

Abstract
Cell type-specific changes in neuronal excitability have been proposed to contribute to the selective degeneration of corticospinal neurons in amyotrophic lateral sclerosis (ALS) and to neocortical hyperexcitability, a prominent feature of both inherited and sporadic variants of the disease, but the mechanisms underlying selective loss of specific cell types in ALS are not known. We analyzed the physiological properties of distinct classes of cortical neurons in the motor cortex of hSOD1G93A mice of both sexes and found that they all exhibit increases in intrinsic excitability that depend on disease stage. Targeted recordings and in vivo calcium imaging further revealed that neurons adapt their functional properties to normalize cortical excitability as the disease progresses. Although different neuron classes all exhibited increases in intrinsic excitability, transcriptional profiling indicated that the molecular mechanisms underlying these changes are cell type specific. The increases in excitability in both excitatory and inhibitory cortical neurons show that selective dysfunction of neuronal cell types cannot account for the specific vulnerability of corticospinal motor neurons in ALS. Furthermore, the stage-dependent alterations in neuronal function highlight the ability of cortical circuits to adapt as disease progresses. These findings show that both disease stage and cell type must be considered when developing therapeutic strategies for treating ALS.
SIGNIFICANCE STATEMENT It is not known why certain classes of neurons preferentially die in different neurodegenerative diseases. It has been proposed that the enhanced excitability of affected neurons is a major contributor to their selective loss. We show using a mouse model of amyotrophic lateral sclerosis (ALS), a disease in which corticospinal neurons exhibit selective vulnerability, that changes in excitability are not restricted to this neuronal class and that excitability does not increase monotonically with disease progression. Moreover, although all neuronal cell types tested exhibited abnormal functional properties, analysis of their gene expression demonstrated cell type-specific responses to the ALS-causing mutation. These findings suggest that therapies for ALS may need to be tailored for different cell types and stages of disease.
Keywords: corticocortical neuron, corticospinal neuron, interneuron, intrinsic excitability, RNA sequencing, two-photon imaging
Introduction
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive neurodegenerative disease characterized by progressive loss of corticospinal neurons (CSNs), brainstem motor neurons, and spinal motor neurons, leading to increasing weakness and ultimately to death. A common feature of both sporadic and familial forms of ALS is cortical hyperexcitability (Vucic and Kiernan, 2006; Vucic et al., 2008; Williams et al., 2013; Geevasinga et al., 2015). Patients exhibit cortical hyperexcitability early in the clinical disease course (Vucic et al., 2008; Geevasinga et al., 2015), and the degree of neuronal hyperexcitability is inversely correlated with survival (Kanai et al., 2012; Shibuya et al., 2016). Interventions that decrease neuronal hyperexcitability modestly slow disease progression and increase motor neuron survival (Bensimon et al., 1994; Wainger et al., 2014). However, recent studies in model systems suggesting that increases in neuronal excitability may be protective (Saxena et al., 2013; Leroy et al., 2014) raise the possibility that therapeutic interventions may need to be tailored to cell type or disease state.
Although the pathogenesis of ALS is likely multifactorial, it has been hypothesized that enhanced excitation or persistent depolarization of CSNs contributes to their selective degeneration in both sporadic and inherited forms of ALS (Saxena and Caroni, 2011; Taylor et al., 2016). Previous studies have primarily focused on assessing the function of spinal motor neurons from mouse models or from patient-derived cells, and a number, though not all, have identified increases in excitability during embryonic and neonatal stages (Pieri et al., 2003; Kuo et al., 2004; van Zundert et al., 2008; Elbasiouny et al., 2012; Martin et al., 2013; Leroy et al., 2014; Wainger et al., 2014; Venugopal et al., 2015), followed by progressive loss of action potential output (Delestrée et al., 2014; Devlin et al., 2015). Although altered excitability has been detected in cortical neurons (Pieri et al., 2009; Wainger and Cudkowicz, 2015; Geevasinga et al., 2016; Saba et al., 2016), it is not known whether these changes in excitability are restricted to CSNs or represent more global changes in cortical physiology. Moreover, it is unclear whether these changes progress monotonically or vary with disease progression.
Using hSOD1G93A mice that closely mimic the human disease (Gurney et al., 1994), we discovered that increases in intrinsic excitability were not restricted to CSNs but occurred in all excitatory and inhibitory cell types examined. Although changes in excitability were detected as early as a few days after birth, the intrinsic properties of cortical neurons largely normalized in juvenile mice before these neurons ultimately become hyperexcitable again at end stage, indicating that cortical neurons adapt their responsiveness during the course of disease. Two-photon calcium imaging revealed that increases in intrinsic excitability did not translate into neuronal hyperactivity in vivo, highlighting the ability of cortical circuits to adapt to abnormal cellular function. Despite this normalized activity, CSNs exhibited higher resting calcium levels in vivo, suggesting that they remain functionally compromised. Transcriptional profiling of CSNs and corticocortical neurons (CCNs) in neonatal mice revealed distinct molecular responses, indicating that cortical cell types are differentially affected by the expression of mutant SOD1 even at this earliest time point. Our results also revealed that these functional changes in cortical neurons do not follow a linear progression of increasing hyperexcitability with age. Rather, the properties of cortical neurons were dynamically altered during different stages of disease. These findings provide insights into the cellular mechanisms underlying cortical hyperexcitability, a common phenotype across disease variants, including sporadic, SOD1, and C9orf72 disease, and have important implications for the timing of therapeutic interventions in ALS.
Materials and Methods
Animals.
All experimental procedures were approved by the Johns Hopkins Animal Care and Use Committee or the Institutional Animal Care and Use Committee of Harvard University and were conducted in accordance with the guidelines of the National Institutes of Health and the Society for Neuroscience. hSOD1G93A (B6SJL-Tg(hSOD1*G93A)1Gur/J; https://www.jax.org/strain/002726) and hSOD1WT (B6SJL-Tg(hSOD1*WT)2Gur/J; https://www.jax.org/strain/002297) male mice were regularly obtained from The Jackson Laboratory and bred with the following transgenic strains: Rbp4-Cre (Gerfen et al., 2013; RRID:MMRRC_031125-UCD); loxP-STOP-loxP-tdTomato Cre reporter lines [Madisen et al., 2010; Ai9 (https://www.jax.org/strain/007909) and Ai14 (https://www.jax.org/strain/007908)]; a Gad67-GFP line [Chattopadhyaya et al., 2004; G42 (https://www.jax.org/strain/007677)]; and a Pvalb-Cre line (Hippenmeyer et al., 2005; https://www.jax.org/strain/008069). Mice were housed up to five mice per cage under a 12 h light/dark cycle and given ad libitum access to food and water. For targeted recordings of CSNs and CCNs on postnatal day 4 (P4) to P6 mice, Rbp4-Cre mice were first crossed with LSL-tdTomato mice to generate Rbp4-Cre;tdTomato mice. Subsequently, hSOD1G93A males were crossed with Rbp4-Cre;tdTomato females to generate hSOD1G93A;Rbp4-Cre;tdTomato and Rbp4-Cre;tdTomato mice. The Gad67-GFP line crossed with hSOD1G93A mice was used to target fast-spiking parvalbumin (PV)-positive interneurons for recording. As we used several transgenic lines, we confirmed that the life expectancy of the mutant mice was similar to that of the B6SJL-Tg(hSOD1*G93A)1Gur/J line (hSOD1G93A;Rbp4-Cre;tdTomato: 126.8 ± 3.1 d, n = 15 mice; hSOD1G93A;Gad67-GFP: 130.4 ± 1.7 d, n = 17 mice; hSOD1G93A: 132 ± 1.3 d; Gurney, 1997). Mice of either sex were used in this study. All experiments were performed with the experimenters blinded to the genotypes of the mice.
Stereotaxic injections of neuronal tracers and viral vectors.
Mice were anesthetized with ketamine (50 mg/kg), dexmedetomidine (25 μm/kg) and the inhalation anesthetic isoflurane (1–3%). To label CSNs, a small laminectomy (C5–C6) was performed over the left cervical spinal cord, and 100–200 nl of retrograde neuronal tracer (red Retrobeads or green Retrobeads IX, Lumafluor, or Alexa Fluor 488- or Alexa Fluor 555-conjugated cholera toxin B, Invitrogen) were pressure injected through a glass pipette (tip diameter, 15–25 μm; Drummond), 500 μm lateral to the midline of the exposed spinal cord at a depth of 1000 μm. To label CCNs, a retrograde neuronal tracer was injected in 9–16 sites in the left motor cortex (600–1800 μm anterior and 1500–2500 μm lateral to bregma; 800 μm below the cortical surface). The analgesic buprenorphine (0.05 mg/kg) was administered to all animals postoperatively. To compare the morphology of CSNs and CCNs at different experimental ages (see Fig. 6B,D), a canine adenoviral vector carrying a construct for Cre recombinase (CAV-Cre; Optogenetics and Transgenic Technology Core, National Institute on Drug Abuse, Bethesda, MD) was injected into the spinal cord or motor cortex of Cre-dependent tdTomato reporter mice in a manner similar to that of the tracers to visualize CSNs or CCNs, respectively (Kremer et al., 2000; Soudais et al., 2001). Mice were killed 2–10 d after tracer injections and 20–30 d after injections of CAV-Cre.
Figure 6.

Cre expression in L5b of Rbp4-Cre mice distinguishes corticospinal and corticocortical neurons. A, Low- and high-magnification images of biocytin-filled L5b tdTomato-positive cells from a P5 Rbp4-Cre;tdTomato mouse. B, Schematic showing the experimental setup (left) and low- and high-magnification images of a corticospinal neuron (right) revealed by injecting a canine adenoviral vector carrying a construct for Cre recombinase (CAV-Cre) into the cervical spinal cord of a P90 tdTomato Cre-reporter mouse. C, Low- and high-magnification images of a biocytin-filled L5b tdTomato-negative neuron from a P5 Rbp4-Cre;tdTomato mouse. D, Schematic showing the experimental setup (left) and low- and high-magnification images of a corticocortical neuron (right) revealed by injecting CAV-Cre into the contralateral motor cortex of a P90 tdTomato Cre-reporter mouse. E, F, Summary data showing the input resistance and sag amplitude for L5b tdTomato-positive and tdTomato-negative neurons recorded from P5–P6 Rbp4-Cre;tdTomato mice (E, n = 6 tdTomato-positive neurons from 3 mice; n = 17 tdTomato-negative neurons from 7 mice; input resistance: L5b tdTomato-positive neurons, 220.2 ± 39.4 MΩ; L5b tdTomato-negative neurons, 460.3 ± 24.7 MΩ; p = 0.0009, Mann–Whitney test; Sag amplitude: L5b tdTomato-positive neurons, 4.9 ± 1.1 mV; tdTomato-negative neurons, 1.8 ± 0.2 mV; p = 0.0037, Mann–Whitney test) and P90–P100 retrogradely labeled CSNs and CCNs (F; n = 26 CSNs from 8 mice; n = 23 CCNs from 8 mice; input resistance: CSNs, 50.8 ± 2.7 MΩ; CCNs, 79.5 ± 3.5 MΩ; p < 0.0001, Mann–Whitney test; Sag amplitude: CSNs, −0.8 ± 0.1 mV; CCNs, −0.4 ± 0.1 mV; p < 0.0001, Mann–Whitney test). Scale bars: A, C, left, 100 μm; A, C, middle, 50 μm; A, C, right, 30 μm; B, D, left, 100 μm; B, D, right, 50 μm.
Brain slice preparation and cell identification.
Acute brain slices were generated for in vitro electrophysiological recording largely as previously described except for the slice orientation (Kim et al., 2014). Mice were anesthetized with isoflurane and decapitated, and the brains were rapidly removed and chilled in ice-cold sucrose solution containing the following (in mm): 76 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, and 7 MgSO4, pH 7.3. After an initial coronal cut, brains were mounted on the cut face tilted anteriorly at a 15° angle and acute brain slices (300 μm) were prepared using a vibratome (VT-1200s, Leica). Slices were then incubated in warm (32–35°C) sucrose solution for 30 min and then transferred to warm (32–34°C) artificial CSF (aCSF) composed of (in mm): 125 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 20 glucose, 2 CaCl2, 0.4 ascorbic acid, 2 pyruvic acid, and 4 L-(+)-lactic acid, pH 7.3, 315 mOsm, and allowed to cool to room temperature (RT). All solutions were continuously bubbled with 95% O2/5% CO2.
For whole-cell recordings, slices were transferred to a submersion chamber on an upright microscope (AxioExaminer, Zeiss; objectives: 5×, 0.16 numerical aperture (NA); and 40×, 1.0 NA) fitted for infrared differential interference contrast and fluorescence microscopy. Slices were continuously superfused (2–4 ml/min) with warm oxygenated aCSF (32–34°C). Neurons were visualized with a digital camera (Sensicam QE, Cooke) using either transmitted light or epifluorescence. In slices from P26–P129 mice, CSNs or CCNs were identified based on the presence of the fluorescent retrograde tracers. In slices from P4–P6 mice, CSNs or CCNs were identified based on the presence of the tdTomato fluorescence in layer 5b (L5b) of motor cortex of Rbp4-Cre;tdTomato mice. PV interneurons were identified based on their green fluorescent protein (GFP) expression in Gad67-GFP mice.
Whole-cell recordings and analysis.
Glass recording electrodes (2–4 MΩ) were filled with an internal solution containing the following (in mm): 2.7 KCl, 120 KMeSO4, 9 HEPES, 0.18 EGTA, 4 MgATP, 0.3 NaGTP, and 20 phosphocreatine(Na), pH 7.3, 295 mOsm. Biocytin (0.25% weight/volume) was added to the internal solution. Whole-cell patch-clamp recordings were obtained using a Multiclamp 700B amplifier (Molecular Devices) and digitized using an ITC-18 data acquisition interface (Instrutech) controlled by software written in Igor Pro (WaveMetrics; RRID:SCR_000325). The mean (±SD) series resistance was 10.9 ± 5.4 MΩ (n = 675 neurons; all <30 MΩ) and was not compensated for. All electrophysiological recordings were performed in the presence of the following blockers of glutamate and GABA receptors: 5 μm NBQX (AMPA receptor antagonist), 5 μm (RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (NMDA receptor antagonist), and 10 μm 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide (SR95531; GABAA receptor antagonist; all from Tocris Bioscience). This concentration of SR95531 blocks synaptic as well as extrasynaptic GABA receptors (Farrant and Nusser, 2005). The resting membrane potential was measured after whole-cell configuration was achieved. Neurons exhibiting a resting membrane potential greater than −60 mV in slices from P90–P129 mice were excluded from the analysis. As neurons in slices from P4–P6 and P26–P40 mice were more depolarized, a cutoff of −55 mV was used. One cell was eliminated from the rheobase analysis as it began firing spontaneous action potentials. The input resistance was determined by measuring the voltage change in response to a 1 s hyperpolarizing current step (−25 to −100 pA for cortical neurons from P4–P6 mice and −100 pA for cortical neurons from P26–P129 mice). The amplitude of the sag response was calculated as the difference between the minimum membrane potential following the initial current step and the steady-state membrane potential at the end of the 1 s step (P5–P6, −50 pA; P90–P100, −100 pA; see Fig. 6E,F). The current–spike frequency relationship was measured with a range of depolarizing current steps presented in pseudorandom order (1 s long, 40 pA increments, 5 s interstimulus intervals). Each current intensity was tested a total of three times. For each current intensity, the total number of action potentials exceeding 0 mV generated during each step was measured and then averaged across the three trials. The rheobase was determined by first probing the response of the neuron with 1-s-long depolarizing steps (5 s interstimulus intervals) to define a small range of current steps that bounded the rheobase. The response of the neurons was then tested within this range using 1-s-long depolarizing steps with 1 pA increments. Action potential properties were measured from single spikes evoked by rheobase current injections. Signals were low-pass filtered at 10 kHz and sampled at 20–100 kHz.
Anatomical identification of cell types.
To compare the morphology of tdTomato-positive and tdTomato-negative neurons in neonatal Rbp4-Cre;tdTomato mice, neurons were filled with biocytin during whole-cell recordings in acute slices. Slices were then fixed in 4% paraformaldehyde (PFA) in 0.1 m PBS for at least 3 h and were then rinsed with PBS, incubated in 2% Triton X-100 (Sigma-Aldrich) in PBS for 1 h at RT, incubated with Alexa Fluor 488-conjugated streptavidin (1:200; catalog #S32354, Invitrogen; RRID:AB_2315383) for 48 h at 4°C, rinsed again, and mounted in VECTASHIELD (Vector Laboratories). The morphology of CSNs and CCNs in adult mice transfected with the CAV-Cre virus in P61–P64 tdTomato reporter mice was revealed by first fixing the brains in 4% PFA in PBS. Then 100 μm coronal sections were cut in the same plane as for the electrophysiological recordings (coronal sections tilted 15° anteriorly) using a vibratome (VT-1000s, Leica). Sections were then rinsed in PBS, incubated with blocking buffer containing 0.25% Triton X-100 (Sigma-Aldrich) and 5% normal goat serum (Invitrogen) in PBS for 1 h at RT, and then incubated with rabbit anti-RFP antibody (1:500; catalog #632496, Clontech Laboratories; RRID:AB_10013483) overnight at 4°C. The sections were then rinsed again in PBS, incubated with Alexa Fluor 568-conjugated goat anti-rabbit secondary antibody (1:300; catalog #A-11011, Invitrogen; RRID:AB_143157) for 2 h at RT, rinsed again in PBS, and mounted in VECTASHIELD. Images of CSNs and CCNs were taken with a confocal microscope (LSM 510, Zeiss) using either 10× (0.3 NA) or 20× (0.8 NA) objectives. Images were analyzed, and the contrast and brightness were globally adjusted in ImageJ (National Institutes of Health; RRID:SCR_003070) and LSM Image Browser (Zeiss; RRID:SCR_014344).
Two-photon in vivo imaging.
To express GCaMP6s (GENIE Project, Janelia Research Campus, Howard Hughes Medical Institute, Janelia Farms, VA) selectively in CSNs, a CAV-Cre viral vector was injected into the spinal cord as described above for the neuronal tracers and an adeno-associated viral vector, AAV9-Syn-Flex-GCaMP6s (University of Pennsylvania Vector Core, Philadelphia, PA), was injected into the left motor cortex. The GCaMP6s virus was injected using the following coordinates (from bregma) to allow for easier placement of the imaging window: AP, −0.2 mm; ML, 1.5 mm; DV, 0.8 mm. The viruses were injected at P51–P72. Cranial windows were implanted using methods previously described (Hughes et al., 2013). Briefly, mice were anesthetized with isoflurane (5% induction; 1.5–2% maintenance), and body temperature was maintained at 37°C with a thermostat-controlled heating pad. The skin overlying the skull was retracted, and the skull was cleaned and dried. A 2 × 2 mm craniotomy was performed using a high-speed drill with the craniotomy centered at the following coordinates (from bregma): AP, −1 mm; ML, 1.5 mm. Following the craniotomy, a 2 × 2 mm glass coverslip (No. 1, VWR) was placed within the craniotomy and sealed using dental cement (C&B Metabond, Parkell). A custom-designed aluminum head plate was then affixed to the skull using dental cement to allow for head stabilization. The entire procedure required 40–60 min. Animals regained normal locomotion and were allowed to recover for a minimum of 3.5 h before in vivo imaging was commenced. The head of the mouse was immobilized by attaching the head plate to a custom-machined stage, and images were collected during awake resting periods. Images were collected using a Zeiss model 710 microscope using a mode-locked Ti:Sapphire laser (Ultra II, Coherent) tuned to 920 nm. The average power at the sample did not exceed 30 mW. Images were acquired at 5.6 frames/s with a 256 × 256 pixel resolution at a depth of 250 μm from the pial surface.
Because the cell bodies of CSNs are located deep in L5b, it was difficult to clearly image somatic calcium events. Therefore, we focused on imaging apical dendritic stalks, which show a strong linear correlation between AP firing and calcium activity (Hill et al., 2013). For neurons with multiple apical dendritic stalks at the imaging plane (250 μm beneath the pial surface), calcium signaling events were analyzed blinded to cell identity, and correlation coefficients were subsequently determined using a MATLAB (R2015a, MathWorks; RRID:SCR_001622) function, corrcoef, for dendritic stalks from the same neuron identified by visualization of the joining of multiple branches into a single stalk (hSOD1G93A, n = 18 cells; control, n = 17 cells) or a soma (hSOD1G93A, n = 2 cells; control, n = 1 cell). Calcium signals in apical dendritic stalks of the same neuron were highly correlated (hSOD1G93A: 0.981 ± 0.009, n = 20 cells from 7 mice; control: 0.989 ± 0.009, n = 18 cells from 10 mice), indicating that these events represent cell-wide calcium events and likely represent somatic action potentials (Hill et al., 2013). All CSNs were therefore imaged at ∼250 μm from the pial surface. In cells with multiple dendritic stalks, time series represent an average of the dendritic stalks from an individual neuron. CSNs were imaged at P92–P134 when CSNs from hSOD1G93A mice exhibited significantly increased intrinsic excitability, as measured by comparing the current–spike frequency relationships and the maximum action potential frequency (Fmax) values relative to those of control mice (Fig. 1D,F).
Figure 1.

Corticospinal and corticocortical neurons are hyperexcitable in symptomatic hSOD1G93A mice. A, Schematic of the experimental design. Alexa Fluor 555-conjugated cholera toxin B (red CTB) was injected into the left cervical spinal cord, and Alexa Fluor 488-conjugated cholera toxin B (green CTB) was injected into the left motor cortex to label CSNs and CCNs, respectively. B, Confocal images of retrogradely labeled CSNs (magenta) and CCNs (green) in L5b of motor cortex. C, Representative voltage traces recorded from CSNs from P90–P129 hSOD1G93A and WT mice evoked by 360, 480, and 600 pA current steps. D, The current–spike frequency relationship measured from CSNs from hSOD1G93A and WT mice (hSOD1G93A, n = 64 cells from 19 mice; WT, n = 46 cells from 12 mice; p < 3.58 × 10−11, Wald test). E, The rheobase measured from CSNs (hSOD1G93A, 259.45 ± 11.46 pA; WT, 297.98 ± 15.27 pA; p = 0.101, Mann–Whitney test). F, The Fmax measured from CSNs (hSOD1G93A, 37.62 ± 1.36 Hz; WT, 32.04 ± 1.8 Hz; p = 0.0198, Mann–Whitney test). G, H, Representative voltage traces (G), and the current–spike frequency relationship (H) recorded from CCNs from P90–P129 hSOD1G93A and WT mice (hSOD1G93A, n = 50 cells from 19 mice; WT, n = 36 cells from 11 mice; p < 2 × 10−16, Wald test). I, The rheobase measured from CCNs (hSOD1G93A, 201.92 ± 10.53 pA; WT, 318.83 ± 14.76 pA; p < 0.0001, Mann–Whitney test). J, The Fmax measured from CCNs (hSOD1G93A, 41.3 ± 2.18 Hz; WT, 27 ± 2.13 Hz; p < 0.0001, Mann–Whitney test).
Image analysis.
Image analysis was performed using Origin (versions 7 and 8, OriginLab; RRID: SC_014212), FIJI (1.46r; RRID:SCR_002285), MiniAnalysis (Synaptosoft; RRID:SCR_014441), Excel 2011 (Microsoft), and MATLAB (R2015a, MathWorks). Images were registered using the Turboreg plugin of FIJI. An average projection of the field of view across the entire time series was used to place regions of interest (ROIs) centered on dendritic stalks. F, representing the mean fluorescence value of all the pixels within the ROI, was calculated for each time point. To remove slow changes in the raw fluorescence time series, the eighth percentile of the fluorescence distribution in a 15 s window was subtracted for each time point in MATLAB (Dombeck et al., 2007). Then the baseline fluorescence of the ROI, F0, was estimated as the 30th percentile of the fluorescence distribution of the entire time series and used to calculate ΔF/F0 = ((F − F0)/F0). Calcium signaling events were detected using the MiniAnalysis software. Event characteristics were further analyzed using custom scripts in Excel. To calculate the time course of the decay of individual calcium events, traces of isolated events were fit with an exponential decay function (ExpDecay2; Origin). Statistical analyses were performed using Origin.
To calculate baseline dendritic calcium, a 10 s epoch was manually identified midway through the time-series acquisition (2.5–3.5 min) without a calcium signaling event. The average intensity of this epoch was calculated, and the resulting image was smoothed with a Gaussian filter (σ = 1). A Shanbhag threshold was used to automatically define an ROI within the dendritic process. The baseline fluorescence for each dendrite was determined as the mean fluorescence value of all of the pixels within the ROI during the 10 s epoch for the tdTomato and the GCaMP6s signals.
Neuronal cell-type labeling, enrichment, and RNA isolation.
hSOD1G93A and hSOD1WT P5–P7 mice were used for expression profiling. For each biological replicate, the cortex was isolated and dissociated into a single-cell suspension as previously described (Molyneaux et al., 2015). Cells were then immediately fixed in 4% PFA, incubated in RNase-free conditions with antibodies against the nuclear markers CTIP2 and SATB2: rat anti-CTIP2 (1:100; catalog #ab18465, Abcam; RRID:AB_2064130), mouse anti-SATB2 (1:250; catalog #ab51502, Abcam; RRID:AB_882455), Alexa Fluor 546 goat anti-rat (1:1000; catalog #A11081, Invitrogen; RRID:AB_141738), Alexa Fluor 488 goat anti-mouse (1:1000; catalog #A11011, Invitrogen; RRID:AB_2534069), and FACS sorted as previously described to isolate SATB2-positive and CTIP2-positive neurons (Molyneaux et al., 2015). RNA was recovered from the FACS-purified cells as previously described (Molyneaux et al., 2015), quantified by spectral absorbance on the NanoDrop, and qualitatively assessed on the Agilent Bioanalyzer RNA-6000 Pico chip. RNA integrity numbers for all samples ranged between 8.3 and 10.
Sequencing library preparation and analysis.
For each enriched sample, 10 ng of total RNA was reverse transcribed to synthesize cDNA using the Smart seq2 V4 kit (Clontech). Amplified cDNA was used as input for the high-throughput RNA sequencing library preparation using the Nextera XT library kit (Illumina). Individual libraries were indexed, pooled, and sequenced to a mean depth of 5.466 × 107-125 bp paired-end reads per sample. Reads were aligned to the mouse genome (mm9) using TopHat2 (Kim et al., 2013) with the following additional parameters: --no-coverage-search -max-multihits = 10 -p 16. Gene expression was quantified using cuffquant (Trapnell et al., 2013) against a previously assembled reference transcriptome consisting of UCSC genes supplemented with mouse cortical long noncoding RNAs (Molyneaux et al., 2015). Differential expression testing was performed using cuffdiff version 2.2.1 (RRID:SCR_001622), and data were analyzed and visualized using cummeRbund (Trapnell et al., 2013). Additional custom scripts and visualizations were also developed using the cummeRbund framework in R/Bioconductor.
For each cell type, genes were ranked according to their Cuffdiff2 test statistic, which integrates both confidence in the differential expression between hSOD1G93A and hSOD1WT as well as the direction and magnitude of the differential effect. These rankings were then used as input for a preranked Gene Set Enrichment Analysis (GSEA; Subramanian et al., 2005) against the Reactome database (MSigDB version 5.1 c2.cp.Reactome), a publicly available collection of annotated gene sets, to identify pathways and processes with coordinated significant differential enrichment. Gene sets were called significant with a Benjamini–Hochberg corrected p value of <0.05 (Kolmogorov–Smirnov test). The network map of significant GSEA results was assembled using the Enrichment Map tool in Cytoscape version 3.4.0 (RRID:SCR_003032; Merico et al., 2010).
Electrophysiology data analysis.
Data analysis was performed in Igor Pro (WaveMetrics), Excel (Microsoft), SigmaPlot (Systat Software; RRID:SCR_003210), Origin (OriginLab), MiniAnalysis (Synaptosoft), Clampfit (Molecular Devices), MATLAB (MathWorks), and R Project for Statistical Computing (RRID:SCR_001905) using the mgcv package (Wood, 2011). The rheobase, input resistance, resting membrane potential, spike properties, and calcium measurements recorded from hSOD1G93A and control mice were compared with Mann–Whitney tests. To compare current–frequency relationships of neurons recorded from hSOD1G93A and control mice, we used two approaches. First, we independently fit the data for each cell type to a generalized additive model where the relationship between frequency and current was estimated as a smooth spline and further parameterized by genotype and disease stage. Using these models, we tested for an independent effect of genotype on frequency for each comparison. Second, we compared the maximum action potential frequency (Fmax) for CSNs and CCNs. Unlike CSNs and CCNs, the response of PV neurons saturated at the highest current injections (Fig. 2D). Therefore, for PV neurons, we compared the frequency for current steps that elicited 50% of the Fmax of the cells (F50). To test for an effect of age on excitability, we used a similar generalized additive model to fit the data for each cell type across all three time points assessed and tested the interaction effect of genotype and age on the relationship between current and frequency using the Wald test. In addition, we used a similar approach to compare the change in rheobase values across time points for PV neurons using log-transformed data. The data points for all individual cells, the mean and the SEM are shown in plots of rheobase, Fmax, and F50 as well as for the measurements related to the in vivo imaging experiments. A generalized additive model was used to fit the current–frequency data and to generate the 95% CIs shown in each graph. Data were considered significant for p < 0.05. In the figures, the statistical significance is expressed as follows: *p < 0.05; **p < 0.01; or ***p < 0.001. Numbers in bold in the tables indicate p values < 0.05.
Figure 2.

PV-positive inhibitory interneurons are hyperexcitable in symptomatic hSOD1G93A mice. A, Schematic of the experimental design. CSNs in Gad67-GFP mice were retrogradely labeled with Alexa Fluor 555 cholera toxin B (red CTB). B, Confocal images of GFP-expressing PV-positive interneurons (green) and retrogradely labeled CSNs (magenta) in L5b of motor cortex. C, D, Representative voltage responses (C) and the current–spike frequency relationship (D) measured from PV neurons in P90–P101 mice (hSOD1G93A;Gad67-GFP, n = 21 cells from 8 mice; Gad67-GFP, n = 25 cells from 9 mice; p < 2.99 × 10−10, Wald test,). E, The rheobase measured from PV interneurons (hSOD1G93A, 193.52 ± 29.88 pA; WT, 284.12 ± 23.44 pA; p = 0.0074, Mann–Whitney test). F, The F50 measured from PV interneurons (hSOD1G93A, 119 ± 13.94 Hz; WT, 43.64 ± 13.45 Hz; p = 0.0054, Mann–Whitney test).
Data availability.
The data that support the findings of this study are available from the corresponding author upon request. All primary data from the RNA-sequencing (RNA seq) experiments are deposited on the National Center for Biotechnology Information (NCBI) Short Read Archive with NCBI GEO Accession Number GSE101391, and all scripts and workflows for RNA-sequencing analysis are available in conjunction with the preprocessed data upon request. Processed data supporting the findings can be found in an extended data file associated with Figure 8.
Figure 8.

Gene set enrichment analysis implicates distinct molecular mechanisms in corticospinal and corticocortical neurons from hSOD1G93A mice. A, Schematic of the experimental procedure for selective purification of two different molecularly identified cortical projection neuron populations. Total RNA from CTIP2-positive corticospinal neurons and SATB2-positive corticocortical neurons was purified for RNA-seq analysis using cell type-specific antibodies. B, Ctip2 and Satb2 expression profiles for molecularly identified cell types from hSOD1G93A and hSOD1WT mice (n = 4 mice/group). C, Enrichment map of significantly enriched Reactome gene sets from hSOD1G93A vs hSOD1WT RNA sequencing analyses in CTIP2-positive corticospinal neurons (CSNs) and SATB2-positive corticocortical neurons (CCNs). Node color corresponds to gene set enrichment (red) or depletion (blue) in hSOD1G93A CSNs relative to hSOD1WT CSNs. The border color represents gene set enrichment or depletion results for CCNs. White indicates no significant enrichment or depletion for a given gene set for that cell type. Node areas correspond to relative gene set sizes, and line thicknesses (tan) indicate the degree of overlap in gene composition between connected sets. Gene sets were determined to be significantly enriched or depleted using a preranked gene set enrichment analysis (Kolmogorov–Smirnov test, p < 0.05, Benjamini–Hochberg corrected). Supporting data are found in Figure 8-1 available at https:/10.1523/JNEUROSCI.0811-17.2017.f8-1.
Results
Intrinsic hyperexcitability is not restricted to corticospinal neurons
It has been proposed that cell type-specific changes in neuronal excitability contribute to neurodegeneration in ALS (Roselli and Caroni, 2015). However, whether functional abnormalities are restricted to CSNs or involve additional cortical cell types remains unclear. Because the neocortex is composed of different classes of cortical projection neurons and inhibitory interneurons, with distinct electrophysiological, molecular, and morphological characteristics (Sugino et al., 2006; Ascoli et al., 2008; Oswald et al., 2013; Harris and Shepherd, 2015; Tremblay et al., 2016), we addressed this question by comparing the intrinsic electrophysiological properties of different classes of cortical neurons in wild-type (WT) and hSOD1G93A mice. To assess the intrinsic excitability of CSNs and CCNs, the two major classes of cortical projection neurons found in L5b of motor cortex (Hattox and Nelson, 2007; Miller et al., 2008; Suter et al., 2013), we retrogradely labeled the two cell types with two different fluorescent tracers (Fig. 1A,B). We then targeted these cells for whole-cell patch-clamp recording in acute brain slices from P90 to P129 mice, an age range when mice show significant loss of CSNs and overt clinical symptoms of the disease (Zang and Cheema, 2002; Ozdinler et al., 2011; Vinsant et al., 2013). The intrinsic electrophysiological properties of CSNs and CCNs were measured in the presence of antagonists of ionotropic glutamate and GABA receptors. Although it has been postulated that chronic depolarization of CSNs promotes their degeneration, we found no difference in the resting membrane potential of either CSNs or CCNs between hSOD1G93A and WT mice (Table 1). However, the current–frequency relationship for both cell types revealed that CSNs and CCNs from hSOD1G93A mice were significantly more excitable than those from WT mice (Fig. 1C–J). Comparisons of the current–frequency data indicated that the current–frequency curves of CSNs and CCNs from hSOD1G93A mice were significantly different from those from control mice (Fig. 1D,H) and that the maximum frequency elicited by the current steps (Fmax) was significantly greater for both CSNs and CCNs from hSOD1G93A mice than those from controls (Fig. 1F,J). These data indicate that both CSNs and CCNs in symptomatic mice are hyperexcitable.
Table 1.
Electrophysiological properties of cortical neurons recorded from symptomatic (P90–P129) hSOD1G93A or control mice
| p value (Mann–Whitney test) | |||
|---|---|---|---|
| CSN (P90–P129) | WT (46 cells, 12 mice) | hSOD1G93A (64 cells, 19 mice) | |
| Resting membrane potential (mV) | −74.39 ± 0.52 | −72.91 ± 0.57 | 0.0735 |
| Input resistance (MΩ) | 51.19 ± 1.9 | 56.7 ± 1.73 | 0.0147 |
| Spike threshold (mV) | −37.4 ± 0.66 | −36.03 ± 0.77 | 0.3271 |
| Spike amplitude (mV) | 68.04 ± 1.09 | 66.95 ± 1.24 | 0.9283 |
| Spike half-width (ms) | 0.53 ± 0.01 | 0.55 ± 0.01 | 0.1868 |
| CCN (P90–P129) | WT (36 cells, 11 mice) | hSOD1G93A (50 cells, 19 mice) | |
| Resting membrane potential (mV) | −81.57 ± 0.68 | −79.15 ± 0.86 | 0.0767 |
| Input resistance (MΩ) | 79.3 ± 2.64 | 109.13 ± 5.38 | <0.0001 |
| Spike threshold (mV) | −33.44 ± 0.7 | −34.94 ± 0.63 | 0.1868 |
| Spike amplitude (mV) | 67.14 ± 1.46 | 63.68 ± 1.15 | 0.0257 |
| Spike half-width (ms) | 0.64 ± 0.01 | 0.71 ± 0.01 | <0.0001 |
| PV (P90–P101) | WT (25 cells, 9 mice) | hSOD1G93A (21 cells, 8 mice) | |
| Resting membrane potential (mV) | −73.84 ± 0.97 | −73.35 ± 1.39 | 0.9648 |
| Input resistance (MΩ) | 135.68 ± 8.61 | 183.78 ± 22.80 | 0.0609 |
| Spike threshold (mV) | −36.86 ± 1.15 | −35.49 ± 1.03 | 0.7491 |
| Spike amplitude (mV) | 53.31 ± 1.75 | 47.51 ± 2.19 | 0.0497 |
| Spike half-width (ms) | 0.29 ± 0.02 | 0.33 ± 0.02 | 0.0039 |
Although both cell types exhibited increases in intrinsic excitability, the underlying cellular mechanisms differed. CSNs showed no significant differences in rheobase, while the rheobase of CCNs was significantly decreased (Fig. 1E,I). In addition, the waveform of the action potential was significantly altered in CCNs but not in CSNs (Table 1). The input resistance of both cell types, however, was significantly increased in hSOD1G93A mice (Table 1). These data suggest that although both classes of excitatory cortical projection neurons are hyperexcitable in later stages of disease, the cellular mechanisms that underlie these increases in excitability are distinct.
Inhibitory cortical interneurons are hyperexcitable
Because cortical activity is powerfully regulated by inhibitory interneurons, it has been postulated that dysfunction of these neurons contributes to cortical hyperexcitability in ALS (Turner and Kiernan, 2012; Geevasinga et al., 2016; King et al., 2016). PV-expressing interneurons comprise the largest class of inhibitory interneuron in the cortex. Although some studies, but not all, have detected PV cell loss in ALS (Nihei et al., 1993; Minciacchi et al., 2009; Ozdinler et al., 2011), it is not known whether their electrophysiological properties are altered in the disease. To test whether reduced excitability of PV interneurons contributes to disinhibition of cortical projection neurons, we measured the intrinsic properties of PV interneurons selectively labeled with GFP in hSOD1G93A;Gad67-GFP (hSOD1G93A) and Gad67-GFP (WT) P90–P101 mice (Fig. 2A,B). Contrary to this prediction, we found that the intrinsic excitability of PV interneurons was increased in ALS mice. Although the mean resting membrane potential of PV interneurons from mutant and control mice was similar (Table 1), the rheobase was lower in PV neurons from hSOD1G93A mice relative to control mice (Fig. 2E). Furthermore, as with CSNs and CCNs, a comparison of the current–spike frequency relationships of hSOD1G93A and WT PV interneurons indicated that PV interneurons from hSOD1G93A mice were significantly hyperexcitable (Fig. 2D,F). Together, these data reveal functional abnormalities in excitatory and inhibitory cortical neurons in symptomatic hSOD1G93A mice, with all cell types tested exhibiting increases in intrinsic excitability.
Corticospinal neuron activity is normalized in vivo in hSOD1G93A mice
To assess the impact of these increases in intrinsic neuronal excitability that we measured in P90–P129 mice on the activity of CSNs in vivo, we monitored the activity of individual CSNs in awake hSOD1G93A and control mice of similar age using in vivo two-photon imaging after virus-mediated delivery of the genetically encoded calcium indicator GCaMP6s to CSNs (Fig. 3A,B). Robust calcium transients were observed in CSNs in both hSOD1G93A and control mice (P92–P134), with individual CSNs exhibiting diverse activity patterns (Fig. 3C,D). However, there was no significant difference in the overall frequency of activity between hSOD1G93A and control mice (Fig. 3E) or in the distribution of interevent intervals (Fig. 3F). Although GCaMP6s offers greater sensitivity, the slow time course of this sensor relative to the time course of individual action potentials (Chen et al., 2013) could mask differences in the pattern of neural activity underlying individual calcium events. Any such differences would be reflected in the amplitude and time course of the individual transients. However, we found no difference in the amplitude or the time course of individual events measured from symptomatic hSOD1G93A and control mice (Fig. 3G,H). These results indicate that despite their increased intrinsic excitability in late-stage disease, CSNs are not hyperactive in the intact brains of hSOD1G93A mice under these conditions, highlighting the ability of cortical circuits to counterbalance the abnormal physiological properties of these neurons.
Figure 3.

The activity of corticospinal neurons measured in vivo is similar in symptomatic hSOD1G93A and control mice. A, Canine adenovirus carrying a Cre recombinase construct was injected into the left spinal cord of hSOD1G93A;lox-STOP-lox-tdTomato (hSOD1G93A) or lox-STOP-lox-tdTomato (WT) mice. An adeno-associated virus (AAV) carrying a Cre-dependent GCaMP6s construct was injected into the right motor cortex. Only a subset of CSNs transfected with both viruses expressed both tdTomato and GCaMP6s. B, Morphology of an imaged CSN. Scale bar, 50 μm. C, Representative traces of calcium events recorded from CSNs in an hSOD1G93A and a WT animal. D, Averaged isolated calcium events from hSOD1G93A and WT mice. E, F, The mean frequency of events (E; hSOD1G93A: 12.4 ± 2.0 events/5 min epoch, n = 43 cells from 12 mice; control: 9.3 ± 1.3 events/5 min epoch, n = 55 cells from 14 mice; p = 0.2139, Mann–Whitney test) and the mean interevent interval (F; hSOD1G93A: 32.0 ± 5.3 s, n = 40 cells from 9 mice; WT: 50.7 ± 8.7 s, n = 46 cells from 14 mice; p = 0.1064, Mann–Whitney test). G, H, The mean amplitude (G; hSOD1G93A: 52.3 ± 6.2 ΔF/F, n = 43 cells from 12 mice; WT: 41.4 ± 4.4 ΔF/F, n = 55 cells from 14 mice; p = 0.2026, Mann–Whitney test) and the time course of the event decay (H; hSOD1G93A: 1.8 ± 0.1 s, n = 40 cells from 11 mice; WT: 2.2 ± 0.2 s, n = 49 cells from 13 mice; p = 0.5092, Mann–Whitney test).
Elevated intracellular calcium in corticospinal neurons from symptomatic mice
Abnormalities in calcium homeostasis have been widely implicated in the pathogenesis of ALS (Grosskreutz et al., 2010; Leal and Gomes, 2015). Although the effects of increases in intrinsic excitability may be compensated for by changes in synaptic input, degeneration could be promoted in hSOD1G93A mice if neurons are impaired in their ability to sustain low intracellular calcium levels. To explore this possibility, we measured the ratio of GCaMP6s fluorescence to that of the calcium-insensitive fluorescent reporter tdTomato in CSNs in vivo, allowing normalization across cells. This ratio was significantly elevated in hSOD1G93A mice relative to controls, indicating that basal calcium levels in the apical dendrites of CSNs are elevated in ALS mice (Fig. 4A–C). These results reveal that calcium homeostasis in CSNs is impaired in hSOD1G93A mice, despite normalization of their activity patterns.
Figure 4.

Basal intracellular calcium levels in corticospinal neurons measured in vivo are significantly elevated in hSOD1G93A mice. A, Images of cross sections of the apical dendritic stalks of two CSNs in control (WT) and in hSOD1G93A mice showing the tdTomato signal (left), the GCaMP6s signal (middle), and the two overlaid (right). Scale bar, 2 μm. B, The cumulative plot of the mean intensity of the tdTomato fluorescence (hSOD1G93A: 33.9 ± 3.8 a.u., n = 40 neurons from 12 mice; WT: 38.6 ± 4.2 a.u., n = 52 neurons from 16 mice; p = 0.3884, Mann–Whitney test). C, The cumulative plot of the ratio of the GCaMP6s intensity to the tdTomato intensity (hSOD1G93A: 1.6 ± 0.2, n = 40 neurons from 12 mice; WT: 1.1 ± 0.2, n = 52 neurons from 16 mice; p = 0.0011, Mann–Whitney test).
Changes in intrinsic excitability as disease progresses
These results indicate that neuronal dysfunction extends beyond corticospinal neurons in the cortices of hSOD1G93A mice with advanced disease. Prior studies have reported that cortical projection neurons are hyperexcitable earlier in the disease process (Pieri et al., 2009; Saba et al., 2016), but whether selective abnormalities in CSN function precede the abnormalities we detected in corticocortical and PV inhibitory interneurons is not known. To determine whether CSN dysfunction occurs before abnormalities in CCNs and PV interneurons, or whether hyperexcitability evolves similarly across these cortical cell types, we assessed the intrinsic properties of CSNs, CCNs, and PV interneurons at two earlier ages. At an age when mice exhibit subtle clinical symptoms and cellular abnormalities begin to appear in CSNs, but frank CSN neurodegeneration has not yet started (P26–P40; Zang and Cheema, 2002; Ozdinler et al., 2011; Vinsant et al., 2013; Fogarty et al., 2015, 2016; Saba et al., 2016), we found no differences in the rheobase for CSNs, CCNs, and PV interneurons between hSOD1G93A and control mice, and the differences in the current–frequency relationships were attenuated relative to what was observed in advanced disease (Fig. 5).
Figure 5.

The intrinsic excitability in cortical neurons in presymptomatic hSOD1G93A mice. A, E, Representative voltage traces recorded from CSNs (A) and CCNs (E) from P26–P40 hSOD1G93A;Gad67-GFP (hSOD1G93A) and Gad67-GFP (WT) mice evoked by 240, 360, and 480 pA current steps, as indicated. B, F, The current–spike frequency relationship measured from CSNs (B; hSOD1G93A: n = 56 cells from 12 mice; WT: n = 33 cells from 11 mice; p < 0.0119, Wald test) and CCNs (F; hSOD1G93A, n = 13 cells from 5 mice; WT, n = 25 cells from 7 mice; p < 0.0779, Wald test,) from hSOD1G93A and WT mice. C, G, The rheobase measured from CSNs (C; hSOD1G93A, 271.95 ± 8.73 pA; WT, 275.3 ± 14.27 pA; p = 0.6434, Mann–Whitney test) and CCNs (G; hSOD1G93A, 282.85 ± 18.12 pA; WT, 297.56 ± 19.1 pA; p = 0.7233, Mann–Whitney test). D, H, The Fmax measured from CSNs (D; hSOD1G93A, 30.95 ± 1.01 Hz; WT, 29.59 ± 1.5 Hz; p = 0.337, Mann–Whitney test) and CCNs (H; hSOD1G93A, 28.18 ± 2.85 Hz; WT, 23.27 ± 2.43 Hz; p = 0.2801, Mann–Whitney test). I, J, Representative voltage responses (I) and the current–spike frequency relationship (J) measured from PV interneurons in P26–P35 mice (hSOD1G93A, n = 18 cells from 6 mice; WT, n = 17 cells from 7 mice; p < 0.00375, Wald test). K, The rheobase measured from PV interneurons (hSOD1G93A, 285.83 ± 41.41 pA; WT, 271.29 ± 41.91 pA; p = 0.692, Mann–Whitney test). L, The F50 measured from PV interneurons (hSOD1G93A, 96.5 ± 16.47 Hz; WT, 77.08 ± 15.15 Hz; p = 0.3843, Mann–Whitney test).
To determine whether hyperexcitability emerges progressively with disease, we also recorded from CSNs, CCNs, and PV interneurons in young mice (P5–P6). To target CSNs and CCNs for recording in slices from P5–P6 mice, we first developed a method for identifying these two cell types at a time point when the promiscuous projections to the pyramidal tract found early in development preclude the use of retrograde tracers (O'Leary and Koester, 1993). We found that the pattern of Cre expression in a transgenic mouse line, Rbp4-Cre (Gerfen et al., 2013), could be used to distinguish CSNs from CCNs in L5b (Fig. 6). After retrogradely labeling CSNs and CCNs in juvenile Rbp4-Cre;tdTomato mice (P21–P34) when the projections to the pyramidal tract have been eliminated (O'Leary and Koester, 1993), we found that none of the retrogradely labeled CCNs in L5b expressed tdTomato (0 ± 0%, n = 3 mice, 0 of 220 total cells), while >90% of retrogradely labeled CSNs expressed tdTomato (91.9 ± 3.5%, n = 3 mice, 66 of 70 total cells). Furthermore, the dendritic morphologies and the electrophysiological properties of tdTomato-positive and tdTomato-negative neurons in neonatal Rbp4-Cre mice were similar to those of CSNs and CCNs in adult mice, respectively (Fig. 6). For example, biocytin-filled L5b tdTomato-positive cells from P5 Rbp4-Cre;tdTomato mice (Fig. 6A; n = 34 cells from 16 mice) had large apical dendritic tufts similar to CSNs from adult mice (Fig. 6B; n = 54 cells from 16 mice), while L5b tdTomato-negative neurons from P5 Rbp4-Cre;tdTomato mice had slim apical tufts (Fig. 6C; n = 9 cells from 4 mice), as seen for CCNs from adult mice (Fig. 6D; n = 13 cells from 7 mice). Interestingly, CSNs in neonatal hSOD1G93A mice had a significantly lower mean rheobase relative to CSNs in control mice (Fig. 7C). Furthermore, a comparison of both the current–frequency curves and the Fmax values of the current–frequency relationship indicated that CSNs were significantly hyperexcitable (Fig. 7A,B,D). Although the rheobase was not elevated for CCNs (Fig. 7G), the same analyses of the current–frequency relationship indicated that CCNs were also significantly hyperexcitable (Fig. 7E,F,H). The significantly decreased mean rheobase and the analysis of the current–frequency relationship demonstrated that PV interneurons from mutant mice were also hyperexcitable (Fig. 7I–L). These data indicate that neuronal hyperexcitability extends beyond CSNs in the cortex of neonatal hSOD1G93A mice. These increases in excitability were specific to the hSOD1G93A mutation and not due solely to overexpression of the SOD1 transgene, as we found no significant differences in the rheobase of CSNs or CCNs recorded from neonatal mice that overexpress wild-type human SOD1 (hSOD1WT) and control mice (rheobase CSNs: hSOD1WT: 80.67 ± 8.19 pA, n = 21 cells from 7 mice; WT: 74.94 ± 6.43 pA, n = 33 cells from 10 mice; p = 0.2713, Mann–Whitney test; Rheobase CCNs: hSOD1WT: 50.29 ± 3.44 pA, n = 21 cells from 7 mice; WT: 49.7 ± 4.39 pA, n = 33 cells from 10 mice; p = 0.3628, Mann–Whitney test). Moreover, the Fmax was not different between CSNs or CCNs recorded from hSOD1WT and control mice (CSNs: hSOD1WT, n = 21 cells from 7 mice; WT, n = 33 cells from 10 mice; p = 0.4009, Mann–Whitney test; CCNs: hSOD1WT, n = 21 cells from 7 mice; WT, n = 33 cells from 10 mice; p = 0.5485, Mann–Whitney test), and a generalized additive model found no effect of genotype on the current–frequency relationships for CSNs and CCNs (p < 0.174, Wald test). Thus, the overexpression of mutant SOD1 induces hyperexcitability broadly in cortical neurons in neonatal mice, although the precise cellular mechanisms may differ across cell types (see Table 3).
Figure 7.

Cortical neurons are hyperexcitable in neonatal hSOD1G93A mice. A, E, Representative voltage traces recorded from CSNs (A) and CCNs (E) neurons from neonatal hSOD1G93A;Rbp4-Cre;tdTomato (hSOD1G93A) and Rbp4-Cre;tdTomato (WT) mice evoked by 40, 120, and 200 pA current steps. B, F, The current–spike frequency relationship measured from CSNs (B; P5–P6; hSOD1G93A, n = 29 cells from 9 mice; WT, n = 53 cells from 10 mice; p < 1.13 × 10−12, Wald test,) and CCNs (F; P5–P6; hSOD1G93A, n = 15 cells from 5 mice; WT, n = 24 cells from 8 mice; p < 1.82 × 10−6, Wald test,) from neonatal hSOD1G93A and WT mice. C, G, The rheobase measured from CSNs (C; hSOD1G93A, 53.03 ± 4.48 pA; WT, 70.55 ± 3.93 pA; p = 0.0008, Mann–Whitney test) and CCNs (G; hSOD1G93A, 39.43 ± 4.71 pA; WT, 43.2 ± 5.15 pA; p = 0.7123, Mann–Whitney test). D, H, The Fmax measured from CSNs (D; hSOD1G93A, 39.79 ± 1.5 Hz; WT, 34.17 ± 1.31 Hz; p = 0.0017, Mann–Whitney test) and CCNs (H; hSOD1G93A, 31.56 ± 4.51 Hz; WT, 20.31 ± 3.04 Hz; p = 0.0293, Mann–Whitney test). I, Representative voltage traces recorded from PV interneurons from neonatal hSOD1G93A;Gad67-GFP;Rbp4-Cre;tdTomato (hSOD1G93A) and Gad67-GFP;Rbp4-Cre;tdTomato (WT) mice evoked by 20, 40, and 80 pA current steps. J, The current–spike frequency relationship for PV neurons from hSOD1G93A and WT neonatal mice (P6; hSOD1G93A, n = 23 cells from 11 mice; WT, n = 19 cells from 7 mice; p < 0.00556, Wald test). K, The rheobase measured from PV interneurons (hSOD1G93A, 14.9 ± 1.81 pA; WT, 27.65 ± 3.37 pA; p = 0.0028, Mann–Whitney test). L, The F50 measured from PV interneurons (hSOD1G93A, 52.11 ± 2.13 Hz; WT, 47.12 ± 2.31 Hz; p = 0.1415, Mann–Whitney test).
Table 3.
Electrophysiological properties of cortical neurons recorded from neonatal (P5–P6) hSOD1G93A or control mice
| p value (Mann–Whitney test) | |||
|---|---|---|---|
| CSN (P5–P6) | WT (53 cells, 15 mice) | hSOD1G93A (29 cells, 9 mice) | |
| Resting membrane potential (mV) | −66.77 ± 0.51 | −67.26 ± 0.77 | 0.5552 |
| Input resistance (MΩ) | 170.32 ± 7.06 | 180.67 ± 6.93 | 0.0801 |
| Spike threshold (mV) | −35.83 ± 0.46 | −37.73 ± 0.52 | 0.0183 |
| Spike amplitude (mV) | 69.47 ± 0.78 | 71.38 ± 0.99 | 0.1031 |
| Spike half-width (ms) | 1.59 ± 0.03 | 1.62 ± 0.03 | 0.3681 |
| CCN (P5–P6) | WT (24 cells, 8 mice) | hSOD1G93A (15 cells, 5 mice) | |
| Resting membrane potential (mV) | −69.91 ± 0.84 | −69.17 ± 1.19 | 0.7052 |
| Input resistance (MΩ) | 505.8 ± 32.41 | 559.59 ± 42.3 | 0.2565 |
| Spike threshold (mV) | −32.62 ± 1.22 | −30.96 ± 1.44 | 0.8107 |
| Spike amplitude (mV) | 49.99 ± 1.44 | 49 ± 1.94 | 0.8974 |
| Spike half-width (ms) | 1.88 ± 0.11 | 1.77 ± 0.09 | 0.825 |
| PV (P6) | WT (19 cells, 7 mice) | hSOD1G93A (23 cells, 11 mice) | |
| Resting membrane potential (mV) | −68.22 ± 1.37 | −66.82 ± 1.23 | 0.5111 |
| Input resistance (MΩ) | 653.69 ± 53.57 | 709.64 ± 53.45 | 0.5441 |
| Spike threshold (mV) | −32.27 ± 1.09 | −36.45 ± 1.13 | 0.0203 |
| Spike amplitude (mV) | 45.5 ± 2.27 | 47.08 ± 1.71 | 0.7489 |
| Spike half-width (ms) | 0.96 ± 0.03 | 1.09 ± 0.04 | 0.0198 |
We next tested whether this hyperexcitability phenotype emerges progressively during the disease course by applying a generalized additive model fit to the current–frequency relationships assessed at the three time points for each cell type. Models were additionally parameterized by both genotype and disease stage to estimate baseline effects. We found that hSOD1G93A CSNs measured at P26–P40 were significantly less hyperexcitable than those measured during symptomatic disease (P90–P129; p < 0.000715, Wald test), while there was no significant difference in the hyperexcitability of hSOD1G93A CSNs measured during the neonatal (P5–P6) and the symptomatic (P90–P129) stages (p < 0.0809, Wald test). Likewise, the modeled current–frequency relationships of CCNs showed that hSOD1G93A CCNs were significantly less hyperexcitable during the presymptomatic stage than in symptomatic disease (P26–P40 vs P90–P129, p < 2.61 × 10−11, Wald test), while there was no significant difference in the hyperexcitability of hSOD1G93A CCNs measured during the neonatal and symptomatic periods (P5–P6 vs P90–P129, p < 0.2068, Wald test). A similar comparison of the current–frequency relationships of PV interneurons indicated that hSOD1G93A PV neurons were most hyperexcitable during symptomatic disease (P26–P35 versus P90–P129, p < 0.00381; P6 vs P90–P129, p < 0.04605, Wald test), although the analysis suggested that the partial effect of age was greater for PV neurons from P26–P35 hSOD1G93A mice than for those from P6 mice, which is consistent with our results for CSNs and CCNs. Furthermore, an analysis of the rheobase measurements indicated that PV neurons were more hyperexcitable during the neonatal and symptomatic stages than during the presymptomatic stage. There was no significant difference in the reduction in rheobase measurements when comparing the neonatal and symptomatic disease stages (P90–P101 vs P6, p = 0.5419, Wald test), while the rheobase was significantly less reduced during presymptomatic disease than symptomatic disease (P26–P35 vs P90–P101, p = 0.0329, Wald test). Together, these findings indicate that physiological abnormalities in cortical neurons do not follow a linear progression toward enhanced excitability with increasing severity of disease, but that physiological abnormalities emerge early, are comparatively normalized, and then re-emerge in later stages of disease.
Cell type-specific changes in gene expression in hSOD1G93A mice
Although functional alterations were observed in multiple classes of cortical neurons in hSOD1G93A mice, the precise electrophysiological changes exhibited by each cell type tested were distinct (Tables 1, 2, 3; Figs. 1, 2, 5, 7), suggesting that cell type-specific molecular changes may be induced in response to hSOD1G93A expression. For example, although the mean Fmax was significantly increased for both CSNs and CCNs from P5–P6 hSOD1G93A mice relative to control mice, the mean rheobase was significantly decreased only for CSNs and not for CCNs (Fig. 7). To determine whether different classes of cortical neurons mount distinct responses to the expression of mutant SOD1, we assessed the gene expression profiles of CSNs and CCNs in P5–P7 mice, a time point early in the disease course when changes in neuronal excitability were detected. We focused on this time point to emphasize initial changes in the disease, as additional compensatory mechanisms are likely engaged in late-stage mice. To generate an unbiased assessment of changes in gene expression, total RNA isolated from dissociated cortical neurons immunostained for CTIP2 (Bcl11b), a marker for CSNs, and SATB2, a marker for CCNs, was purified from four biological replicates (Fig. 8A,B; Molyneaux et al., 2015). RNA-seq analysis identified 476 genes that were differentially expressed (DE) in CSNs and 339 genes that were differentially expressed in CCNs between these groups of hSOD1G93A and hSOD1WT mice [Cuffdiff2; 5% false discovery rate (FDR)]. Of these DE genes, only 33 were common to both sets of DE genes, suggesting that the transcriptional responses differed across the two neuronal subtypes. Common changes in gene expression included the downregulation of a calcium channel subunit (Cacnb4) that has been proposed to link neuronal excitability to gene transcription (Zamponi et al., 2015) and a GABA receptor subunit (Gabra4) that contributes to tonic inhibition (Farrant and Nusser, 2005). However, not all shared DE genes were coordinately regulated. For example, the calcium-binding protein, calbindin (Calb1), was upregulated in hSOD1G93A CSNs and downregulated in hSOD1G93A CCNs relative to controls. Together, these results highlight that there are a limited number of common gene expression changes shared between these two cortical neuron types in response to expression of hSOD1G93A and suggest that differences in excitability observed early in the disease may result from cell type-specific transcriptional responses.
Table 2.
Electrophysiological properties of cortical neurons recorded from presymptomatic (P26–P40) hSOD1G93A or control mice
| p-value (Mann-Whitney test) | |||
|---|---|---|---|
| CSN (P26–P40) | WT (33 cells, 11 mice) | hSOD1G93A (56 cells, 12 mice) | |
| Resting membrane potential (mV) | −71.5 ± 1.34 | −68.72 ± 0.84 | 0.0474 |
| Input resistance (MΩ) | 58.48 ± 2.63 | 60.72 ± 2.28 | 0.6866 |
| Spike threshold (mV) | −35.49 ± 1.76 | −34.92 ± 1.01 | 0.8418 |
| Spike amplitude (mV) | 76.88 ± 1.47 | 75.68 ± 1.08 | 0.9831 |
| Spike half-width (ms) | 0.66 ± 0.01 | 0.66 ± 0.02 | 0.8784 |
| CCN (P26–P40) | WT (25 cells, 7 mice) | hSOD1G93A (13 cells, 5 mice) | |
| Resting membrane potential (mV) | −73.53 ± 1.41 | −75.4 ± 2.09 | 0.3401 |
| Input resistance (MΩ) | 88.32 ± 4.9 | 97.92 ± 6.32 | 0.1962 |
| Spike threshold (mV) | −29.48 ± 2.08 | −27.15 ± 1.92 | 0.4237 |
| Spike amplitude (mV) | 66.56 ± 1.64 | 65.46 ± 2.42 | 0.8777 |
| Spike half-width (ms) | 0.76 ± 0.03 | 0.71 ± 0.03 | 0.2354 |
| PV (P26–P35) | WT (17 cells, 7 mice) | hSOD1G93A (18 cells, 6 mice) | |
| Resting membrane potential (mV) | −76.87 ± 2.22 | −72.58 ± 2.22 | 0.1248 |
| Input resistance (MΩ) | 127.11 ± 12.5 | 117.32 ± 10.44 | 0.6321 |
| Spike threshold (mV) | −40.47 ± 2.13 | −37.69 ± 2.57 | 0.6323 |
| Spike amplitude (mV) | 56.07 ± 2.66 | 51.88 ± 2.85 | 0.1924 |
| Spike half-width (ms) | 0.36 ± 0.03 | 0.3 ± 0.01 | 0.2204 |
Although there was little overlap in genes differentially expressed in control and mutant neurons between these two neuronal types, it is possible that similar molecular pathways are affected in both neuronal types. To test this hypothesis, we conducted GSEA using all expressed genes in CSNs and CCNs between hSOD1G93A and hSOD1WT mice. For each cell type, genes were ranked according to their Cuffdiff2 test statistic, and this was used as input for a preranked GSEA against Reactome gene sets (MSigDB version 5.1 C2.CP Reactome). There was minimal overlap among the specific gene sets enriched for the two cell types (Fig. 8C). Both cell types exhibited significantly increased the expression of genes related to the translation and the organization of the extracellular matrix in the hSOD1G93A mutant mice, although in many cases different gene sets related to these cellular functions were identified in each type. Only CSNs exhibited broad modulation in gene sets associated with ion channels, GABAergic signaling, receptor signaling, and axon guidance. Processes implicated in DNA repair were significantly downregulated in CSNs. Conversely, genes involved in the respiratory cycle, mitochondrial translation, and calcium-dependent events were specifically downregulated in CCNs. Although both cell types demonstrated significant transcriptional differences in response to the expression of hSOD1G93A, these differences did not point to a common cellular response contributing to hyperexcitability, suggesting that distinct cellular mechanisms underlie the hyperexcitability phenotypes of CSNs and CCNs in this ALS mouse model.
Although we identified changes in intrinsic electrophysiological properties in both CSNs and CCNs, the GSEA implicated gene sets associated with ion channels only in CSNs. To further understand the electrophysiological phenotypes that we identified, we asked whether the expression of individual ion channel genes that shape the intrinsic properties of neurons were differentially modulated in CSNs and CCNs. Both sodium and potassium channel subunits were significantly differentially expressed in CSNs and CCNs relative to controls, but the specific subunits identified were completely distinct for the two cell types (Fig. 9). Furthermore, although some changes in ion channel expression, such as increases Scn1b expression, may be consistent with the increases in intrinsic excitability that we measured (Brackenbury and Isom, 2011), others such as with Hcn2, may represent cell type-specific compensatory mechanisms working to counterbalance the increases in excitability (Shah, 2014). The differences in these changes between CSNs and CCNs reinforce the conclusion that the molecular mechanisms underlying the changes in intrinsic excitability in the two cell types in hSOD1G93A mice are distinct.
Figure 9.
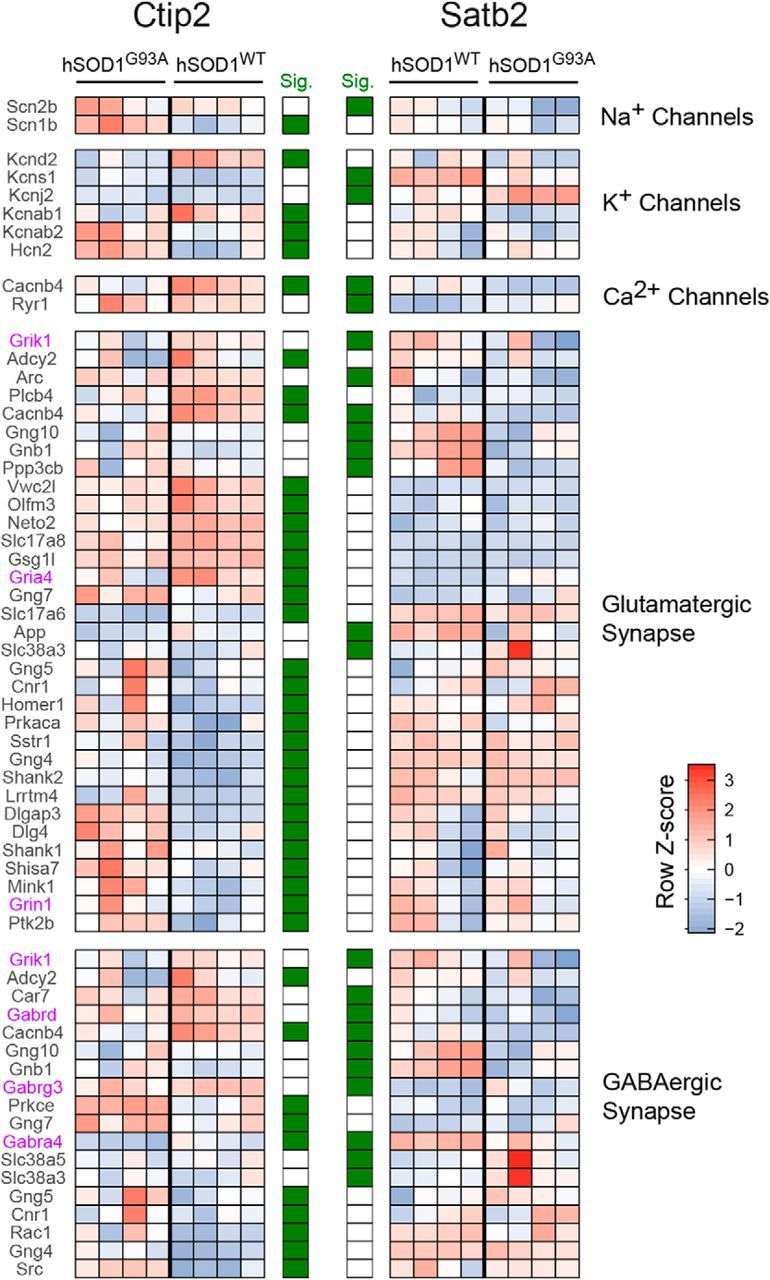
Differential modulation of genes for ion channels and synaptic proteins in corticospinal and corticocortical neurons in an ALS mouse model. Heatmap of significantly differentially expressed genes from select, physiologically relevant gene sets. Genes were selected as significant from differential expression analyses between hSOD1G93A and hSOD1WT in either CSN-enriched (CTIP2-positive) or CCN-enriched (SATB2-positive) cellular populations. Row z scores across all 16 samples are used to highlight the changes in expression profiles for each gene across the four conditions. Genes called significant using cuffdiff2 (5% FDR, Benjamini–Hochberg corrected) for CSN or CCN populations are marked in green in the significance columns. Genes for glutamate or GABA receptor subunits are shown in purple. Gene sets were hand curated from publicly available annotations including Reactome, MGI, and HUGO. Supporting data are found in Figure 8-1 available at https:/10.1523/JNEUROSCI.0811-17.2017.f8-1.
In addition to differences in ion channel expression, changes in the expression of specific glutamate receptor subunits (Gria4, Grin1, and Grik1) in either CSNs or CCNs as well as downregulation of the vesicular glutamate transporters Vglut2 (Slc17a6) and Vglut3 (Slc17a8) in CSNs (Fig. 9), all suggest the modulation of excitatory synaptic transmission in hSOD1G93A neurons. Similarly, the downregulation of GABA receptor subunits primarily associated with extrasynaptic tonic inhibition, including Gabra4, which was downregulated in both CSNs and CCNs from hSOD1G93A mice, and Gabrd, which was downregulated only in CCNs (Farrant and Nusser, 2005), suggest an altered inhibitory tone in hSOD1G93A mice. As seen in the GSEA results, there was little overlap among genes associated with glutamatergic and GABAergic synaptic transmission that were differentially expressed in CSNs and CCNs (Fig. 9). Together, these RNA-seq experiments indicate that hSOD1G93A expression alters largely different genes in CSNs and CCNs early in the disease process and suggest that cortical dysfunction reflects a constellation of changes, both in the intrinsic properties of cortical neurons and the pathways related to synaptic transmission.
Discussion
Our results demonstrate that the expression of an ALS-causing mutant form of SOD1 leads to increases in intrinsic excitability in diverse classes of cortical excitatory and inhibitory neurons. Moreover, abnormalities in CSN function did not precede those in other cell types but rather evolved in parallel during the course of the disease. Cortical neurons were initially hyperexcitable in neonatal animals and largely normalized in juvenile mice before becoming hyperexcitable again, indicating that they adapt their responsiveness during disease progression. Cell type-specific in vivo calcium imaging of cortical neuron activity showed that these increases in intrinsic excitability did not directly translate into hyperactivity in vivo, suggesting that cortical circuit plasticity also plays an important role in the response of the cortex to cellular dysfunction. Furthermore, cell type-specific transcriptional profiling early in the disease showed that different cortical cell types engage distinct cellular mechanisms in response to the expression of this ALS-linked mutation. Our results are inconsistent with the hypothesis that the intrinsic electrophysiological properties of CSNs in ALS are selectively altered, leading to abnormal CSN activity and their subsequent degeneration. Rather, our results show that the properties of all cortical neuron types tested are dynamically altered during disease progression and suggest that their distinct molecular responses to the hSOD1G93A mutation may lead to cell type-specific vulnerability.
By probing functional changes in cortical neurons throughout the disease course, we show that cortical dysfunction is not a fixed state in ALS, but rather that cortical neurons adapt dynamically during disease progression. Consistent with prior studies (Pieri et al., 2009; Saba et al., 2016), we detected increases in intrinsic excitability in cortical neurons early in disease. Thus, cortical dysfunction occurs early, although additional studies will be required to determine whether cortical and spinal neuron dysfunction occurs in parallel or whether one causes the other in this mouse model (Eisen et al., 1992; Chou and Norris, 1993). Unexpectedly, these increases in intrinsic excitability normalized as disease progressed before returning in end-stage disease. This is in contrast to studies of spinal motor neurons in mouse models and patient-derived motor neurons indicating that motor neuron excitability decreases with disease progression or days in culture (Delestrée et al., 2014; Devlin et al., 2015). Although patients with known ALS-causing mutations may exhibit cortical hyperexcitability shortly before the development of clinical symptoms, measures of cortical excitability are typically normal when patients are asymptomatic (Vucic et al., 2008; Geevasinga et al., 2015). Whether patients exhibit a period of abnormal cortical excitability in childhood, for example, similar to the changes we detected in this mouse model, remains unknown, but could represent an early diagnostic for the evaluation of at-risk patients.
Our studies identified functional changes in PV interneurons, suggesting that abnormalities in cortical inhibition contribute to cortical dysfunction. Clinical studies have suggested that cortical hyperexcitability is in part due to impaired inhibitory mechanisms (Turner and Kiernan, 2012; Geevasinga et al., 2016; King et al., 2016) and the loss of inhibitory PV interneurons (Nihei et al., 1993) and cortical GABA (Foerster et al., 2013), although not all studies have detected such changes (Ince et al., 1993; Maekawa et al., 2004; Attarian et al., 2009). Inhibitory neuron dysfunction has also been implicated in a TDP-43 mouse model (Zhang et al., 2016). Although PV interneuron loss has not been documented in hSOD1G93A mice (Minciacchi et al., 2009; Ozdinler et al., 2011), our findings indicate that PV interneuron function changes concurrently with that of cortical projection neurons in hSOD1G93A mice.
Our results also indicate that additional synaptic mechanisms may normalize cortical activity during the course of the disease. Although CSNs exhibited significant increases in intrinsic excitability in symptomatic disease, CSNs were not hyperactive when monitored in vivo in the awake mouse during this same stage, highlighting the ability of the cortex to maintain homeostasis. The possibility that changes in synaptic inputs offset increases in intrinsic excitability is consistent with studies showing changes in dendritic spines and synaptic input to cortical neurons in ALS (Fogarty et al., 2015, 2016; Saba et al., 2016), as well as with the gene expression differences in neurotransmitter receptors and synaptic proteins that we detected using cell type-specific RNA-seq approaches. However, the timing of these changes relative to changes in intrinsic excitability remains to be clarified (Spalloni et al., 2011; Jara et al., 2012; Fogarty et al., 2015, 2016; Saba et al., 2016). Changes in dendritic morphology or spine density of neurons in motor cortex have not been reported before the third week of life (Fogarty et al., 2015, 2016), suggesting that increased intrinsic excitability precedes detectable morphological changes. Although some have reported electrophysiological signatures of increased excitatory input, most studies have found that dendritic arbor size and spine density progressively decrease in motor cortex of hSOD1G93A mice (Spalloni et al., 2011; Jara et al., 2012; Fogarty et al., 2015, 2016; Saba et al., 2016) and of patients (Hammer et al., 1979; Udaka et al., 1986). Combined with our findings, these results suggest that increases in intrinsic excitability later in disease may counterbalance decreased synaptic input to generate comparable in vivo activity of CSNs in symptomatic hSOD1G93A and control mice. Together, these results highlight the different plasticity mechanisms engaged during the course of the disease.
The onset of clinically evident disease may arise when the limits of homeostatic processes are reached. Studies of hSOD1G93A mice have identified a number of cellular abnormalities in embryonic and neonatal mice as well as transient behavioral abnormalities (Vinsant et al., 2013). It is possible that early in the disease process, neuronal circuits compensate for an initial insult, while later in the disease process, when cell loss and inflammation manifest, neurons are no longer able to adapt to underlying pathological changes, resulting in progressive, late-stage disease (van Zundert et al., 2012). Understanding the impact of these changes on cell survival has important implications for the timing of therapeutic strategies seeking to alter the excitability of neurons in ALS.
Although CSNs were not hyperactive in vivo during symptomatic disease, we found that CSNs nonetheless exhibited abnormal function in the intact ALS mouse. The elevated basal calcium levels we detected, which may lead directly to increased neurotransmitter release and a cycle of increasing excitotoxicity and calcium dysregulation, are consistent with the dysregulation of calcium handling long implicated in ALS (Grosskreutz et al., 2010; Saxena and Caroni, 2011; Leal and Gomes, 2015) and are reminiscent of persistent increases in intracellular calcium levels identified in spinal motor neuron terminals in patients (Siklós et al., 1996). Our results motivate testing whether targeting the increases in intracellular calcium we detected in CSNs in vivo represents an effective alternative to targeting changes in excitability directly.
Although different cortical cell types exhibit functional changes with similar consequences for neuronal excitability in the hSOD1G93A mouse, our studies indicate that the molecular mechanisms underlying these changes are cell type specific. Transcriptional profiling at single-population resolution provides the first snapshot of the transcriptional responses of different neuronal types to mutant SOD1 and revealed cell type-specific responses to hSOD1G93A expression. Both GSEA and more focused analyses of gene expression confirmed the many distinct cell type-specific differences in gene expression occurring in this ALS mouse model. Furthermore, although several genes implicated in ALS pathogenesis were differentially expressed between hSOD1G93A and hSOD1WT CSNs or CCNs, the changes did not occur in parallel between the two cells types: Chrna2, Chrna4, and Chgb were selectively downregulated in CSNs, while Tuba4a was downregulated and Epha3 was upregulated in CCNs (Marangi and Traynor, 2015). These results were initially unexpected in view of the overall similarity in the functional consequences for excitability that we detected in the different cell types. However, that gene expression changes induced by the hSOD1G93A mutation differed between CSNs and CCNs is consistent with the hypothesis that hyperexcitability differences are due to distinct cell type-specific cellular responses to the hSOD1G93A variant. Thus, although the overall functional consequences of the hSOD1G93A variant were similar across different cell types, the precise underlying cellular mechanisms we measured differed across cell types (Tables 1, 2, 3; Figs. 1, 2, 5, 7).
Although the causes of ALS are likely multifactorial and can arise from mutations in different genes (Marangi and Traynor, 2015), studies in human patients indicate that cortical hyperexcitability is a common feature of both sporadic and familial ALS. Indeed, patients with ALS arising from mutations in SOD1 and C9orf72, as well as patients with no known mutations, exhibit cortical hyperexcitability (Vucic and Kiernan, 2006; Vucic et al., 2008; Williams et al., 2013; Geevasinga et al., 2015). Our findings using a mouse model demonstrating stage-dependent alterations in neuronal function in ALS mice highlight the dynamic changes that occur in cortical circuits during neurodegeneration and have important implications for identifying cell- and stage-specific therapeutic targets. The approaches taken here to evaluate the cell types affected, the time course of these changes, and the impact on neuronal activity in the intact nervous system provide a means to assess common responses to distinct ALS mutations in additional mouse models and to develop therapeutic strategies tailored to specific alterations experienced by cortical neurons.
Footnotes
This work was supported by Target ALS (S.P.B., D.E.B, P.A., L.A.G.), the Robert Packard Center for ALS Research (S.P.B.), and the National Institutes of Health (Grants NS-050274 and NS-098819). J.K. was supported by a National Research Foundation of Korea Fellowship (NRF-2011-357-E00005). E.G.H. is supported by the Boettcher Webb-Waring Foundation. S.P.B. is supported by a Klingenstein-Simons Fellowship in the Neurosciences. We thank Dohee Kim, Daniel F. Gill, Brandie A. Morris, Elliot Stryker, Lainey Bukowiec, and Lisle Blackbourn for technical assistance; Eric J. Kremer (Institut de Génétique Moléculaire de Montpellier), Brandon K. Harvey (National Institute on Drug Abuse), and the National Institute on Drug Abuse Optogenetics and Transgenic Technology Core for CAV-Cre viral constructs; and Vivek Jayaraman, Douglas S. Kim, Loren L. Looger, and Karel Svoboda from the GENIE Project; and Janelia Research Campus, Howard Hughes Medical Institute, for the GCaMP6s construct.
The authors declare no competing financial interests.
References
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsáki G, Cauli B, Defelipe J, Fairén A, Feldmeyer D, Fishell G, Fregnac Y, Freund TF, Gardner D, Gardner EP, Goldberg JH, Helmstaedter M, Hestrin S, Karube F, et al. (2008) Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9:557–568. 10.1038/nrn2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attarian S, Pouget J, Schmied A (2009) Changes in cortically induced inhibition in amyotrophic lateral sclerosis with time. Muscle Nerve 39:310–317. 10.1002/mus.21137 [DOI] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 330:585–591. 10.1056/NEJM199403033300901 [DOI] [PubMed] [Google Scholar]
- Brackenbury WJ, Isom LL (2011) Na channel β subunits: overachievers of the ion channel family. Front Pharmacol 2:53. 10.3389/fphar.2011.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ (2004) Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci 24:9598–9611. 10.1523/JNEUROSCI.1851-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SM, Norris FH (1993) Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 16:864–869. 10.1002/mus.880160810 [DOI] [PubMed] [Google Scholar]
- Delestrée N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D (2014) Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 592:1687–1703. 10.1113/jphysiol.2013.265843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S, Miles GB (2015) Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun 6:5999. 10.1038/ncomms6999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW (2007) Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron 56:43–57. 10.1016/j.neuron.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A, Kim S, Pant B (1992) Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve 15:219–224. 10.1002/mus.880150215 [DOI] [PubMed] [Google Scholar]
- Elbasiouny SM, Quinlan KA, Eissa TL, Heckman CJ (2012) Electrophysiological abnormalities in SOD1 transgenic models in amyotrophic lateral sclerosis: the commonalities and differences. In: Amyotrophic lateral sclerosis (Maurer MH, ed). Rijeka, Croatia: InTech. [Google Scholar]
- Farrant M, Nusser Z (2005) Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6:215–229. 10.1038/nrn1625 [DOI] [PubMed] [Google Scholar]
- Foerster BR, Pomper MG, Callaghan BC, Petrou M, Edden RA, Mohamed MA, Welsh RC, Carlos RC, Barker PB, Feldman EL (2013) An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol 70:1009–1016. 10.1001/jamaneurol.2013.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MJ, Noakes PG, Bellingham MC (2015) Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurosci 35:643–647. 10.1523/JNEUROSCI.3483-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MJ, Mu EW, Noakes PG, Lavidis NA, Bellingham MC (2016) Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Acta Neuropathol Commun 4:77. 10.1186/s40478-016-0347-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geevasinga N, Menon P, Nicholson GA, Ng K, Howells J, Kril JJ, Yiannikas C, Kiernan MC, Vucic S (2015) Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol 72:1268–1274. 10.1001/jamaneurol.2015.1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geevasinga N, Menon P, Özdinler PH, Kiernan MC, Vucic S (2016) Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat Rev Neurol 12:651–661. 10.1038/nrneurol.2016.140 [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, Heintz N (2013) GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80:1368–1383. 10.1016/j.neuron.2013.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskreutz J, Van Den Bosch L, Keller BU (2010) Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47:165–174. 10.1016/j.ceca.2009.12.002 [DOI] [PubMed] [Google Scholar]
- Gurney ME. (1997) The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci 152 [Suppl 1]:S67–S73. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775. 10.1126/science.8209258 [DOI] [PubMed] [Google Scholar]
- Hammer RP Jr, Tomiyasu U, Scheibel AB (1979) Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp Neurol 63:336–346. 10.1016/0014-4886(79)90129-8 [DOI] [PubMed] [Google Scholar]
- Harris KD, Shepherd GM (2015) The neocortical circuit: themes and variations. Nat Neurosci 18:170–181. 10.1038/nn.3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattox AM, Nelson SB (2007) Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J Neurophysiol 98:3330–3340. 10.1152/jn.00397.2007 [DOI] [PubMed] [Google Scholar]
- Hill DN, Varga Z, Jia H, Sakmann B, Konnerth A (2013) Multibranch activity in basal and tuft dendrites during firing of layer 5 cortical neurons in vivo. Proc Natl Acad Sci U S A 110:13618–13623. 10.1073/pnas.1312599110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S (2005) A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3:e159. 10.1371/journal.pbio.0030159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Kang SH, Fukaya M, Bergles DE (2013) Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 16:668–676. 10.1038/nn.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince P, Stout N, Shaw P, Slade J, Hunziker W, Heizmann CW, Baimbridge KG (1993) Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol Appl Neurobiol 19:291–299. 10.1111/j.1365-2990.1993.tb00443.x [DOI] [PubMed] [Google Scholar]
- Jara JH, Villa SR, Khan NA, Bohn MC, Ozdinler PH (2012) AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis 47:174–183. 10.1016/j.nbd.2012.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai K, Shibuya K, Sato Y, Misawa S, Nasu S, Sekiguchi Y, Mitsuma S, Isose S, Fujimaki Y, Ohmori S, Koga S, Kuwabara S (2012) Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 83:734–738. 10.1136/jnnp-2011-301782 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Matney CJ, Blankenship A, Hestrin S, Brown SP (2014) Layer 6 corticothalamic neurons activate a cortical output layer, layer 5a. J Neurosci 34:9656–9664. 10.1523/JNEUROSCI.1325-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AE, Woodhouse A, Kirkcaldie MT, Vickers JC (2016) Excitotoxicity in ALS: overstimulation, or overreaction? Exp Neurol 275:162–171. 10.1016/j.expneurol.2015.09.019 [DOI] [PubMed] [Google Scholar]
- Kremer EJ, Boutin S, Chillon M, Danos O (2000) Canine adenovirus vectors: an alternative for adenovirus-mediated gene transfer. J Virol 74:505–512. 10.1128/JVI.74.1.505-512.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bär PR, Anelli R, Heckman CJ, Kroese AB (2004) Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 91:571–575. 10.1152/jn.00665.2003 [DOI] [PubMed] [Google Scholar]
- Leal SS, Gomes CM (2015) Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability. Front Cell Neurosci 9:225. 10.3389/fncel.2015.00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Lamotte d'Incamps B, Imhoff-Manuel RD, Zytnicki D (2014) Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife 3:e04046. 10.7554/eLife.04046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S, Al-Sarraj S, Kibble M, Landau S, Parnavelas J, Cotter D, Everall I, Leigh PN (2004) Cortical selective vulnerability in motor neuron disease: a morphometric study. Brain 127:1237–1251. 10.1093/brain/awh132 [DOI] [PubMed] [Google Scholar]
- Marangi G, Traynor BJ (2015) Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res 1607:75–93. 10.1016/j.brainres.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Cazenave W, Cattaert D, Branchereau P (2013) Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 54:116–126. 10.1016/j.nbd.2013.02.011 [DOI] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, Bader GD (2010) Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5:e13984. 10.1371/journal.pone.0013984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MN, Okaty BW, Nelson SB (2008) Region-specific spike-frequency acceleration in layer 5 pyramidal neurons mediated by Kv1 subunits. J Neurosci 28:13716–13726. 10.1523/JNEUROSCI.2940-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi D, Kassa RM, Del Tongo C, Mariotti R, Bentivoglio M (2009) Voronoi-based spatial analysis reveals selective interneuron changes in the cortex of FALS mice. Exp Neurol 215:77–86. 10.1016/j.expneurol.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Goff LA, Brettler AC, Chen HH, Hrvatin S, Rinn JL, Arlotta P (2015) DeCoN: genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron 85:275–288. 10.1016/j.neuron.2014.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihei K, McKee AC, Kowall NW (1993) Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients. Acta Neuropathol 86:55–64. 10.1007/BF00454899 [DOI] [PubMed] [Google Scholar]
- O'Leary DD, Koester SE (1993) Development of projection neuron types, axon pathways, and patterned connections of the mammalian cortex. Neuron 10:991–1006. 10.1016/0896-6273(93)90049-W [DOI] [PubMed] [Google Scholar]
- Oswald MJ, Tantirigama ML, Sonntag I, Hughes SM, Empson RM (2013) Diversity of layer 5 projection neurons in the mouse motor cortex. Front Cell Neurosci 7:174. 10.3389/fncel.2013.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdinler PH, Benn S, Yamamoto TH, Güzel M, Brown RH Jr, Macklis JD (2011) Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G93A transgenic ALS mice. J Neurosci 31:4166–4177. 10.1523/JNEUROSCI.4184-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C (2003) Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 351:153–156. 10.1016/j.neulet.2003.07.010 [DOI] [PubMed] [Google Scholar]
- Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C (2009) Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol 215:368–379. 10.1016/j.expneurol.2008.11.002 [DOI] [PubMed] [Google Scholar]
- Roselli F, Caroni P (2015) From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85:901–910. 10.1016/j.neuron.2014.12.063 [DOI] [PubMed] [Google Scholar]
- Saba L, Viscomi MT, Caioli S, Pignataro A, Bisicchia E, Pieri M, Molinari M, Ammassari-Teule M, Zona C (2016) Altered functionality, morphology, and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb Cortex 26:1512–1528. 10.1093/cercor/bhu317 [DOI] [PubMed] [Google Scholar]
- Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71:35–48. 10.1016/j.neuron.2011.06.031 [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96. 10.1016/j.neuron.2013.07.027 [DOI] [PubMed] [Google Scholar]
- Shah MM. (2014) Cortical HCN channels: function, trafficking and plasticity. J Physiol 592:2711–2719. 10.1113/jphysiol.2013.270058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya K, Park SB, Geevasinga N, Menon P, Howells J, Simon NG, Huynh W, Noto Y, Götz J, Kril JJ, Ittner LM, Hodges J, Halliday G, Vucic S, Kiernan MC (2016) Motor cortical function determines prognosis in sporadic ALS. Neurology 87:513–520. 10.1212/WNL.0000000000002912 [DOI] [PubMed] [Google Scholar]
- Siklós L, Engelhardt J, Harati Y, Smith RG, Joó F, Appel SH (1996) Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann Neurol 39:203–216. 10.1002/ana.410390210 [DOI] [PubMed] [Google Scholar]
- Soudais C, Laplace-Builhe C, Kissa K, Kremer EJ (2001) Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J 15:2283–2285. 10.1096/fj.01-0321fje [DOI] [PubMed] [Google Scholar]
- Spalloni A, Origlia N, Sgobio C, Trabalza A, Nutini M, Berretta N, Bernardi G, Domenici L, Ammassari-Teule M, Longone P (2011) Postsynaptic alteration of NR2A subunit and defective autophosphorylation of alphaCaMKII at threonine-286 contribute to abnormal plasticity and morphology of upper motor neurons in presymptomatic SOD1G93A mice, a murine model for amyotrophic lateral sclerosis. Cereb Cortex 21:796–805. 10.1093/cercor/bhq152 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB (2006) Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci 9:99–107. 10.1038/nn1618 [DOI] [PubMed] [Google Scholar]
- Suter BA, Migliore M, Shepherd GM (2013) Intrinsic electrophysiology of mouse corticospinal neurons: a class-specific triad of spike-related properties. Cereb Cortex 23:1965–1977. 10.1093/cercor/bhs184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539:197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31:46–53. 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, Rudy B (2016) GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91:260–292. 10.1016/j.neuron.2016.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Kiernan MC (2012) Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler 13:245–250. 10.3109/17482968.2011.636050 [DOI] [PubMed] [Google Scholar]
- Udaka F, Kameyama M, Tomonaga M (1986) Degeneration of Betz cells in motor neuron disease. A Golgi study. Acta Neuropathol 70:289–295. 10.1007/BF00686086 [DOI] [PubMed] [Google Scholar]
- van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH Jr, Constantine-Paton M, Bellingham MC (2008) Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28:10864–10874. 10.1523/JNEUROSCI.1340-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Izaurieta P, Fritz E, Alvarez FJ (2012) Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Cell Biochem 113:3301–3312. 10.1002/jcb.24234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal S, Hsiao CF, Sonoda T, Wiedau-Pazos M, Chandler SH (2015) Homeostatic dysregulation in membrane properties of masticatory motoneurons compared with oculomotor neurons in a mouse model for amyotrophic lateral sclerosis. J Neurosci 35:707–720. 10.1523/JNEUROSCI.1682-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C (2013) Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav 3:431–457. 10.1002/brb3.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC (2006) Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129:2436–2446. 10.1093/brain/awl172 [DOI] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA, Kiernan MC (2008) Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131:1540–1550. 10.1093/brain/awn071 [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Cudkowicz ME (2015) Cortical hyperexcitability in amyotrophic lateral sclerosis: C9orf72 repeats. JAMA Neurol 72:1235–1236. 10.1001/jamaneurol.2015.2197 [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ (2014) Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7:1–11. 10.1016/j.celrep.2014.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, Nicholson GA (2013) Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry 84:931–935. 10.1136/jnnp-2012-304529 [DOI] [PubMed] [Google Scholar]
- Wood SN. (2011) Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J R Stat Soc Series B Stat Methodol 73:3–36. 10.1111/j.1467-9868.2010.00749.x [DOI] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015) The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev 67:821–870. 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang DW, Cheema SS (2002) Degeneration of corticospinal and bulbospinal systems in the superoxide dismutase 1(G93A G1H) transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 332:99–102. 10.1016/S0304-3940(02)00944-8 [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang L, Liang B, Schroeder D, Zhang ZW, Cox GA, Li Y, Lin DT (2016) Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat Neurosci 19:557–559. 10.1038/nn.4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. All primary data from the RNA-sequencing (RNA seq) experiments are deposited on the National Center for Biotechnology Information (NCBI) Short Read Archive with NCBI GEO Accession Number GSE101391, and all scripts and workflows for RNA-sequencing analysis are available in conjunction with the preprocessed data upon request. Processed data supporting the findings can be found in an extended data file associated with Figure 8.
Figure 8.
Gene set enrichment analysis implicates distinct molecular mechanisms in corticospinal and corticocortical neurons from hSOD1G93A mice. A, Schematic of the experimental procedure for selective purification of two different molecularly identified cortical projection neuron populations. Total RNA from CTIP2-positive corticospinal neurons and SATB2-positive corticocortical neurons was purified for RNA-seq analysis using cell type-specific antibodies. B, Ctip2 and Satb2 expression profiles for molecularly identified cell types from hSOD1G93A and hSOD1WT mice (n = 4 mice/group). C, Enrichment map of significantly enriched Reactome gene sets from hSOD1G93A vs hSOD1WT RNA sequencing analyses in CTIP2-positive corticospinal neurons (CSNs) and SATB2-positive corticocortical neurons (CCNs). Node color corresponds to gene set enrichment (red) or depletion (blue) in hSOD1G93A CSNs relative to hSOD1WT CSNs. The border color represents gene set enrichment or depletion results for CCNs. White indicates no significant enrichment or depletion for a given gene set for that cell type. Node areas correspond to relative gene set sizes, and line thicknesses (tan) indicate the degree of overlap in gene composition between connected sets. Gene sets were determined to be significantly enriched or depleted using a preranked gene set enrichment analysis (Kolmogorov–Smirnov test, p < 0.05, Benjamini–Hochberg corrected). Supporting data are found in Figure 8-1 available at https:/10.1523/JNEUROSCI.0811-17.2017.f8-1.