

Abstract
A method for a directed stereoselective guanidinylation of alkenes is described. The guanidine unit can be delivered as an intact fragment by a hydroxy or carboxy group, usually with a high level of stereocontrol. After the guanidine delivery, the directing group can be cleaved under exceptionally mild conditions, typically by alcoholysis in the presence of acetic acid. Broad functional group tolerance and mild reaction conditions for the cycloguanidilation suggest applications in medicinal chemistry and natural products synthesis.
Graphical Abstract
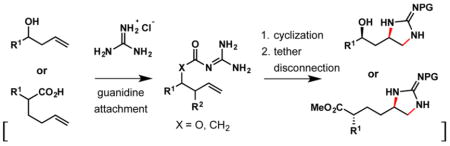
Guanidines are challenging targets for synthesis even by the standards of other nitrogen-containing compounds.1 General methods for the introduction of the guanidyl group into complex structures, let alone stereoselective methods, are limited and typically involve guanidylation of a preinstalled amino group. These classic transformations include a coupling of primary amines with cyanamide,2 pyrrazolylcarrboxamidines,3 S-alkylisothiourea reagents,4 or trifluoromethane-sulfonyl guanidine reagents (Goodman reagent).5 Implementation of these strategies in the synthesis of complex guanidine-containing compounds and natural products thus can require extended synthetic routes, in some cases reducing the overall efficiency of the synthesis.
The recently emerging methods for vicinal diamination of alkenes provide an intriguing alternative for the synthesis of functionalized guanidines.6 Several inter- and intramolecular diaminations of unactivated alkenes with ureas, sulfamides, and guanidines catalyzed by palladium,7 nickel,8 copper,9 and gold complexes10 as well as halogenating reagents11 have been reported. These promising approaches are yet to demonstrate a broad scope and applicability for stereoselective synthesis of cyclic guanidines with desired functionalization. The requirement for tethering of guanidine or another dinitrogen source by a simple alkyl chain complicates further functionalization of products, and still generally requires guanidilation of a preinstalled amino group at an earlier stage. Our interest in developing efficient strategies for the synthesis of guanidine alkaloids shown in Scheme 1a brought these limitations to our attention and prompted us to develop a method for a stereoselective delivery of the entire intact guanidine group to an unactivated alkene directed by a hydroxy and carboxy group. The method is operationally simple and readily allows for further functionalization of the installed guanidine.
Scheme 1.
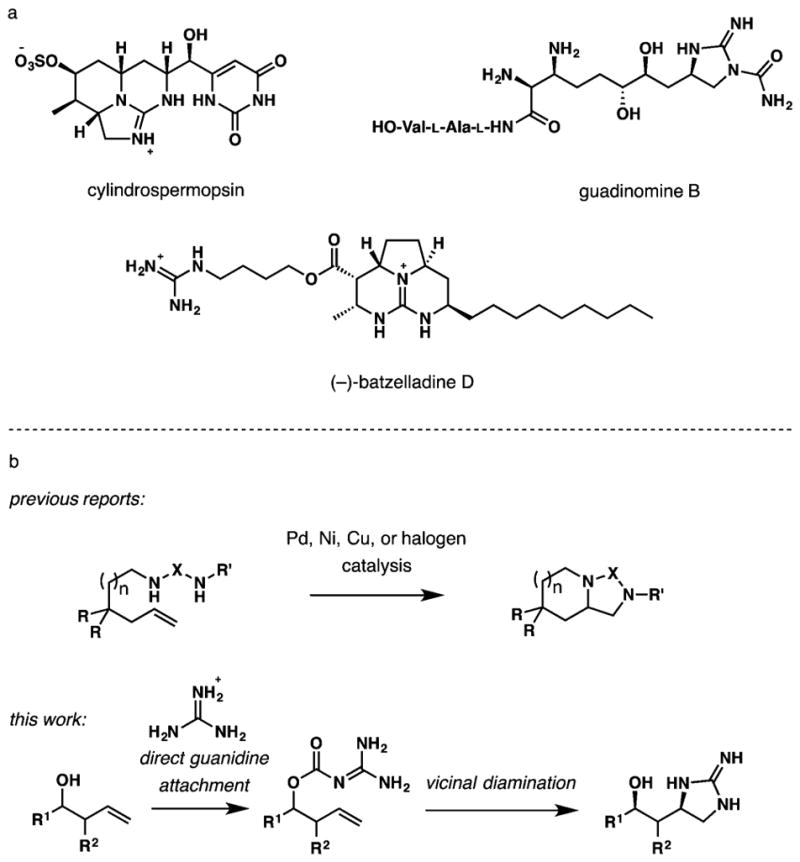
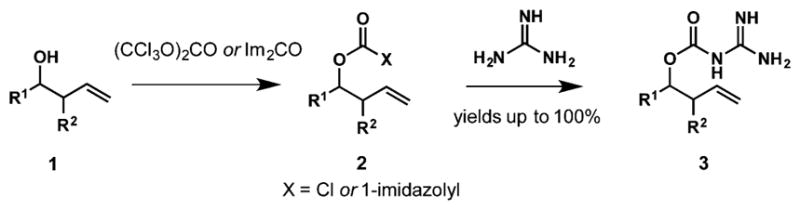
As depicted in Scheme 1b, our reaction design requires attachment of guanidine to a hydroxy group by a carbamate linker followed by electrophilic cyclization onto the alkene. The initial goal was to develop a simple method for the attachment of guanidine to homoallylic alcohols using readily available guanidine hydrochloride. Although different prospective tethers were considered, including silyl, sulfonyl, and methylene, the majority of attempted transformations were unfruitful due to the low stability of intermediates and products. In contrast, we found that carbamates 3 could be prepared in two simple steps from alcohols 1 via an initial conversion to chloroformates with triphosgene or (1-imidazolyl)formats with 1,1′-carbonyl-diimidazole (CDI), followed by treatment with guanidine (Scheme 2). Both methods delivered the material of similar purity and in comparable yields (see Supporting Information); however, CDI was a more benign and user-friendly reagent due to considerably lower toxicity and volatility compared to triphosgene. In addition, when R1 = Ar, CDI enabled a cleaner conversion to 3 without formation of carbonate side products observed with triphosgene. Therefore, for subsequent studies, 1,1′-carbonyldiimidazole had been used to access a set of substrates for intramolecular electrophilic delivery of guanidine to the double bond.
Scheme 2.
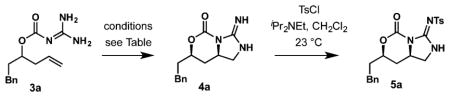
With an effective protocol for the preparation of substrates 3 established, the next goal was to identify optimal conditions for cycloguanidilation. Despite previous success in diamination and cycloguanidylation of alkenyl guanidines mediated by metal catalysis, only low conversions were observed under varying conditions with catalysts such as Pd(OAc)2, NiCl2, or CuI (Table 1, entries 1–5).
Table 1.
Identification of Optimal Reagent for Cycloguanidylationa
| |||
|---|---|---|---|
| entry | reaction conditions | conversion (%) | yield (%) |
| 1 | 10 mol % Pd(OAc)2, CuI, K2CO3, DMF, 23 °C, 24 h | 0 | 0 |
| 2 | 25 mol % Pd(OAc)2, PhI(OAc)2, CH2Cl2, 23 °C, 24 h | 15 | 0 |
| 3 | 5 mol % Pd(OAc)2, PhI(OAc)2, NMe4Cl, NaOAc, CH2Cl2, 23 °C, 24 h | 30 | 0 |
| 4 | 10 mol % NiCl2, PhI(OAc)2, NaOAc, DMF, 23 °C, 24 h | 0 | 0 |
| 5 | 10 mol % CuI, K2CO3, 10 mol % 2,2′-bipyridine, O2, DMF, 60 °C, 24 h | 0 | 0 |
| 6 | 3 equiv of t-BuOCl, CH2Cl2, 0 °C, 20 min | 100b | 0 |
| 7 | Py2IBF4, toluene, 23 °C or reflux, 24 h | 65 | 0 |
| 8 | 2.1 equiv of I2, NaHCO3, CH2Cl2, 23 °C, 6 h | 15 | 0 |
| 9 | 2.1 equiv of NIS, NaHCO3, CH2Cl2, 23 °C, 1 h | 100 | 62 |
| 10 | 2.1 equiv of NIS, NaHCO3, CH3CN, 0 °C, 5 h | 100 | 84 |
See Supporting Information for additional details; isolated yields of 5a after tosylation are reported. Conversion measured by 1H NMR analysis of a crude mixture of products.
Dichloroguanidine is isolated.
Halogen-based reagents were examined next. Although conversion with iodine was still low (entry 8), it was substantially improved with tert-butylhypochlorite and dipyridineiodonium tetrafluoroborate (entries 6, 7). tert-Butyl hypochlorite effected N-chlorination only, affording a fairly unreactive dichloroguanidine product. The optimal reagent has been determined to be N-iodosuccinimide (NIS), which under optimal conditions (NaHCO3, CH3CN, 0 °C) enabled 100% conversion and delivered the product in 84% isolated yield (entry 10). Importantly, complete diastereoselectivity was observed, meeting our goal of stereocontroled delivery of guanidine to the unactivated double bond directed by a preinstalled hydroxy group.
With the optimized set of reagents, the reaction scope was examined with a variety of aryl and alkyl substituted substrates derived from homoallylic alcohols (Figure 1). The substrate scope was designed to test compatibility with a range of reactive functional groups suitable for applications in medicinal chemistry and natural products synthesis. All products were isolated after guanidine sulfonylation to ease purification. We first established that N-nosylation, N-tosylation, and N-(4-methoxyphenyl)sulfonylation afford comparable isolated yields with 4a (products 5a–c) and proceeded with tosylation for the remainder of the substrate scope studies. Compounds comprising different linear and branched alkyl substituents readily undergo cyclization, producing expected guanidines 5d–f in high yields as single diastereomers. The introduction of benzyl or silyl ethers as well as ester groups in the substrate side chain does not hamper the overall efficiency of guanidylation, affording compounds 5g–j after tosylation.
Figure 1.

Substrate scope for the cycloguanidylation reaction. a The reaction was performed at 0 °C for 2 h. b The reaction was performed at 0 °C for 5 h and then at 23 °C for another 5 h. c Products were isolated after methanolysis.
In order to further explore the chemoselectivity of the cycloguanidylation method, substrates comprising the distant terminal alkyne and olefin moiety were examined. In both cases, application of standard conditions initially resulted in reduced yields of cyclic guanidine products; however, decreasing the reaction time to 2 h allowed the isolatation of compounds 5k and 5l in substantially improved yields (70% and 50% respectively). Substrates derived from substituted benzyl alcohols were found to be unexpectedly less reactive compared to their aliphatic counterparts and require extended reaction times to reach full conversion. Nevertheless, phenyl (5m), naphtyl (5n), 4-bromophenyl (5o), 2-fluorophenyl (5p), and 4-cyanophenyl (5q) substrates were isolated after tosylation in practical yields and excellent stereoselectivity. At the same time, cyclization of the guanidine bearing a 4-methoxyphenyl group produced compound 5r in only 12% yield, accompanied by significant amounts of decomposition products, presumably arising via a carbocationic pathway. The cyclization of substrates derived from both syn- and anti-3-methyl-1-alkene-4-ols was also investigated. The reaction of the latter with NIS produced cyclic product 5s in 64% yield as a 2:1 mixture of diastereomers. Notably, the reaction of the syn-isomer gave rise to 5t in 70% yield as a single diastereomer.
Substrates incorporating acylated primary amines were shown to undergo the cyclization under standard conditions; however, their separation from succinimide was troublesome. On the other hand, clean products were obtained in good yields after subsequent methanolysis (see below) as corresponding methyl carbonates 5u and 5v.
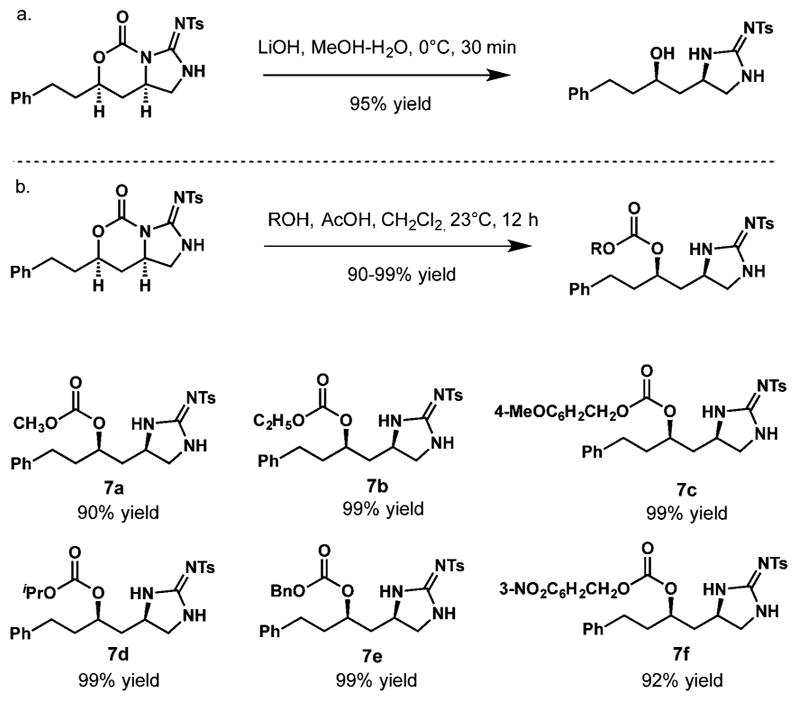
Next, two strategies to modify the carbonyl tether were explored. The rapid hydrolysis of compound 5a mediated by aqueous lithium hydroxide readily furnished alcohol 6 in 95% yield (Scheme 3a). In addition, we discovered unprecedented and unexpected reactivity, whereby cyclic guanidine-derived carbamates reacted under exceptionally mild conditions with different alcohols resulting in regioselective ring opening of the carbamate to furnish corresponding carbonates 7 (Scheme 3b). The reaction requires no extra additives, but can be accelerated by addition of acetic acid. A range of simple aliphatic and aromatic alcohols can be utilized for this transformation providing orthogonally protected compounds 7a–f in excellent yields. t-BuOH was found to be the only unreactive alcohol, presumably due to steric hindrance.
Scheme 3.
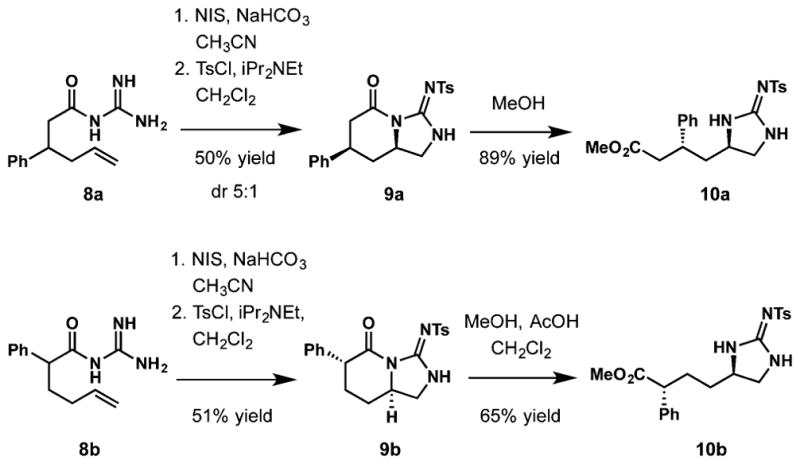
Encouraged by these results, we then envisioned that carboxylic acids might also serve as a directing group for the cycloguanidilation reaction. 2-Phenyl- and 3-phenylhexenoic acids were converted to acylguanidine substrates 8a and 8b by treatment with carbonyldiimidazole and guanidine (Scheme 4). Subsequent exposure to NIS under the standard conditions resulted in a clean cyclization; however, an attempt to isolate the product prior to tosylation led to a rapid polymerization of the material. A successful isolation was realized by treatment of the reaction mixture with p-toluenesulfonyl chloride, which allowed isolation of the cyclic guanidines 9a and 9b in 50% and 51% yield, respectively. These cyclic products were subjected to methanolysis, furnishing guanidyl esters 10a and 10b in good yields.
Scheme 4.
While the precise mechanistic underpinnings related to the first C–N bond forming event are presently unclear, we hypothesize that the stereochemical outcome is governed by transannular interactions in the developing six-membered ring, maximizing the number of equatorial substituents.
In closing, we developed a method for directed stereo-selective guanidylation of alkenes. Guanidine can be delivered as an intact unit by a hydroxy or carboxy group, usually with a high level of stereocontrol. We demonstrated that, after guanidine delivery, the directing group can be cleaved under exceptionally mind conditions, typically by alcoholysis in the presence of acetic acid. Broad functional group tolerance and mild reaction conditions for the cycloguanidylation and subsequent tether removal or modification suggest applications in medicinal chemistry and natural products synthesis.
Supplementary Material
Acknowledgments
Dr. Hongjun Zhou (UC Santa Barbara) is acknowledged for the continued assistance with NMR spectroscopy. This work was supported by the NIH (NIGMS, R01-077379, and NIEHS R03 ES025345-01). K.Y. was supported by an NSF Graduate Fellowship (DGE-1144085). J.C. is a recepient of the Rickborn–Johnson Fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b02778.
Experimental procedures, characterization of new compounds, and spectral data (PDF)
References
- 1.(a) Mailyan AK, Eickhoff JA, Minakova AS, Gu Z, Lu P, Zakarian A. Chem Rev. 2016;116:4441. doi: 10.1021/acs.chemrev.5b00712. [DOI] [PubMed] [Google Scholar]; (b) Ma Y, De S, Chen C. Tetrahedron. 2015;71:1145. doi: 10.1016/j.tet.2014.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Berlinck RGS, Romminger S. Nat Prod Rep. 2016;33:456. doi: 10.1039/c5np00108k. [DOI] [PubMed] [Google Scholar]; (d) Berlinck RGS, Burtoloso ACB, Kossuga MH. Nat Prod Rep. 2008;25:919. doi: 10.1039/b507874c. [DOI] [PubMed] [Google Scholar]; (e) Mulcahy JV, Du Bois J. J Am Chem Soc. 2008;130:12630. doi: 10.1021/ja805651g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fleming JJ, McReynolds MD, Du Bois J. J Am Chem Soc. 2007;129:9964. doi: 10.1021/ja071501o. [DOI] [PubMed] [Google Scholar]; (g) Bhonde VR, Looper RE. J Am Chem Soc. 2011;133:20172. doi: 10.1021/ja2098063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Davis TL. Org Synth. 1927;7:46. [Google Scholar]; (b) Dukat M, Abdel-Rahman A, Ismaiel A, Ingher S, Teitler M, Gyermek L, Glennon RA. J Med Chem. 1996;39:4017. doi: 10.1021/jm9603936. [DOI] [PubMed] [Google Scholar]; (c) Tsubokura K, Iwata T, Taichi M, Kurbangalieva A, Fukase K, Nakao Y, Tanaka K. Synlett. 2014;25:1302. [Google Scholar]; (d) Li J, Neuville L. Org Lett. 2013;15:6124. doi: 10.1021/ol4029622. [DOI] [PubMed] [Google Scholar]; (e) Looper RE, Haussener TJ, Mack JBC. J Org Chem. 2011;76:6967. doi: 10.1021/jo201264j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Bernatowicz MS, Wu Y, Matsueda GR. Tetrahedron Lett. 1993;34:3389. [Google Scholar]; (b) Drake B, Patek M, Lebl M. Synthesis. 1994;1994:579. [Google Scholar]; (c) Katriztky AR, Parris RL, Allin SM, Steel PS. Synth Commun. 1995;25:1173. [Google Scholar]
- 4.(a) Poss MA, Iwanowicz E, Reid JA, Lin J, Gu Z. Tetrahedron Lett. 1992;33:5933. [Google Scholar]; (b) Iwanowicz E, Poss MA, Lin J. Synth Commun. 1993;23:1443. [Google Scholar]; (c) Kim KS, Qian L. Tetrahedron Lett. 1993;34:7677. [Google Scholar]; (d) Robinson S, Roskamp EJ. Tetrahedron. 1997;53:6697. [Google Scholar]; (e) Guo ZX, Cammidge AN, Horwell DC. Synth Commun. 2000;30:2933. [Google Scholar]; (f) Chandrakumar NS. Synth Commun. 1996;26:2613. [Google Scholar]; (g) Yong YF, Kowalski JA, Lipton MA. J Org Chem. 1997;62:1540. [Google Scholar]; (h) Yong YF, Kowalski JA, Lipton MA. Tetrahedron Lett. 1999;40:53. [Google Scholar]; (i) Gers T, Kunce D, Markowski P, Izdebski J. Synthesis. 2004;2004:37. doi: 10.1002/psc.585. [DOI] [PubMed] [Google Scholar]; (j) Lammin SG, Pedgrift BL, Ratcliffe AJ. Tetrahedron Lett. 1996;37:6815. [Google Scholar]; (k) Levallet C, Lerpiniere J, Ko SY. Tetrahedron. 1997;53:5291. [Google Scholar]
- 5.(a) Feichtinger K, Zapf C, Sings HL, Goodman M. J Org Chem. 1998;63:3804. [Google Scholar]; (b) Feichtinger K, Sings HL, Baker TJ, Matthews K, Goodman MJ. J Org Chem. 1998;63:8432. [Google Scholar]
- 6.(a) De Jong S, Nosal DG, Wardrop DJ. Tetrahedron. 2012;68:4067. doi: 10.1016/j.tet.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Muñiz K, Martínez C. J Org Chem. 2013;78:2168. doi: 10.1021/jo302472w. [DOI] [PubMed] [Google Scholar]
- 7.(a) Streuff J, Hövelmann CH, Nieger M, Muñiz K. J Am Chem Soc. 2005;127:14586. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (b) Bar GLJ, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2005;127:7308. doi: 10.1021/ja051181d. [DOI] [PubMed] [Google Scholar]; (c) Du H, Zhao B, Shi Y. J Am Chem Soc. 2007;129:762. doi: 10.1021/ja0680562. [DOI] [PubMed] [Google Scholar]; (d) Du H, Yuan W, Zhao B, Shi Y. J Am Chem Soc. 2007;129:7496. doi: 10.1021/ja072080d. [DOI] [PubMed] [Google Scholar]; (e) Xu L, Du H, Shi Y. J Org Chem. 2007;72:7038. doi: 10.1021/jo0709394. [DOI] [PubMed] [Google Scholar]; (f) Muñiz K, Hövelmann CH, Streuff J. J Am Chem Soc. 2008;130:763. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]; (g) Muñiz K, Streuff J, Chávez P, Hövelmann CH. Chem - Asian J. 2008;3:1248. doi: 10.1002/asia.200800148. [DOI] [PubMed] [Google Scholar]; (h) Hövelmann CH, Streuff J, Brelot L, Muñiz K. Chem Commun. 2008:2334. doi: 10.1039/b719479j. [DOI] [PubMed] [Google Scholar]; (i) Iglesias Á, Pérez EG, Muñiz K. Angew Chem, Int Ed. 2010;49:8109. doi: 10.1002/anie.201003653. [DOI] [PubMed] [Google Scholar]; (j) Muñiz K, Kirsch J, Chávez P. Adv Synth Catal. 2011;353:689. [Google Scholar]; (k) Sibbald PA, Michael FE. Org Lett. 2009;11:1147. doi: 10.1021/ol9000087. [DOI] [PubMed] [Google Scholar]; (l) Chávez P, Kirsch J, Streuff J, Muñiz K. J Org Chem. 2012;77:1922. doi: 10.1021/jo202507d. [DOI] [PubMed] [Google Scholar]
- 8.Muñiz K, Streuff J, Hövelmann CH, Núñez A. Angew Chem, Int Ed. 2007;46:7125. doi: 10.1002/anie.200702160. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]; (b) Zabawa TP, Chemler SR. Org Lett. 2007;9:2035. doi: 10.1021/ol0706713. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sequeira FC, Turnpenny BW, Chemler SR. Synfacts. 2010;2010:1291. [Google Scholar]; (d) Wen Y, Zhao B, Shi Y. Org Lett. 2009;11:2365. doi: 10.1021/ol900808z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhao B, Peng X, Zhu Y, Ramirez TA, Cornwall RG, Shi Y. J Am Chem Soc. 2011;133:20890. doi: 10.1021/ja207691a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang YF, Zhu X, Chiba S. J Am Chem Soc. 2012;134:3679. doi: 10.1021/ja2120629. [DOI] [PubMed] [Google Scholar]; (g) Shen K, Wang Q. Chem Sci. 2015;6:4279. doi: 10.1039/c5sc00897b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu RH, Wei D, Han B, Yu W. ACS Catal. 2016;6:6525. [Google Scholar]
- 10.(a) Iglesias A, Muñiz K. Chem - Eur J. 2009;15:10563. doi: 10.1002/chem.200901199. [DOI] [PubMed] [Google Scholar]; (b) de Haro T, Nevado C. Angew Chem, Int Ed. 2011;50:906. doi: 10.1002/anie.201005763. [DOI] [PubMed] [Google Scholar]
- 11.(a) Muñiz K, Hövelmann CH, Campos-Gómez E, Barluenga J, González JM, Streuff J, Nieger M. Chem - Asian J. 2008;3:776. doi: 10.1002/asia.200700373. [DOI] [PubMed] [Google Scholar]; (b) Li H, Widenhoefer RA. Tetrahedron. 2010;66:4827. doi: 10.1016/j.tet.2010.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chávez P, Kirsch J, Hövelmann CH, Streuff J, Martinez-Belmonte M, Escudero-Adán EC, Martin E, Muñiz K. Chem Sci. 2012;3:2375. [Google Scholar]; (d) Röben C, Souto JA, Escudero-Adán EC, Muñiz K. Org Lett. 2013;15:1008. doi: 10.1021/ol3034884. [DOI] [PubMed] [Google Scholar]; (e) Danneman MW, Hong KB, Johnston JN. Org Lett. 2015;17:2558. doi: 10.1021/acs.orglett.5b01177. [DOI] [PubMed] [Google Scholar]; (f) Mizar P, Laverny A, El-Sherbini M, Farid U, Brown M, Malmedy F, Wirth T. Chem - Eur J. 2014;20:9910. doi: 10.1002/chem.201403891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hong KB, Johnston JN. Org Lett. 2014;16:3804. doi: 10.1021/ol501693j. [DOI] [PubMed] [Google Scholar]; (h) Ortiz GX, Kang B, Wang Q. J Org Chem. 2014;79:571. doi: 10.1021/jo4022666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.