to proceed for 48 h in total for transfected cells or 7 d for transduced neurons and secreted mHtt was collected for the last 16 h. Concentrated media and cell lysates were analyzed by Western blotting using anti-Htt, anti-Flag, or anti-GFP antibodies. Tubulin was used as a loading control. Knock down of syt7 was verified by probing with anti-syt 7 antibody (bottom). Right, Ratio of mHtt in the media and cell lysates expressed as a percentage of control. n = 3, p = 0.0283, 0.0035, 0.0001, and 0.0055. B, Calcium chelation leads to increased Htt punctae. Left, Neuro2A-mHtt cells were treated with DMSO or 0.2 μm BAPTA AM for 4 h. Cells were then pre-permeabilized, fixed, immunolabeled with anti-Htt Ab, and analyzed by confocal microscopy. Nuclei were visualized using DAPI staining. Right, Integrated density of mHtt signal per cell expressed as a percentage of control. Scale bar, 10 μm. n = 3, p = 0.021. C, Ablation of LE/Lys results in reduced secretion of mHtt. Top, Neuro2A-mHtt cells were pulsed with HRP (ablation/abl) or plain OptiMEM (ctrl). HRP was chased for 90, 30, or 0 min to label LE/Lys, intermediate compartments of the endocytic pathway, or early endosomes, respectively. Cells were then treated with DAB, incubated for 30 min in preconditioned OptiMEM and concentrated media, and cell lysates were analyzed by Western blotting using anti-Htt antibody. Tubulin was used as a loading control. Bottom left, Ratio of mHtt in the media and cell lysates expressed as a percentage of control. Control for each time point is assigned a 100% value. n = 4, 3, and 3; p = 0.0014, 0.4754, and 0.9708, respectively. Bottom right, GCase activity was measured in the media conditioned on the cells with and without ablated LE/Lys (90 min chase) and the values were normalized to the total amount of proteins in the cell lysate. n = 4; p = 0.0001. Error bars indicate SD. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., Not significant.
Preferential targeting of mHtt over wtHtt to LE/Lys
We hypothesized that preferential secretion of mHtt compared with wtHtt from LE/Lys might be due to preferential targeting of mHtt to LE/Lys. To assess directly the targeting of mHtt versus wtHtt to LE/Lys, we first determined the relative amounts of the two proteins in the LE/Lys-containing light membranes upon cell fractionation. We found that wtHtt is significantly less enriched in the light membrane fraction compared with the mutant (Fig. 4A). Moreover, when we used a cytosol-washout procedure followed by immunolabeling with anti-Htt antibody and confocal microscopy to detect intravesicular Htt, we found ∼40% less punctate wtHtt compared with mHtt (Fig. 4B). Finally, the colocalization of punctate wtHtt with Lamp1 was reduced by ∼30% compared with the mutant protein (Fig. 4B). Together, these data show higher relative abundance of mHtt in Lamp1-positive vesicles, suggesting that preferential targeting of mHtt to LE/Lys might be the underlying reason for its elevated secretion compared with the wild-type protein.
Figure 4.

mHtt is targeted preferentially to LE/Lys compared with wtHtt. A, Enrichment of mHtt over wtHtt in the light membrane fraction. Neuro2A-mHtt and Neuro2A-wtHtt cells were homogenized and subjected to floatation in sucrose density gradients. Then, 2 μg of light membrane (LM) (left) and cytosolic (cyt) proteins (center) were analyzed by Western blotting using anti-Htt antibody. Lamp1 and tubulin were used as loading controls for the light membranes and cytosols, respectively. Right, Ratio of light membrane and cytosolic Htt expressed as a percentage of mHtt. n = 3, p = 0.0222. B, Increased targeting of mHtt over wtHtt to Lamp1-positive vesicles. Left, Neuro2A-mHtt (top) or Neuro2A-wtHtt (bottom) cells were pre-permeabilized, fixed, immunolabeled with anti-Htt and anti-Lamp1 antibodies, and analyzed by confocal microscopy. DAPI was used to visualize nuclei. Scale bar, 10 μm. Top right, Htt integrated density per cell expressed as a percentage of mHtt. n = 4, p = 0.0001. Bottom right, Htt/Lamp1 colocalization. n = 3, p = 0.0036. Error bars indicate SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Neutral sphingomyelinase (NS) inhibition reduces mHtt secretion without accompanying intracellular accumulation of mHtt and cytotoxicity
Previous studies suggested that secreted mHtt can be propagated in the CNS, potentially seeding aggregation and causing toxicity in remote brain areas (Babcock and Ganetzky, 2015; Jeon et al., 2016). These findings imply that inhibition of mHtt secretion might prevent the progression of non-cell-autonomous pathology in HD. To explore possibilities for drug-mediated inhibition of mHtt release from LE/Lys, we tested several compounds that modulate structure and/or function of the endolysosomal pathway in a nontoxic manner. Using this approach, we found that the NS inhibitor GW4869 was the most efficient in reducing secretion of both the 571 aa fragment and/or full-length mHtt without lowering intracellular levels of the proteins or inducing cytotoxicity in Neuro2A cells and rat primary cortical neurons (Fig. 5A). Neutral sphingomyelinase inhibitor also inhibited the secretion of endogenous mHtt in STHdhQ111/Hdh+ and STHdhQ111/HdhQ111 (Fig. 5A).
Figure 5.
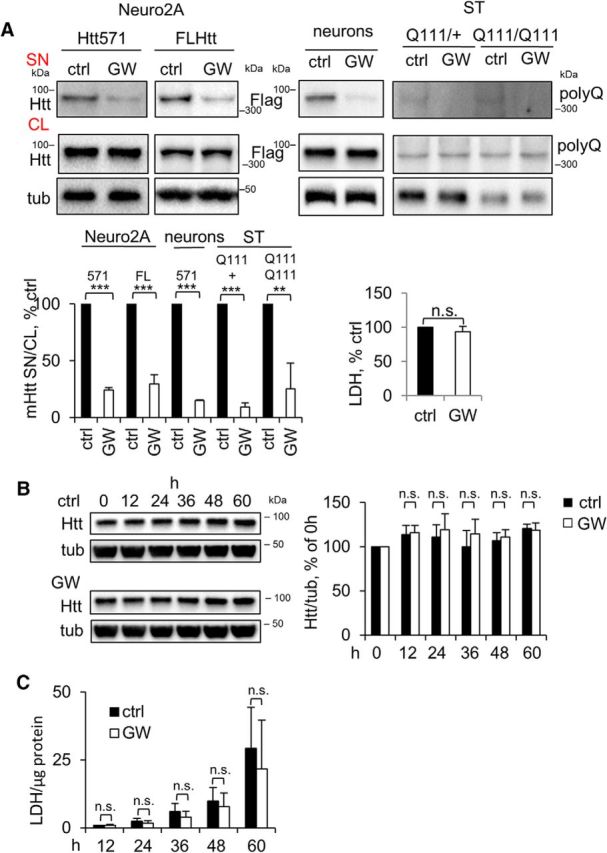
Inhibition of NS reduces the secretion of mHtt without intracellular accumulation of mHtt and increased cytotoxicity. A, Reduced secretion of mHtt by inhibition of NS. Left, Neuro2A-mHtt cells, naive Neuro2A cells transfected with Flag-tagged full-length mHtt (FL Htt), primary cortical neurons 7 d after transduction with Htt571/72Q-Flag lentivirus, and mHtt hetero-(Q111/+) and homozygous (Q111/Q111) striatal cells were treated with DMSO (ctrl) or 5 μm GW4869 (GW) for 16 h. Concentrated media and cell lysates were analyzed by Western blotting using anti-Htt, anti-Flag, or anti-polyQ antibodies. Tubulin was used as a loading control. Bottom left, Ratio of mHtt in the media and cell lysates expressed as a percentage of control. n = 3, p = 0.0001, 0.0001, 0.0001, 0.0001, and 0.0048. Bottom right, LDH assay was performed on 30 μl of the media from mHtt-Neuro2A cells before concentrating. n = 3, p = 0.1021. B, Left, Neuro2A-mHtt cells were treated with DMSO (ctrl) or GW4869 (GW) for the indicated periods of time. Cells were lysed and 10 μg protein equivalents were analyzed by Western blotting. Tubulin was used as a loading control. Right, Ratios of Htt and tubulin expressed as a percentage of 0 h time point value. n = 4. p = 0.7424, 0.4773, 0.2886, 0.5062, and 0.7390. C, LDH cytotoxicity assay was performed on 50 μl of the media for each time point and the values were normalized to the total cellular protein amount. n = 4. p = 0.9564, 0.2814, 0.3196, 0.5858, and 0.5483. Error bars indicate SD. n.s., Not significant.
We also investigated whether secretion of mHtt might play a role in elimination of mHtt from the cells, potentially through lysosomal exocytosis as a clearance mechanism for accumulated toxic lysosomal substrates (Medina et al., 2011). We thus treated cells with GW4869 to inhibit mHtt secretion and analyzed the intracellular levels of mHtt. However, we found no significant intracellular accumulation of mHtt after prolonged treatment with GW4869 (Fig. 5B), suggesting that mHtt secretion does not lead to a detectable decrease in intracellular levels of the mutant protein. We also did not find any difference in cytotoxicity in treated versus control cells, which would reflect potential accumulation of a toxic protein (Fig. 5C). Together, these data suggest that mHtt lysosomal exocytosis is unlikely to contribute to mHtt clearance in a major way. However, we cannot exclude a possibility that a small degree of mtHtt exocytosis over many years plays a role in clearance of the mutant protein in HD patients.
mHtt secretion is regulated by phosphatidylinositol 3-kinase (PI3K)
Another potent approach to reducing mHtt secretion was through inhibition of PI3K. We found that the PI3K inhibitors Ly294002 and 3-MA were efficient in reducing secretion of both the 571 aa fragment and full-length mHtt in Neuro2A cells, striatal cells, and rat primary cortical neurons (Fig. 6A). Because PI3K regulates lipid composition in the endocytic pathway through the generation of 3′-phosphoinositides, some of which are specifically enriched in endosomes, this finding further supports our data that the endocytic pathway is involved in the secretion of mHtt (Figs. 3, 4). To confirm the efficacy of the PI3K inhibitors (Ly294002 and 3-MA), we took advantage of the fact that phosphatidylinositol 3-phosphate recruits FYVE domain-containing proteins specifically to endosomal membranes (Raiborg et al., 2013). Indeed, both treatments disrupted the normal, punctate pattern of EGFP-2xFYVE fluorescence (Fig. 6B). mHtt secretion was also diminished significantly by shRNA-mediated silencing of Vps34 (Fig. 6C), a class III PI3K, in agreement with the data obtained upon chemical inhibition of the enzyme (Fig. 6A).
Figure 6.

Inhibition of PI3K reduces the secretion of mHtt. A, Reduced secretion of mHtt by inhibition of PI3K. Top, Neuro2A-mHtt cells were treated for 3 h with DMSO (ctrl), 10 mm 3-MA, or 25 μm Ly294002 (Ly); naive Neuro2A cells were transfected with FL Htt, primary cortical neurons were transduced with Htt571/72Q-Flag lentivirus and STHdh+/Hdh+ were transfected with Htt 571/72Q-GFP. Cells were then treated for 3 h with DMSO or Ly294002. Concentrated media and cell lysates were analyzed by Western blotting using anti-Htt, anti-Flag, or anti-GFP antibodies. Tubulin was used as a loading control. Bottom left, Ratio of mHtt in the media and cell lysates expressed as a percentage of control. n = 3, p = 0.0126, 0.0011, 0.0012, 0.0031, and 0.0003. Bottom right, The LDH assay was performed on 30 μl of the media from Neuro2A-mHtt cells before concentrating. n = 3, p = 0.4760 and 0.6717. B, 3-MA or Ly294002 treatment disrupts vesicular targeting of pEGFP-2xFYVE efficiently. Neuro2A-mHtt cells were transfected with pEGFP-2xFYVE. Twenty hours after transfection, the cells were treated for 3 h with DMSO (ctrl), 10 mm 3-MA, or 25 μm Ly294002 (Ly), fixed, and analyzed by confocal microscopy. Nuclei were visualized using DAPI staining. Scale bar, 10 μm. C, Knock down of Vps34 leads to reduced secretion of mHtt. Left, Cells were transfected with scrambled shRNA or shRNAs against Vps34 and incubated for 3–4 d. Media were replaced with OptiMEM 16 h before harvesting of the media and cell lysis. Concentrated media and the cell lysates were analyzed by Western blotting using anti-Htt antibody. Tubulin was used as a loading control. Knock down of Vps34 was verified by probing with anti-Vps34 antibody (bottom). Right, Ratio of mHtt in the media and cell lysates expressed as a percentage of control. n = 3, p = 0.0338, 0.0161, and 0.0170. Error bars indicate SD. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., Not significant.
NS and PI3K regulate vesicular targeting of mHtt
Next, we examined the mechanism of Ly294002- and GW4869-mediated inhibition of mHtt release. If the compounds interfered with the targeting of mHtt to LE/Lys, then we would expect a relative decrease in the light membrane/vesicular mHtt, whereas in the case of inhibited secretion, mHtt would more likely accumulate in this compartment. To address this question, we examined the intensity of punctate mHtt, as well as the relative abundance of mHtt in the light membrane fraction, upon treatments with GW4869 and Ly294002. Both compounds led to a decrease in punctate mHtt (Fig. 7A), as well as decreased levels of mHtt in the light membrane fraction relative to the cytosol (Fig. 7B), suggesting that inhibition of both PI3K and NS hampers vesicular targeting of mHtt and consequently the export of the protein from the cell.
Figure 7.

NS and PI3K inhibition impair vesicular targeting of mHtt. A, Punctate mHtt is reduced by inhibition of NS or PI3K. Neuro2A-mHtt cells were treated for 16 h with DMSO (ctrl) or 5 μm GW4869 (GW) (left) and for 3 h with DMSO or 25 μm Ly294002 (Ly) (right), followed by pre-permeabilization, fixation, immunolabeling with anti-Htt antibody, and analysis by confocal microscopy. DAPI was used to visualize nuclei. Scale bar, 10 μm. Right, Integrated density of mHtt signal per cell expressed as a percentage of control. n = 3, p = 0.0001 and 0.0067. B, mHtt in the light membrane fraction is reduced by inhibition of NS or PI3K. Left, Neuro2A-mHtt cells were treated for 16 h with DMSO (ctrl) or 5 μm GW4869 (GW) and for 3 h with DMSO (ctrl) or 25 μm Ly294002 (Ly). PNS were subjected to floatation in the sucrose density gradient and 2 μg of cytosolic (cyt) and light membrane (LM) protein equivalents were analyzed by Western blotting using anti-Htt antibody. Lamp1 and tubulin were used as LM and cyt markers, respectively. Center, Ratios of mHtt in the LM and cyt expressed as a percentage of control. n = 3, p = 0.0111 and 0.0001. Right, Ratios of mHtt and Lamp1 in the LM expressed as a percentage of control. n = 3, p = 0.0001 and 0.0001. Error bars indicate SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
The goal of this study was to elucidate the cellular mechanism of mHtt secretion. We performed a comprehensive, unbiased analysis of secretory pathways and identified the unconventional lysosomal pathway as an important mechanism of mHtt secretion. We observed elevated secretion of mHtt compared with the wtHtt, which is likely a consequence of the specific targeting of mHtt to LE/Lys. Moreover, we showed that mHtt vesicular targeting and secretion can be decreased efficiently by inhibitors of PI3K and NS.
Several recent studies pointed to the relevance of mHtt secretion and propagation for the HD pathogenesis. For instance, prion-like propagation of mHtt has been suggested in a Drosophila model of HD (Pearce et al., 2015). This study showed that glial cells phagocytose neuronal mHtt aggregates, which then induce prion-like conversion of soluble wtHtt within glial cytoplasm. mHtt spreading has also been found in models in which human wild-type neurons were integrated into brain slices of HD mouse (Pecho-Vrieseling et al., 2014). These human neurons not only internalized mHtt, but also exhibited morphological aberrations. Conversely, an in vivo model has been designed by implanting fibroblasts or iPSCs derived from HD patients into wild-type neonatal mouse brains, which resulted in recapitulation of HD-associated phenotypes in host animals (Jeon et al., 2016). Finally, a 588 aa N-terminal fragment mHtt was expressed selectively in a subset of neurons in Drosophila to reveal spreading of the protein and accompanying non-cell-autonomous pathology in remote brain areas (Babcock and Ganetzky, 2015).
The above studies largely focused on the fate of extracellular mHtt and detrimental effects of its propagation, whereas the mechanism of mHtt secretion has not been analyzed in great detail. mHtt propagation has thus far been linked to extracellular vesicles (Jeon et al., 2016), tunneling nanotubes (Costanzo et al., 2013), or synaptic transmission (Pecho-Vrieseling et al., 2014); however, based on the spreading pattern in a Drosophila model, Babcock and Ganetzky (2015) suggested that mHtt transmission is not limited to neuronal circuits. Our work focused on the initial stages of mHtt propagation to identify the secretion mechanism and examine strategies to manipulate intracellular targeting and release of mHtt.
We first demonstrated the preferential secretion of mHtt because its extracellular levels are elevated compared with the wild-type protein, consistent with the in vivo data showing that mHtt is propagated more efficiently to remote brain areas in a Drosophila model (Babcock and Ganetzky, 2015). Furthermore, mHtt secretion was temperature dependent, suggesting the involvement of active transport. Next, we eliminated the possibility that mHtt follows the conventional secretory pathway and found that extracellular vesicles are not a significant source of the secreted protein, although a small portion was found in exosomal fraction, in agreement with prior studies (Jeon et al., 2016; Zhang et al., 2016). Conversely, mHtt secretion depended on the presence of functional LE/Lys, as shown by the endosome ablation assay and on synaptotagmin 7, a key regulator of lysosomal secretion. Together with the localization of mHtt within Lamp1-positive compartments, these data establish LE/Lys secretion as a significant route for mHtt export from the cell.
We found that mHtt localizes preferentially to LE/Lys compared with wtHtt and that this targeting depends on PI3K and NS. How is mHtt delivered to the LE/Lys lumen? Previous studies indicated that mHtt is subjected to lysosomal degradation via autophagic pathway (Ravikumar et al., 2002; Qin et al., 2003). This would be consistent with our data showing that short treatment with the autophagy inhibitor Ly294002 reduces the levels of lysosomal mHtt. Another study showed that mHtt is targeted to and degraded in the lysosome via chaperone-mediated autophagy (Qi and Zhang, 2014). In addition, it is tempting to speculate that mHtt crosses the LE/Lys limiting membrane through polyglutamine-sequence-mediated membrane penetration (Kegel et al., 2009b; Ren et al., 2009; Burke et al., 2013). This could also explain the preferential LE/Lys targeting of mHtt compared with the wild-type protein. Interestingly, mHtt and wtHtt differ in their affinities for lipids/membranes, especially for phosphoinositides (Kegel et al., 2009a; Kegel-Gleason, 2013). Because we show here that PI3K inhibition reduces the presence of mHtt in the light membrane fraction and in the extracellular medium (Figs. 6, 7), it is possible that the specific binding of mHtt to 3′-phosphoinositides is an important factor for its targeting to LE/Lys. In either of those scenarios, it is possible that a portion of lysosomal mHtt escapes degradation through lysosomal exocytosis.
Lysosomal exocytosis of misfolded proteins may contribute to a disease pathogenesis by promoting clearance of degradation-resistant lysosomal cargo, which would be beneficial for cells harboring mutant proteins. To address this possibility in our experimental system, we took advantage of our most potent inhibitor of mHtt secretion, GW4869. Interestingly, we found neither obvious accumulation of mHtt within the cells nor increased cytotoxicity over extended period of treatment (Fig. 5B,C). This may be due to limited secretion of mHtt or possibly to degradation of the retained mutant protein in cells. Regardless, our data suggest that preventing mHtt secretion is unlikely to have harmful effects on the cells of origin. At the same time, we cannot exclude a possibility that a small degree of mtHtt exocytosis over many years in HD patients contributes to clearance of the mutant protein.
Alternatively, secretion may contribute to toxic propagation of aggregated proteins and development of non-cell-autonomous pathologies. Prion-like self-propagation of misfolded proteins is a common phenomenon in neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and ALS (Soto, 2012; Jucker and Walker, 2013). Proteins implicated in these diseases are secreted via various mechanisms. For instance, in Alzheimer's disease, Aβ can exit the cell via exosomes (Rajendran et al., 2006) and secretory autophagy (Nilsson et al., 2013), whereas Tau follows exosomal (Saman et al., 2012; Simón et al., 2012) and ectosomal secretory pathways (Dujardin et al., 2014). ALS-implicated SOD1 is associated with exosomes (Grad et al., 2014). α-Synuclein is another extensively studied secreted protein relevant to Parkinson's disease and its release depends on exosomes and secretory autophagy (Emmanouilidou et al., 2010; Ejlerskov et al., 2013), similar to Aβ. Understanding the mechanistic details of a misfolded protein secretion is crucial for developing efficient therapeutic strategies. Our study identified two compounds, Ly294002 and GW4869, that interfere with mHtt vesicular targeting/secretion, suggesting a potential strategy for modulating mHtt secretion in neurons.
Footnotes
This work was supported by the National Institutes of Health (NIH Grant R01NS080331 to D.K.). Structured illumination microscopy was performed at the Northwestern University Center for Advanced Microscopy generously supported by the National Cancer Institute Cancer Center Support Grant P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center using a Nikon N-SIM system purchased through the support of NIH Grant 1S10OD016342-01. We thank Jeffrey Savas from Northwestern University for the Flag-Htt plasmid, Yvette Wong from Northwestern University for critical reading of the manuscript, and Karine Laulagnier from Grenoble Institute of Neuroscience and Tatjana Paunesku from Northwestern University for helpful discussions.
The authors declare no competing financial interests.
References
- Abounit S, Zurzolo C (2012) Wiring through tunneling nanotubes–from electrical signals to organelle transfer. J Cell Sci 125:1089–1098. 10.1242/jcs.083279 [DOI] [PubMed] [Google Scholar]
- Andrews NW. (2000) Regulated secretion of conventional lysosomes. Trends Cell Biol 10:316–321. 10.1016/S0962-8924(00)01794-3 [DOI] [PubMed] [Google Scholar]
- Aniento F, Gruenberg J (2003) Subcellular fractionation of tissue culture cells. Curr Protoc Immunol Chapter 8:Unit 8.1C. 10.1002/0471142735.im0801cs57 [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. 10.1038/nature02998 [DOI] [PubMed] [Google Scholar]
- Babcock DT, Ganetzky B (2015) Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc Natl Acad Sci U S A 112:E5427–E5433. 10.1073/pnas.1516217112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC (2013) Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947–959. 10.1016/j.neuron.2013.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ (2015) Huntington disease. Nat Rev Dis Primers 1:15005. 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- Burke KA, Hensal KM, Umbaugh CS, Chaibva M, Legleiter J (2013) Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner. Biochim Biophys Acta 1828:1953–1961. 10.1016/j.bbamem.2013.04.025 [DOI] [PubMed] [Google Scholar]
- Chen S, Ferrone FA, Wetzel R (2002) Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A 99:11884–11889. 10.1073/pnas.182276099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Lacroix S, Cisbani G, Vallières N, Saint-Pierre M, St-Amour I, Tolouei R, Skepper JN, Hauser RA, Mantovani D, Barker RA, Freeman TB (2014) Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol 76:31–42. 10.1002/ana.24174 [DOI] [PubMed] [Google Scholar]
- Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 126:3678–3685. 10.1242/jcs.126086 [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127:59–69. 10.1016/j.cell.2006.09.015 [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL (2011) ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol 12:362–375. 10.1038/nrm3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S, Bégard S, Caillierez R, Lachaud C, Delattre L, Carrier S, Loyens A, Galas MC, Bousset L, Melki R, Aurégan G, Hantraye P, Brouillet E, Buée L, Colin M (2014) Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One 9:e100760. 10.1371/journal.pone.0100760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296:2238–2243. 10.1126/science.1072613 [DOI] [PubMed] [Google Scholar]
- Ejlerskov P, Rasmussen I, Nielsen TT, Bergström AL, Tohyama Y, Jensen PH, Vilhardt F (2013) Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 288:17313–17335. 10.1074/jbc.M112.401174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K (2010) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30:6838–6851. 10.1523/JNEUROSCI.5699-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A 111:3620–3625. 10.1073/pnas.1312245111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon I, Cicchetti F, Cisbani G, Lee S, Li E, Bae J, Lee N, Li L, Im W, Kim M, Kim HS, Oh SH, Kim TA, Ko JJ, Aubé B, Oueslati A, Kim YJ, Song J (2016) Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol 132:577–592. 10.1007/s00401-016-1582-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D (2009) Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137:60–72. 10.1016/j.cell.2009.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51. 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Sapp E, Alexander J, Valencia A, Reeves P, Li X, Masso N, Sobin L, Aronin N, DiFiglia M (2009a) Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J Neurochem 110:1585–1597. 10.1111/j.1471-4159.2009.06255.x [DOI] [PubMed] [Google Scholar]
- Kegel KB, Schewkunow V, Sapp E, Masso N, Wanker EE, DiFiglia M, Goldmann WH (2009b) Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem Biophys Res Commun 387:472–475. 10.1016/j.bbrc.2009.07.039 [DOI] [PubMed] [Google Scholar]
- Kegel-Gleason KB. (2013) Huntingtin interactions with membrane phospholipids: strategic targets for therapeutic intervention? J Huntingtons Dis 2:239–250. 10.3233/JHD-130068 [DOI] [PubMed] [Google Scholar]
- Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M (1999) Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci 19:964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J (1992) brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol 116:1071–1080. 10.1083/jcb.116.5.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laulagnier K, Schieber NL, Maritzen T, Haucke V, Parton RG, Gruenberg J (2011) Role of AP1 and Gadkin in the traffic of secretory endo-lysosomes. Mol Biol Cell 22:2068–2082. 10.1091/mbc.E11-03-0193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Miller EA, Goldberg J, Orci L, Schekman R (2004) Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol 20:87–123. 10.1146/annurev.cellbio.20.010403.105307 [DOI] [PubMed] [Google Scholar]
- Mahajan SS, Little MM, Vazquez R, Wilson AC (2002) Interaction of HCF-1 with a cellular nuclear export factor. J Biol Chem 277:44292–44299. 10.1074/jbc.M205440200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J, McEachern KA, Kyros JA, Nietupski JB, Budzinski T, Ziegler RJ, Yew NS, Sullivan J, Scaria A, van Rooijen N, Barranger JA, Cheng SH (2002) Demonstration of feasibility of in vivo gene therapy for Gaucher disease using a chemically induced mouse model. Mol Ther 6:179–189. 10.1006/mthe.2002.0650 [DOI] [PubMed] [Google Scholar]
- Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW (2000) Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol 148:1141–1149. 10.1083/jcb.148.6.1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. 10.1016/j.cell.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 21:421–430. 10.1016/j.devcel.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W. (2011) The unconventional secretory machinery of fibroblast growth factor 2. Traffic 12:799–805. 10.1111/j.1600-0854.2011.01187.x [DOI] [PubMed] [Google Scholar]
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC (2013) Abeta secretion and plaque formation depend on autophagy. Cell Rep 5:61–69. 10.1016/j.celrep.2013.08.042 [DOI] [PubMed] [Google Scholar]
- Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR (2015) Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun 6:6768. 10.1038/ncomms7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, Esposito MS, Botta P, Goldstein C, Bernhard M, Galimberti I, Müller M, Lüthi A, Arber S, Bouwmeester T, van der Putten H, Di Giorgio FP (2014) Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci 17:1064–1072. 10.1038/nn.3761 [DOI] [PubMed] [Google Scholar]
- Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V (2015) Secretory autophagy. Curr Opin Cell Biol 35:106–116. 10.1016/j.ceb.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Zhang XD (2014) Role of chaperone-mediated autophagy in degrading Huntington's disease-associated huntingtin protein. Acta Biochim Biophys Sin 46:83–91. 10.1093/abbs/gmt133 [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M (2003) Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet 12:3231–3244. 10.1093/hmg/ddg346 [DOI] [PubMed] [Google Scholar]
- Rabouille C, Malhotra V, Nickel W (2012) Diversity in unconventional protein secretion. J Cell Sci 125:5251–5255. 10.1242/jcs.103630 [DOI] [PubMed] [Google Scholar]
- Raiborg C, Schink KO, Stenmark H (2013) Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J 280:2730–2742. 10.1111/febs.12116 [DOI] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simóns K (2006) Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A 103:11172–11177. 10.1073/pnas.0603838103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11:1107–1117. 10.1093/hmg/11.9.1107 [DOI] [PubMed] [Google Scholar]
- Régulier E, Trottier Y, Perrin V, Aebischer P, Déglon N (2003) Early and reversible neuropathology induced by tetracycline-regulated lentiviral overexpression of mutant huntingtin in rat striatum. Hum Mol Genet 12:2827–2836. 10.1093/hmg/ddg305 [DOI] [PubMed] [Google Scholar]
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 11:219–225. 10.1038/ncb1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. 10.1016/S1474-4422(10)70245-3 [DOI] [PubMed] [Google Scholar]
- Rothaug M, Stroobants S, Schweizer M, Peters J, Zunke F, Allerding M, D'Hooge R, Saftig P, Blanz J (2015) LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol Commun 3:6. 10.1186/s40478-014-0182-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF (2012) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287:3842–3849. 10.1074/jbc.M111.277061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón D, García-García E, Royo F, Falcón-Pérez JM, Avila J (2012) Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett 586:47–54. 10.1016/j.febslet.2011.11.022 [DOI] [PubMed] [Google Scholar]
- Song P, Trajkovic K, Tsunemi T, Krainc D (2016) Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J Neurosci 36:2425–2437. 10.1523/JNEUROSCI.2569-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C. (2012) Transmissible proteins: expanding the prion heresy. Cell 149:968–977. 10.1016/j.cell.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel W. (1998) Analysis of the endocytic system by using horseradish peroxidase. Trends Cell Biol 8:503–505. 10.1016/S0962-8924(98)01380-4 [DOI] [PubMed] [Google Scholar]
- Tan Z, Dai W, van Erp TG, Overman J, Demuro A, Digman MA, Hatami A, Albay R, Sontag EM, Potkin KT, Ling S, Macciardi F, Bunney WE, Long JD, Paulsen JS, Ringman JM, Parker I, Glabe C, Thompson LM, Chiu W, Potkin SG (2015) Huntington's disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol Psychiatry 20:1286–1293. 10.1038/mp.2015.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9:2799–2809. 10.1093/hmg/9.19.2799 [DOI] [PubMed] [Google Scholar]
- Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, Weber C, Agid Y, Hirsch EC, Mandel JL (1995) Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 10:104–110. 10.1038/ng0595-104 [DOI] [PubMed] [Google Scholar]
- Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, Miller JR, Zetterberg H, Leavitt BR, Kuhn R, Tabrizi SJ, Macdonald D, Weiss A (2015) Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest 125:1979–1986. 10.1172/JCI80743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Schekman R (2013) Cell biology: unconventional secretion, unconventional solutions. Science 340:559–561. 10.1126/science.1234740 [DOI] [PubMed] [Google Scholar]
- Zhang X, Abels ER, Redzic JS, Margulis J, Finkbeiner S, Breakefield XO (2016) Potential transfer of polyglutamine and CAG-repeat RNA in extracellular vesicles in Huntington's disease: background and evaluation in cell culture. Cell Mol Neurobiol 36:459–470. 10.1007/s10571-016-0350-7 [DOI] [PMC free article] [PubMed] [Google Scholar]