

Graphical abstract

Keywords: Alveolo-capillary barrier, Nanoparticles, Polystyrene, Calu-3, THP-1, HPMEC-ST1.6R
Abstract
As the lung is one of the main routes of exposure to manufactured nanoparticles, we developed an in vitro model resembling the alveolo-capillary barrier for the study of nanoparticle translocation.
In order to provide a relevant and ethical in vitro model, cost effective and easy-to-implement human cell lines were used. Pulmonary epithelial cells (Calu-3 cell line) and macrophages (THP-1 differentiated cells) were cultivated on the apical side and pulmonary endothelial cells (HPMEC-ST1.6R cell line) on the basal side of a microporous polyester membrane (Transwell®). Translocation of non-functionalized (51 and 110 nm) and aminated (52 nm) fluorescent polystyrene (PS) nanobeads was studied in this system.
The use of Calu-3 cells allowed high transepithelial electrical resistance (TEER) values (>1000 Ω cm2) in co-cultures with or without macrophages. After 24 h of exposure to non-cytotoxic concentrations of non-functionalized PS nanobeads, the relative TEER values (%/t0) were significantly decreased in co-cultures. Epithelial cells and macrophages were able to internalize PS nanobeads. Regarding translocation, Transwell® membranes per se limit the passage of nanoparticles between apical and basal side. However, small non-functionalized PS nanobeads (51 nm) were able to translocate as they were detected in the basal side of co-cultures. Altogether, these results show that this co-culture model present good barrier properties allowing the study of nanoparticle translocation but research effort need to be done to improve the neutrality of the porous membrane delimitating apical and basal sides of the model.
1. Introduction
With their new properties, nanoparticles (NPs) open a broad field of novel opportunities for industrials. However, consumers or workers can easily come into contact with these NPs and their effects on human health are still poorly understood [1]. Inhalation is one of the major routes of exposure to NPs present in ambient air and, depending on their unique physico-chemical properties (size, shape, etc.), NPs can reach the alveolar region of the lung [2]. Consequently, NPs could translocate through the alveolo-capillary barrier into the bloodstream, and potentially reach and damage other organs [3], [4].
The alveolo-capillary barrier is vital for the organism as it allows gas exchanges in the course of breathing. This barrier consists of all elements between alveoli and blood micro-vessels including: surfactant, alveolar macrophages, pneumocytes, a basal lamina and the endothelium of blood micro-vessels [5]. Two types of pneumocytes constitute the alveolar epithelium. Type I pneumocytes are large flattened cells covering roughly 90% of the alveolar surface area, playing the main role in the hematosis process [6]. Type II pneumocytes (7% of the alveolar surface area) are cuboidal cells interspersed between type I pneumocytes, synthesizing the alveolar surfactant and acting as progenitors of type I pneumocytes [7], [8]. The alveolar epithelium is highly impermeable, essentially due to the presence of intercellular tight junctions between epithelial cells and, although less significantly, to the decrease of surface tension by surfactant between the air space and the alveolar liquid [9]. Alveolar macrophages are normally quiescent (weak pro-inflammatory secretion and phagocytosis ability) in order to prevent damaging the alveoli [10]. The alveolar endothelium is continuous, non-fenestrated, and constituted by micro-vascular endothelial cells [11].
As these cell types interact to form a functional alveolo-capillary barrier, several in vitro co-culture models using different cellular associations have been developed recently to study the cytotoxic effects of NPs close to the in vivo situation [12], [13], [14], [15]. Rothen-Rutishauser et al. used the A549 alveolar cell line (originating from human lung carcinoma), in association with primary human dendritic cells and macrophages in Transwell® membranes, in order to study the local effects of NPs on the epithelium and alveolar macrophages, on the one hand, and the uptake by dendritic cells (systemic effects), on the other [16], [17]. However, A549 epithelial cells, which are the most widely used and characterized alveolar epithelial model in literature, are not able to form a strong barrier, and are inappropriate for NP translocation studies [12], [18]. NCI-H441 cells (originating from human lung papillary adenocarcinoma) have bronchiolar morphology and develop more elevated TEER values after incubation with dexamethasone when co-cultured with endothelial cells [19], [20]. Hermanns et al. used NCI-H441 cells in co-culture with ISO-HAS-1 endothelial cells on Transwell® membranes to study the interactions with polyethyleneimine NPs [21]. However, Farcal et al., by adding PMA-differentiated THP-1 macrophages on the apical side of the same model, showed a drastic decrease of the transepithelial electrical resistance (TEER) values in co-culture associated with a high release of TNF-α and IL-8 after association with differentiated THP-1 macrophages [4].
To the best of our knowledge, no co-culture model, including macrophages, epithelial and endothelial cells, showing acceptable barrier properties have been previously published. The objective of this work was to develop an in vitro co-culture model with barrier properties mimicking the in vivo situation, and usable to study NP translocation. The bronchial Calu-3 epithelial cell line (originating from a human lung adenocarcinoma) was chosen due its elevated TEER values (600–1000 Ω cm2) when cultivated on Transwell® membranes [22]. These cells were co-cultivated with THP-1 differentiated macrophages on the apical side of a Transwell®, and with micro-vascular endothelial cells (HPMEC-ST1.6R) on the basal side.HPMEC-ST1.6R cells were selected because of their micro-vascular morphology, their pulmonary origin, and due to the constitutive or inducible expression of specific endothelial phenotypic markers (e.g.CD31, von Willebrand factor, etc.) [23]. To assess the effects of NP size and surface chemistry, non-functionalized (51 and 110 nm) and aminated (52 nm) fluorescent polystyrene (PS) nanobeads were tested on co-cultures.
2. Materials and methods
2.1. Nanoparticles
Non-functionalized (PS-NF, primary sizes of 51 and 110 nm) and aminated (PS-NH2, primary size of 52 nm) orange fluorescent polystyrene nanobeads (Magsphere Incorporated) were used. All NPs were suspended at 1 mg/mL in RPMI 1640 culture media without phenol red (Invitrogen) supplemented with 5% (v/v) heat-inactivated fetal calf serum (FCS) (Sigma–Aldrich) and 1% (v/v) penicillin/streptomycin called “complete medium” (Invitrogen). PS-NH2 (52 nm) nanobeads were vortexed in the culture media just before use. Non-functionalized nanobeads were indirectly sonicated 2 min (20 s pause every 20 s) at 30 W with a cuphorn (Sonicator S-4000, Misonix Incorporated) at room temperature. Then, particle size distribution and zeta potential were measured using a Zetasizer® Nano ZS (Malvern). Suspensions and morphology of NPs were also characterized with a transmission electron microscope (TEM) (STEM CM12, lab 6, electron gun 120 kV, Philips).
2.2. Cell culture
A549 (ATCC number: CCL-185™), Calu-3 (ATCC number: HTB-55™), NCI-H441 (ATCC number: HTB-174™), andTHP-1 (ATCC number: TIB-202™) cell lines were obtained from LGC Standards. They were routinely grown in RPMI 1640 medium supplemented with 10%(v/v) heat-inactivated FCS (Sigma–Aldrich) and 1% (v/v) penicillin/streptomycin (Invitrogen). Monocytic THP-1 cells are non-adherent and routinely maintained between 105 and 106 cells/mLin order to avoid any cellular stress. Before their use in our study, they were differentiated (not activated) in adherent macrophages by incubation with 50 nM of PMA (Phorbol 12-myristate 13-acetate) (Sigma–Aldrich, Ref. P1585) during 24 h as previously described [24]. The Institute of Pathology of Mainz in Germany kindly provided HPMEC-ST1.6R endothelial cells. According to Krump-Konvalikova et al. [23], these cells were routinely cultured on tissue culture plasticware pre-coated with gelatin (BD Biosciences) in M199 medium (Sigma–Aldrich) supplemented with 20% (v/v) heat-inactivated FCS, 50 μg/mL endothelial cell growth supplements (Sigma–Aldrich), 25 μg/mL sodium heparin (Sigma–Aldrich) and 1% (v/v) penicillin/streptomycin (Invitrogen).
2.3. Co-cultures
Transwell® membranes (polyester, 12 mm diameter, 0.4 μm pores) (Corning Incorporated) were inverted in a Petri dish and coated by applying 250 L/Transwell® of a 50 μg/mL type IV collagen solution (Sigma–Aldrich) in sterile water on the under surface for 2 h at 37 °C. After removing the collagen solution in excess by aspiration, the under surface was coated by the addition of 100 μL/Transwell® of a 50 μg/mL fibronectin solution (Sigma–Aldrich) in HBSS (without CaCl2 and MgCl2) (Invitrogen) for 1 h at room temperature. Excess fibronectin solution was then removed by aspiration, and HPMEC-ST1.6R endothelial cells (2.5 × 104 cells/cm2) were seeded in a volume of 250 μL and allowed to attach for 2 h at 37 °C, 5% CO2 and 95% humidity. Then the medium was removed by aspiration, and the Transwell® membranes were righted and placed into the medium-containing wells of a 12-well plate. Calu-3 epithelial cells (5 × 104 cells/cm2) were then seeded in the apical side. The day the bi-cultures were seeded (with Calu-3 and HPMEC-ST1.6R cells) was considered as day 0. Bi-cultures were then cultivated in Calu-3 cell growth medium. THP-1 monocytes were separately differentiated at day 6 post-seeding of the bi-cultures in macrophage-like adherent cells according to the protocol described in the previous section. At day 7 post-seeding, when confluent bi-cultures reached sufficient transepithelial electrical resistance (TEER) values (≥1000 Ω cm2), THP-1 macrophages were deposited on the apical side at a concentration of 2 × 104 cells/mL. The medium was changed in both sides every two days. All co-cultures were grown at 37 °C, 5% CO2 and 95% humidity.
2.4. TEER measurements
TEER measurements were performed on cell mono- and co-cultures using an EVOM Voltohmmeter (World Precision Instrument) equipped with an Endohm® 12 chamber as described by [25]. TEER was measured from day 2 to day 11 post-seeding. The Endohm® 12 chamber was pre-filled with pre-warmed (37 °C) RPMI 1640 medium containing 10% (v/v) FCS and 1% (v/v) penicillin/streptomycin. Then, Transwell® membranes were placed into the chamber and resistance measured using the EVOM voltohmmeter. Blank controls, consisting of coated filter membranes without cells were measured. TEER was calculated with the following formula:
where RM is the experimental value of cell mono- or co-cultures, RB the experimental value of the blank control and A the surface area of the filter membrane (1.12 cm2). TEER was expressed in Ω cm2.
2.5. Immunofluorescence
In immunofluorescence assays, cells were rinsed in pre-warmed (37 °C) HBSS (with CaCl2 and MgCl2) and fixed for 15 min at room temperature in 4% (v/v) paraformaldehyde (Sigma–Aldrich). Fixed cells were permeabilized for 3 min with 0.5% (v/v) Triton X100 (Acros Organics) at room temperature and then incubated for 30 min in HBSS (with CaCl2 and MgCl2) containing 2% BSA (Sigma–Aldrich) followed by a 1 h incubation time with respectively primary and secondary antibodies. All antibodies specifications and dilutions are summarized in Table 1. They were diluted with 2% BSA in HBSS containing CaCl2 and MgCl2. Photomicrographs were acquired using a ZeissAxioImager.Z1 fluorescence microscope with an ApoTome (Carl Zeiss).
Table 1.
Summary of primary and secondary antibodies used in this study for immunofluorescence application.
| Primary antibodies |
Secondary antibodies |
||||||
|---|---|---|---|---|---|---|---|
| Human epitopes | Species | Supplier (reference) | Dilution used | Species | Fluorophore (λexc/λem) | Supplier (reference) | Dilution used |
| Occludin | Rabbit | Invitrogen (40–4700) | 1:125 | Goat | Alexa Fluor® (495 nm/519 nm) | Invitrogen (A11008) | 1:2000 |
| E-cadherin | Mouse | Santa Cruz Biotechnology Inc. (sc-73916) | 1:50 | Horse | DyLight® (592 nm/617 nm) | Vector Laboratories (DI2594) | 1:300 |
| VE-cadherin | Mouse | Hycult® biotech (HM2032) | 1:10 | ||||
2.6. Permeability measurements
Permeability measurements were done in the apical to basolateral direction in Transwell® membranes as described by Lemieux et al. [26]. Briefly, the Transwell® membranes were pre-incubated with a pre-warmed (37 °C) HBSS “transport buffer” (with CaCl2 and MgCl2, pH 7.4) (Invitrogen) on both the apical and the basolateral sides. Lucifer Yellow CH dipotassium salt (LY) (Sigma–Aldrich) (0.5 kDa) was then diluted in the HBSS and added to the apical side at a final concentration of 100 μg/mL. After 1 h, aliquots from the basolateral side were collected in a black 96-well micro-plate for determination of the fluorescence leakage of the LY with a cyto-fluorometer adapted to the micro-plate (λexcitation = 485 nm, λemission = 530 nm) (TECAN Infinite M2000). The apparent permeability coefficient (Papp, unit: cm s−1) was calculated as follows:
where dQ/dt is the amount of compound transported per time point (μmol s−1), A is the surface area of the filter (cm2) and C0 the initial donor concentration (μM). The mass balance (R) was calculated as:
where A and D are the amounts of the compounds in the acceptor and donor sides, respectively, and D0 is the amount introduced at t0. The mass balances of all the compounds were between 80 and 120%.
dQ, A and D were determined using a standard curve of LY.
3. Electron microscopy
For transmission electron microscopy (TEM), cell co-cultures were rinsed on Transwell® membranes with pre-warmed (37 °C) HBSS (with CaCl2 and MgCl2) and fixed with 2.5% (v/v) glutaraldehyde in a phosphate buffer (0.1 M, pH 7.4) for 1 h at room temperature. After washing, samples were post-fixed with 1% (v/v) osmium tetroxide (Electron Microscopy Sciences) in distilled water for 1 h 30 min at room temperature in the dark and further dehydrated at different ethanol concentrations. After carrying the filter membranes with cells through propylene oxide as an intermedium, the samples were embedded in Epon resin and submitted to polymerization at 60 °C for 48 h. Ultrathin sections 80 nm thick were cut perpendicular to the filter with an ultra-microtome (Ultracut, Reichert Jung) and placed onto copper grids. Samples were doubly stained with 1% (v/v) uranyl acetate and lead citrate, and ultrastructural analysis was performed with a transmission electron microscope (JEOL 100 S®, Jeol Ltd., 80 kV). For the purpose of scanning electron microscopy (SEM), the cell co-cultures were fixed and post-fixed as described above. After dehydration in graded ethanol and drying using hexamethyldisilazane, samples were placed on stubs and analysed under a scanning electron microscope (FEI Quanta 400®, FEI Company).
3.1. AlamarBlue® viability assay
After 24 h exposure to PS nanobeads, AlamarBlue® assay (Invitrogen) was performed on mono-cultures as previously described [27] and on co-cultures in Transwell® membranes. In mono-cultures, the assay was performed in 96-well micro-plates. In co-cultures, the medium was discarded and filter membranes were washed two times with a pre-warmed (37 °C) HBSS containing CaCl2 and MgCl2. Then 300 μL and 1 mL of a mix containing RPMI 1640 medium without phenol red supplemented with 5% (v/v) FCS and 10% (v/v) AlamarBlue® (Invitrogen) were deposited in the apical and basolateral sides, respectively. After 3 h of incubation at 37 °C in an atmosphere of 5% CO2 and 95% humidity, the apical and basolateral media were pooled on a 24-well plate and the cell viability was estimated by measuring the emitted fluorescence with a cyto-fluorometer (λexcitation = 555 nm, λemission = 585 nm) (TECAN Infinite M2000, Switzerland). The percentage of cell viability for each sample was calculated compared to non-exposed cells (corresponding to 100% viability). The potential interferences of the PS nanobeads with the assay were systematically assessed, and no significant modification in fluorescence was observed. NPs concentrations tested and their correspondance between μg/cm2 and μg/mL units are summarized in Table 2.
Table 2.
Summary of NPs concentrations studied and correspondence between μg/cm2 and μg/mL units.
| Concentrations (μg/cm2) | 1.6 | 8.1 | 16.1 | 32.3 |
| Concentrations (μg/mL) | 7.3 | 14.5 | 29.1 | 58.1 |
3.2. Cellular uptake of PS nanobeads by bi- and tri-cultures
Bi-cultures of Calu-3 and HPMEC-ST1.6R cells, and tri-cultures with macrophages, were exposed to 32.3 μg/cm2 of PS nanobeads for 24 h. For tri-cultures, at day 6 post-seeding, THP-1 cells were differentiated into macrophage-like cells as previously described. Then, THP-1 macrophages were detached using a “cell dissociation buffer enzyme-free and Hank's based” (Invitrogen, Reference 13150-013), and stained with the Cell Proliferation Dye eFluor® 450 (eBioscience) according to the recommendations of the provider. These labelled macrophages were then seeded on the apical side of the bi-cultures for 24 h. Tri-cultures or bi-cultures were then exposed to PS fluorescent nanobeads for 24 h. The cells were rinsed in pre-warmed (37 °C) HBSS (with CaCl2 and MgCl2) (Invitrogen) and fixed for 15 min at room temperature in 4% (v/v) paraformaldehyde (Sigma–Aldrich), washed twice with HBSS and permeabilized for 3 min with 0.5% (v/v) Triton X100 (Acros Organics). The cells were then washed with HBSS and incubated at 37 °C for 20 min with a mix of water, 1:40 AlexaFluor® 635 Phalloidin (Invitrogen), and propidium iodide at 0.1 mg/mL (Invitrogen). Finally, the Transwell® membranes were washed with HBSS containing 0.025% (v/v) TritonX100 before mounting in ProLong® Gold antifade reagent (Invitrogen) to proceed to confocal microscopy visualization.
All picture acquisitions were performed using a Nikon A1R confocal laser scanning microscope system connected to an inverted ECLIPSE Ti (Nikon Corp.). Acquisitions were performed in sequential mode with a 100× objective (NA 1.45) by using optimal spatial resolution settings. Nuclear and cytoplasmic sides were identified with propidium iodide staining (excitation with 561 nm laser, emission collected with a 595/50 nm filter set) and AlexaFluor® 635 phalloidin (excitation with 640 nm laser, emission collected with a 700/50 nm filter set), respectively. Nanobead fluorescence was excited using a 488 nm laser (emission collected with a 525/50 nm filter set). Macrophage fluorescence was excited using a 403 nm laser (emission collected with a 450/50 nm filter set). 3D image reconstructions were performed using Volocity® software (Perkin Elmer).
3.3. Acellular translocation assays
Non-functionalized (PS-NF, primary sizes of 51 and 110 nm) and aminated (PS-NH2, primary size 52 nm) orange fluorescent polystyrene nanobeads were dispersed in RPMI 1640 culture media without phenol red supplemented with 5% (v/v) heat-inactivated FCS and 1% (v/v) penicillin/streptomycin, at a concentration of 16.1 μg/cm2. The apical side of the Transwell® membranes was filled with 300 μL of particle suspensions. The basolateral side was filled with 1 mL of RPMI 1640 culture media without phenol red supplemented with 5% (v/v) heat-inactivated FCS and 1% (v/v) penicillin/streptomycin. The Transwell® plates were then incubated for 24 h at 37 °C, 5% CO2 and 95% humidity. After 24 h, samples from the apical and basolateral sides were collected in a black 96-well micro-plate for determination of the fluorescence with a cyto-fluorometer adapted to the micro-plate (λexcitation = 488 nm, λemission = 550 nm) (TECAN Infinite M2000). The relative fluorescence in each side was calculated in relation to the initial amount of particles used.
3.4. Translocation study of nanobeads across co-cultures
The same PS nanobeads were used for transport experiments in bi- and tri-cultures. The cells were incubated with 1.6, 8.1, 16.1 and 32.3 μg/cm2 of NPs dispersed in RPMI 1640 culture media without phenol red supplemented with 5% (v/v) heat-inactivated FCS and 1% (v/v) penicillin/streptomycin for 24 h. Some of the experiments were performed at 37 °C and others at 4 °C. After 24 h, samples from the apical and basolateral sides were collected in a black 96-well micro-plate for determination of the fluorescence with a cyto-fluorometer adapted to the micro-plate (λexcitation = 488 nm, λemission = 550 nm) (TECAN Infinite M2000). The relative fluorescence in each side was calculated in relation to the autofluorescence of the culture medium.
3.5. Statistical analysis
All quantitative data are represented by the mean of three independent experiments ± SD. The data were analysed by one-way analysis of variance (ANOVA) followed by Dunnett's t-test to compare the different treated groups to the control (α risk = 0.05). To examine the differences between pairs of groups, Student's t-tests (α risk = 0.05) were used.
4. Results
4.1. Choice of the pulmonary epithelial cell line
On the alveolo-capillary barrier, tight junctions play an important functional role. A previous study reported that mono-cultures of human type I pneumocytes developed TEER values of 2180 ± 62 Ω cm2 after 8 days post-seeding on Transwell® membranes [28], [29]. Thus, we measured the TEER development in different pulmonary epithelial cell lines across time, to select cells presenting the highest values (Fig. S1). A549 cells reached maximum TEER values of roughly 18.7 ± 2.5 Ω cm2 after 11 days post-seeding on 12-well Transwell® plates. NCI-H441 cells developed significantly higher TEER values from days 8 to 11 post-seeding (approximately 100.2 ± 25.4 Ω cm2 at day 11 post-seeding). Treatment of NCI-H441 cell mono-cultures by 1 μM DEX from days 3 to 11 post-seeding significantly increased the TEER values to a maximum of 263.9 ± 12.7 Ω cm2 at day 11 post-seeding. Calu-3 cells developed significantly higher TEER values from days 6 (592.5 ± 0.5 Ω cm2) to 11 (1233.5 ± 58.1 Ω cm2) post-seeding compared to other cell lines and were therefore selected for the co-culture model of the alveolo-capillary barrier.
Supplementary Figure S1 related to this article can be found, in the online version, at doi:10.1016/j.toxrep.2014.03.003.

4.2. Barrier properties of bi- and tri-cultures
The endothelial cell line HPMEC-ST1.6R did not develop significant TEER values, showing a maximum TEER value of 11.8 ± 0.8 after day 8 post-seeding (Fig. 1A). The co-culture with Calu-3 epithelial cells showed significantly increased TEER values from days 4 to 11 post-seeding similar to those observed in Calu-3 cell mono-cultures.
Fig. 1.

Barrier properties of mono- and bi-cultures. A represents the time course of TEER development in mono- and bi-cultures of Calu-3 epithelial cells and HPMEC-ST1.6R endothelial cells. All cells were cultured in 12-Transwell® filter plates using RPMI 1640 media supplemented with 10% (v/v) FCS and 1% (v/v) penicillin/streptomycin. Mono-cultures of Calu-3 and HPMEC-ST1.6R cells are represented by green and red curves, respectively, and bi-cultures by the blue curve. Data represent the mean ± SD of four independent experiments. Immunofluorescent labelling of adherent (E-/VE-cadherin) and tight (occludin) junction proteins was performed in bi-cultures on Calu-3 and HPMEC-ST1.6R cells (in white, B–E), at day 8 post-seeding (scale bars = 20 μm).
Bi-cultures were examined 8 days post-seeding by immunofluorescence for the expression of intercellular proteins involved in tight junction (TJ; Occludin) and adherent junction (AJ; E-/VE-cadherin for Calu-3 and HPMEC-ST1.6R respectively) formation. On the apical side of the Transwell® membranes, E-cadherin and occludin appeared intensively and uniformly labelled between Calu-3 epithelial cells (Fig. 1 B and C respectively). On the basal side, VE-cadherin appeared intensively but not uniformly labelled between HPMEC-ST1.6R endothelial cells due to a development of multi-layered structures (Fig. 1 D). Occludin was not labelled on endothelial cells (Fig. 1E).
PMA pre-differentiated THP-1 macrophages were added at day 7 post-seeding on the apical side of the Calu-3 cells in bi-cultures. After 24 h in co-culture, no significant decrease of the TEER values was noted (Fig. 2). In order to verify that these measurements were performed correctly, we also controlled the influence of higher THP-1 macrophages concentration (2 × 105 cells/mL), which induced significant TEER decrease in tri-cultures compared to bi-cultures (data not shown). This result is also consistent with the statistical analysis of the LY Papp values of tri-cultures (Fig. S2). Significantly increased Papp values of the LY across HPMEC-ST1.6R cell mono-cultures compared to Calu-3 cells also confirmed results obtained by TEER measurements in the cell cultures (Fig. 1 A). However, assessment of the LY transport showed significant differences between the Calu-3 mono-cultures and bi-/tri-cultures, contrary to the TEER measurements that did not reveal any differences.
Fig. 2.

Influence of THP-1 differentiated macrophage seeding on TEER of bi-cultures. Prior to adding to bi-cultures, THP-1 monocytes were differentiated during 24 h using 30 ng/mL of PMA. At day 7 post-seeding bi-cultures, THP-1 differentiated macrophages were added at the apical side of Calu-3 cells. TEER measurements were then performed at 2, 4, 6 and 24 h after macrophage seeding. Data represent the mean ± SD of three independent experiments. One-way ANOVA and Dunnett's post-test (tri-cultures vs. bi-cultures comparisons) were performed (**p < 0.01).
Supplementary Figure S2 related to this article can be found, in the online version, at doi:10.1016/j.toxrep.2014.03.003.
4.3. Macrophages change the surface morphology of epithelial cells in co-cultures
At day 8 post-seeding, SEM analysis was performed on both the apical and basolateral sides of the bi-cultures and tri-cultures in order to observe the morphological changes induced by the addition of macrophages (Fig. 3). On the apical side of bi-cultures, numerous microvilli were observed (Fig. 3A). On tri-cultures, THP-1 differentiated macrophages were observed on the apical side of Calu-3 cells (Fig. 3B), but not on the endothelial side. Macrophages were adherent and presented numerous pseudopodia. SEM analysis also revealed epithelial morphological changes with the disappearance of microvilli on the cell apex, and a hilly surface.
Fig. 3.

Representative scanning electron microscopy pictures of the apical side of bi-cultures (A, scale bar = 2 μm) and tri-cultures (B, scale bar = 5 μm). On picture B, the black star indicates the presence of an activated THP-1 macrophage adherent on the apical side of the Calu-3 cells.
4.4. Ultrastructural morphology of tri-culture models
TEM analysis was also performed on both the apical and basolateral sides of the bi- and tri-cultures in order to observe the ultrastructural changes induced by the addition of macrophages (Fig. 4). On bi-cultures, long microvilli (length of approximately 500 nm), desmosomes, and TJ/AJ (Tight junctions/Adherent junctions) were observed on the apex of the Calu-3 epithelial cells (Fig. 4A and B). On tri-cultures, macrophages containing numerous lipid inclusions were observed (Fig. 4C), but not on the endothelial side. The smallest microvilli compared to bi-cultures were also observed, with desmosomes and TJ/AJ still existing in Calu-3 cells of these models (Fig. 4D). On the basal side, long and flattened endothelial cells were observed with a high nucleo-cytoplasmic ratio (Fig. 4E), and numerous perinuclear organelles such as mitochondria. Cytoplasmic extensions of these endothelial cells were also observed superposed, forming thin multi-layers (Fig. 4F). The presence of micro-vesicles on the apical side of these cells also indicates the presence of molecular exchanges occurring on the plasma membrane.
Fig. 4.

Ultrastructural morphology of Calu-3 cells on bi-cultures (A and B) and tri-cultures (C and D). These cells were cultured on Transwell® membranes and then observed at day 8 post-seeding by transmission electron microscopy (TEM). On bi-cultures (Fig. A and B, scale bar = 500 nm), Calu-3 epithelial cells are observed with apical microvilli (m), desmosomes (Des) and tight or adherent junctions (Tj/Aj). On tri-cultures, macrophages (M) with lipid inclusions (i) are located on the apical side ofCalu-3 epithelial cells (Ep) showing small microvilli (m) (C, scale bar = 5 μm). Desmosomes and tight/adherent junctions are also present on the apical side of the epithelial cells (Fig. D, Cyt = cytoplasm, N = nucleus, n = nucleolus, scale bar = 2 μm). Fig. E and F are photographs of the endothelial cells located on the basal side of the Transwell® (v = vesicles, sale bars = 2 and 1 μm respectively).
4.5. Fluorescent polystyrene nanobeads characterization
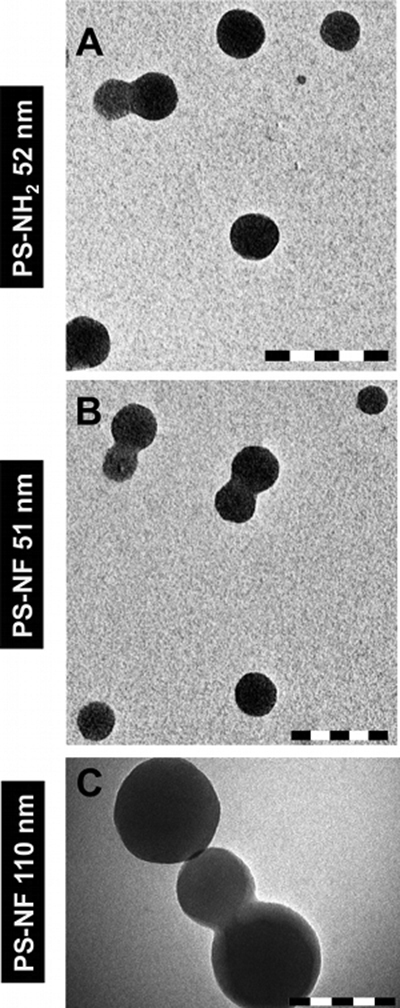
The size distributions of aminated (primary size of 52 nm) and unmodified (primary sizes of 51 and 110 nm) PS nanobeads were characterized after dispersion in the complete culture medium. All suspensions were well mono-dispersed with low polydispersity indexes (<0.7; Malvern's reference value) (Fig. 5). 99.80% of PS-NH2 (52 nm) nanobeads and 91.85% of PS-NF (51 nm) nanobeads were below 100 nm (Fig. 5A and B). 90% of PS-NF (110 nm) nanobeads were below 200 nm (Fig. 5C). All nanobeads had negative zeta potentials. Despite absolute values close to zero, we measured stable size distributions for up to 48 h at room temperature after dispersion (data not shown). NP suspensions were further characterized by TEM and similar sizes were observed with slightly agglomerated round NPs (Fig. S3).
Fig. 5.

Size distribution, zeta potential (ζ) and polydispersity indexes (PDI) of polystyrene nanobeads. Aminated 52 nm, unmodified 51 nm and 110 nm nanobeads (primary sizes) were dispersed in the RPMI 1640 medium (without phenol red supplemented with 5% (v/v) FCS and 1% penicillin/streptomycin), and all parameters were measured by dynamic light scattering (DLS) (Fig. A–C, for each nanobead respectively). Data represent the mean ± SD of three independent experiments.
Supplementary Figure S3 related to this article can be found, in the online version, at doi:10.1016/j.toxrep.2014.03.003.
4.6. Cytotoxicity of polystyrene nanobeads
AlamarBlue® assays showed that unmodified PS nanobeads (51 and 110 nm) did not induce a significant decrease of the cell viability on mono- and co-cultures (data not shown). Aminated PS nanobeads (52 nm) induced significant dose-dependent decreases of cell viability at doses of 16.1 and 32.3 μg/cm2 on Calu-3 epithelial cells (Fig. 6 A; 60.5 ± 3.6% and 46.5 ± 10.4% of viable cells, respectively). Significant decreases of cell viability were also observed at 32.3 μg/cm2 on THP-1 differentiated macrophages and HPMEC-ST1.6R endothelial cells (Fig. 5B and C; 50.2 ± 11.6% and 48.9 ± 15.5% of viable cells, respectively). No significant cytotoxicity was observed on the bi- and tri-cultures (Fig. 5D–F). In order to verify that measurements were performed correctly, 0.01% Triton X-100 was used as a positive control. After 24 h exposure to 0.01% Triton X-100, cell viability was systematically significantly decreased compared to control cells in mono-, bi- and tri-cultures (data not shown).
Fig. 6.

Cytotoxicity of PS-NH2 nanobeads on mono- and co-cultures. Mono-cultures (A–C), bi-cultures of Calu-3 and HPMEC-ST1.6R cells (D), and tri-cultures (E), were exposed for 24 h to PS-NH2 nanobeads, respectively. Cell viability was then measured with the AlamarBlue® assay. Data represent the mean ± SD of three independent experiments. One-way ANOVA and Dunnett's post-test (comparisons vs. control cells not exposed to NPs) were performed (**p < 0.01).
4.7. TEER effects depend on PS nanobead surface chemistry and size
To assess the effects of PS nanobeads on the integrity of bi- and tri-cultures, all co-cultures were exposed for 24 h and TEER was measured at times 0, 2 h, 4 h and 24 h. All results were expressed in relative TEER to the time 0, which corresponds to the first TEER measurement after the equilibration time. In all co-cultures and for all tested nanobeads no significant decreases of relative TEER values were observed after exposure to nanobeads for 2 and 4 h (data not shown). However, a slight and transient significant decrease of relative TEER values was observed after 4 h exposure to PS nanobeads in control cells, and was attributed to the medium change (data not shown). Only PS-NF of both sizes was able to decrease the relative TEER values in the bi- and tri-cultures (Fig. 7). The decrease was more pronounced for the smaller PS-NF (51 nm) than for the larger ones (110 nm) (Fig. 7B and C).
Fig. 7.

Relative TEER of bi- and tri-cultures incubated with polystyrene nanobeads. Co-cultures seeded on Transwell® membranes were incubated with PS-NH2 (52 nm) (A), PS-NF (51 nm) (B), or PS-NF (110 nm) nanobeads (C) during 24 h in RPMI 1640 medium (without phenol red supplemented with 5% (v/v) FCS and 1% penicillin/streptomycin). Bi-cultures (blue bars) and tri-cultures (yellow bars) were tested. The TEER is expressed relative to the time 0 (time after 1 h equilibration in the RPMI 1640 medium without phenol red supplemented with 5% (v/v) FCS and 1% penicillin/streptomycin). Data represent the mean ± SD of three independent experiments. One-way ANOVA and Dunnett's post-test (comparisons vs. control cells not exposed to NPs) were performed (*p < 0.05, **p < 0.01). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
4.8. PS nanobead uptake depends of the surface chemistry
In order to determine the role of macrophages in tri-cultures and the physico-chemical properties of NPs in co-culture uptake, we carried out confocal fluorescence microscopy and three-dimensional reconstructions of the apical and basal sides of the bi- and tri-cultures. Due to the retention of several fluorophores used to perform the multi-labelling, the filter autofluorescence, and the low working distance of the objective used (0.13 mm), it was not possible to perform whole acquisitions from the apical cell layers to the basal endothelial cells.
In bi-cultures, all PS nanobeads were adsorbed or internalized by Calu-3 epithelial cells (Fig. S4A–D). On the basal side, only PS-NH2 (52 nm) and PS-NF (110 nm) nanobeads were observed adsorbed on plasma membranes or internalized by HPMEC-ST1.6R cells (Fig. S4E–H).
Supplementary Figure S4 related to this article can be found, in the online version, at doi:10.1016/j.toxrep.2014.03.003.
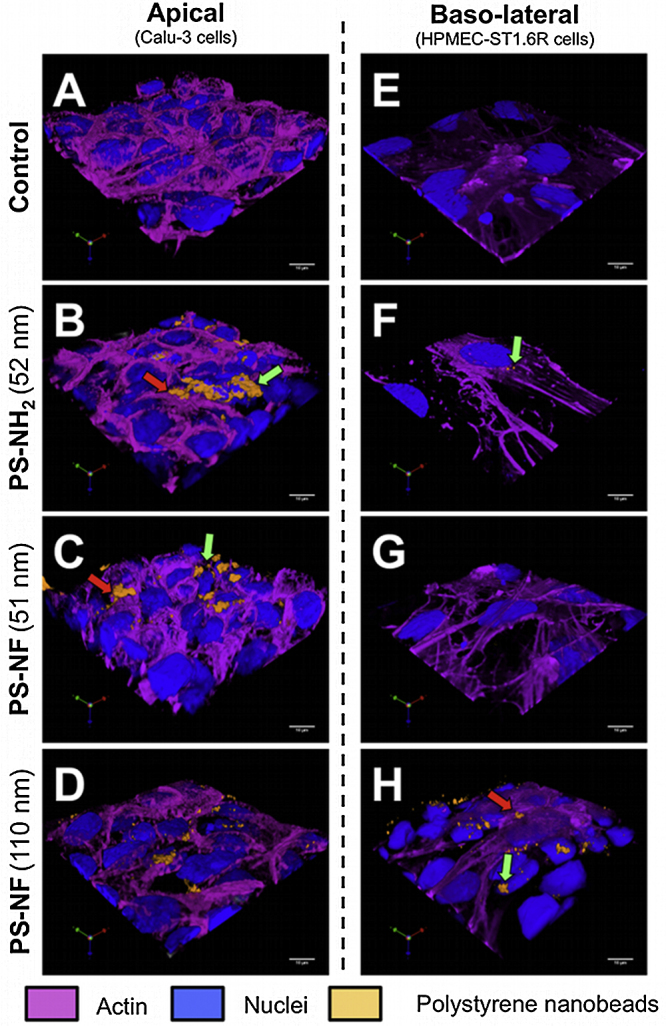
In tri-cultures, all PS nanobeads were internalized by Calu-3 epithelial cells (Fig. 8A–D). PS-NH2 (52 nm) and PS-NF (110 nm) nanobeads were notably internalized by macrophages (Fig. 8B and D). On the basal side, only PS-NF (110 nm) nanobeads were found adsorbed on the plasma membranes of HPMEC-ST1.6R cells (Fig. 8H).
Fig. 8.

Internalization of polystyrene nanobeads by tri-cultures. Three-dimensional models with 10 × 104 THP-1 macrophages/mL, seeded on Transwell® membranes, were incubated during 24 h with 32.3 μg/cm2 of PS-NH2 (52 nm), PS-NF (51 nm), or PS-NF (110 nm) nanobeads. Nuclear and cytoplasmic sides were respectively identified by using propidium iodide staining and Alexa Fluor® 635 phalloidin (in blue and pink on the pictures, respectively). Macrophages were identified using a cell proliferation dye (eBioscience; in white on the pictures). Fluorescent PS nanobeads correspond to the orange fluorescence on the pictures. Three-dimensional image reconstructions and measurements were performed using Volocity® software (Perkin Elmer) on both the apical (A–D) and basolateral (E–H) sides of the Transwell®. These pictures are representative of several observations. Scale bars = 10 μm.
4.9. PS nanobeads are retained by Transwell® membranes
To determine whether or not Transwell® membranes prevent nanobead translocation from the apical to the basolateral side acellular assays were performed with all PS nanobeads. After 24 h incubation, significantly more elevated amounts of fluorescence were measured in the apical sides compared to the basal sides for all tested nanobeads (Fig. 9A). There was no significant difference between the amounts of fluorescence of PS-NH2 (52 nm) and PS-NF (51 nm) nanobeads (Student's t-test, p = 0.0640), whereas these amounts increased very significantly for PS-NF (110 nm) compared to PS-NF (51 nm) nanobeads (Student's t-test, **p = 0.0068). Therefore, the surface chemistry of nanobeads does not significantly modify their translocation, whereas their size does. The sum of the relative fluorescence of the PS-NF (51 nm) nanobeads measured in the two sides (approximately 31%) was below 100%, indicating a possible adsorption of nanobeads on the Transwell® membranes. Three-dimensional acquisitions performed on membranes exposed for 24 h to each nanobead revealed that some of the nanobeads were retained by the randomly distributed pores of Transwell® membranes (Fig. 8B–D).
Fig. 9.

Acellular transport of polystyrene nanobeads across Transwell® membranes. The relative fluorescence (compared to the initial amount of fluorescence deposited) of the nanobeads was measured in both sides of the Transwell® membranes without cells after 24 h incubation in the complete culture medium (A). Data represent the mean ± SD of three independent experiments. Student's t-tests were performed (*p < 0.05) for each group between the fluorescence amounts in the apical and basolateral sides. Three-dimensional image reconstructions of membranes after incubation with PS-NH2 (52 nm), PS-NF (51 nm), or PS-NF (110 nm) nanobeads (16.1 μg/cm2) were performed using IMARIS software (B, C and D, respectively).
Other acellular translocation experiments were performed under identical conditions to compare polyester (1 × 106 pores/cm2) to polycarbonate membranes (4 × 108 pores/cm2). We did not find significant differences between these filter membrane types (data not shown).
4.10. Physico-chemical properties of PS nanobeads influence their translocation across co-cultures
Translocation assays were performed from the apical to the basolateral side in the bi- and tri-cultures during 24 h with each nanobead. Taking into account the retention of nanobeads in the apical side, we only performed a semi-quantitative evaluation of the results, which were expressed in terms of the relative fluorescence compared to the basal autofluorescence of the medium. No significant increase of the relative fluorescence was measured on the basolateral side of all co-cultures for PS-NH2 (52 nm) and PS-NF (110 nm) nanobeads (Fig. 10A and B). Significant increases (concentration dependent) of the relative fluorescence were measured in the basolateral sides of the bi- and tri-cultures (Fig. 10C).
Fig. 10.

Translocation of polystyrene nanobeads across bi- and tri-cultures. Bi- and tri-cultures were exposed to PS-NH2 (52 nm) (A), PS-NF (110 nm) (B), and PS-NF (51 nm) (C) nanobeads for 24 h in RPMI 1640 medium (without phenol red supplemented with 5% (v/v) FCS and 1% penicillin/streptomycin). The relative fluorescence (compared to the autofluorescence of the culture medium) of the nanobeads was then measured in the basolateral sides of the co-cultures. Data represent the mean ± SD of three independent experiments. One-way ANOVA and Dunnett's post-test (comparisons vs. control medium without cells) were performed (**p < 0.01).
4.11. Translocation of PS-NF (51 nm) nanobeads requires passive and active mechanisms
To assess whether or not PS-NF (51 nm) nanobeads translocation is related to passive (paracellular pathways) or to active mechanisms (e.g. endocytosis), translocation assays were performed at 4 °C or 37 °C on the bi- and tri-cultures. Significant decreases of fluorescence were measured in all co-cultures exposed to PS-NF (51 nm) nanobeads for 24 h at 4 °C compared to 37 °C at all concentrations tested (Fig. 11). Therefore, translocation of these nanobeads requires in particular active mechanisms, such as endocytosis, but also passive mechanisms as previously described (Figs. S4 and 7).
Fig. 11.

Translocation mechanisms of PS-NF (51 nm) across bi- and tri-cultures. Bi-cultures (A) and tri-cultures (B) were exposed to PS-NF (51 nm) nanobeads for 24 h in RPMI 1640 medium (without phenol red supplemented with 5% (v/v) FCS and 1% penicillin/streptomycin), at 37 °C or at 4 °C (pink and blue bars, respectively). The relative fluorescence (compared to the autofluorescence of the culture medium) of the nanobeads was then measured in the basolateral sides of the co-cultures. Data represent the mean ± SD of three independent experiments. Student's t-tests were performed (*p < 0.05) for each group between the fluorescence amounts in the basolateral sides after exposure to PS-NF (51 nm) at 4 °C and 37 °C. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
5. Discussion
In this work an in vitro model containing the three main cell types constituting the alveolo-capillary barrier in vivo was developed (epithelial cells [Calu-3 cell line] and macrophages [PMA-differentiated THP-1 cells] in the apical side of a Transwell®, and micro-vascular endothelial cells [HPMEC-ST1.6R endothelial cell line] in the basal side). To address the role of these cells in terms of NP translocation, well-characterized fluorescent PS nanobeads were tested on bi-cultures of epithelial/endothelial cells and on tri-cultures including macrophages. Various particle sizes and surface functionalizations were also tested to determine the role of the physico-chemical properties in terms of NP translocation.
Alveolar epithelial cells play an important part in the establishment of the highly impermeable alveolo-capillary barrier [28]. Measurements of the TEER values in various epithelial cell lines confirmed that A549 failed to develop sufficient TEER values as previously described in literature [12]. NCI-H441 cells also failed to develop high TEER values even in the presence of dexamethasone, which was previously shown to increase the TEER values in this cell line [19], [20]. Calu-3 bronchial epithelial cells developed significantly more elevated TEER values (1233.5 ± 58.1 Ω cm2 at day 11 post-seeding), close to those reported for primary human alveolar epithelial cells (2180 ± 62 Ω cm2 at day 8 post-seeding) [29]. They were then chosen as a surrogate for alveolar epithelial cells in our model. Bi-cultures of Calu-3 and HPMEC-ST1.6R cells showed TEER values not significantly different from Calu-3 cell mono-cultures, indicating the central role played by Calu-3 cells in barrier establishment. The permeability of the Lucifer yellow significantly decreased in the bi-cultures compared to the Calu-3 mono-cultures at day 8 post-seeding. It is unlikely that these differences between TEER measurements and Papp values are due to any modification of barrier properties in HBSS during the LY assay, as TEER values were checked to be unmodified during and after the assay (data not shown). As did Van Itallie et al., we postulated that the permeability of the LY is proportional to tight junction pore number, while small electrolytes are subject to further selectivity by the profile of tight junction proteins (e.g. occludin) [30]. This may explain the differences observed between Papp values in Calu-3 cells and co-cultures. The protein occludin was detected on Calu-3 epithelial cells but not on endothelial cells. Occludin expression by Calu-3 cells was previously described in literature, and this protein is mainly implied in tight junction (TJ) formation in primo-cultures of type II alveolar epithelial cells [31], [32]. Adherent junction (AJ) proteins E- and VE-cadherin were also expressed in bi-cultures by Calu-3 and HPMEC-ST1.6R, respectively, as already described [32], [33]. The presence of TJ/AJ and desmosomes was confirmed by TEM analysis on the apex of Calu-3 cells in bi-cultures, associated with numerous microvilli. Addition of pre-differentiated THP-1 macrophages at day 7 post-seeding on the apical side of the bi-cultures did not induce any significant change in TEER values, nor in LY Papp values. This result differs from that recently obtained by Farcal et al., who also seeded THP-1 differentiated macrophages on the apical side of bi-cultures constituted of NCI-H441 epithelial cells and ISO-HAS-1 endothelial cells and co-incubated with dexamethasone [4]. These authors showed a drastic decrease of TEER values after addition of PMA-differentiated THP-1 macrophages associated with substantial secretion of TNF-α and IL-8, showing that NCI-H441 cells are more sensitive than Calu-3 cells to the addition of PMA-differentiated THP-1 macrophages. We confirmed this by performing co-culture experiments of Calu-3, NCI-H441, or dexamethasone-treated NCI-H441 cells with PMA-differentiated THP-1 macrophages. Significant decreases of TEER values were only observed in NCI-H441/THP-1 co-cultures independently of dexamethasone pre-treatment. Klein and colleagues also reported a higher permeability of their tetraculture system mimicking the alveolo-capillary barrier after adding innate immune system cells THP-1 macrophages and HMC-1 mastocytes [13]. TEM analysis of tri-cultures showed that TJ/AJ and desmosomes were always present but that morphological changes were observed with length reduction and disappearance of microvilli on the apical side of Calu-3 cells. As SEM analysis revealed that macrophages seem to be in an activated shape (numerous pseudopodia), we hypothesize that these cells could trigger slightly elevated pro-inflammatory cytokine release modifying the alveolar morphology. We also suggest that traces of PMA, known to modify protein expression on the apical surface of intestinal Caco-2 cells, may also affect Calu-3 cells microvilli [34]. Consequently, as using macrophages tends to cause some epithelial cellular morphological changes, they might not behave as they do in the lung in vivo. It is a possible limitation of their use in co-culture cell systems. However, further experiments are needed to address the causes and consequences of this modified epithelial morphology.
In a second step, the translocation of fluorescent PS nanobeads of various sizes and surface chemistries were tested on bi- and tri-cultures. These NPs were chosen because they are traceable, easily synthesizable and commercially available in various sizes and with different surface functionalizations [35]. Nanobeads were firstly characterized in the complete culture medium. We showed that suspensions were well mono-dispersed and that size distributions were stable for at least 48 h at room temperature (data not shown). Only PS-NH2 (52 nm) nanobeads were found to be cytotoxic on mono-cultures, whereas unmodified nanobeads (51 and 110 nm) were not. These results, indicating more potent cytotoxic effects of PS-NH2 (52 nm) nanobeads compared to non-functionalized NPs, are consistent with previous results obtained with several cell types in literature [36], [37], [38]. Surprisingly, no cytotoxicity was observed on bi- or on tri-cultures for all tested nanobeads. We previously reported this phenomenon with cytotoxic effects of SiO2 and TiO2 NPs observed after 6 h exposure on monocultures of THP-1 macrophages but not on co-cultures with A549 or NCI-H441 epithelial cells [27]. Several other authors also recently observed decreased cytotoxicity on co-cultures compared to mono-cultures after exposure to diesel exhaust particles or silica NPs [39], [40]. We postulate that this decrease of cytotoxicity in co-cultures may have been due to the higher number of cells in co-culture compared to mono-cultures and the consequent reduced cell surface area accessible to NPs in co-cultures. However, cytotoxicity was also not observed in bi-cultures apically exposed to PS-NH2 nanobeads. One reason may be that intercellular signalling could also play a role in cytotoxicity protection. One experiment to confirm this hypothesis could be to expose epithelial cells previously cultivated with endothelial-conditioned media or vice versa.
Generally, TEER measurements after 24 h exposure to PS nanobeads revealed different sensitivities between co-cultures, which can be classified as follows: tri-cultures > bi-cultures. PS nanobeads may modify barrier properties by acting on junction protein expression, as was previously demonstrated with modulation of the occludin mRNA in 16HBE14o-bronchial cells [41]. Due to possible interferences between LY and PS nanobead fluorescence, we performed only TEER measurements, but complementary investigations on the mRNA expression or immunofluorescence of TJ proteins should be done to confirm this hypothesis. A relation with oxidative stress and barrier properties has been described by various authors. Increased translocation of quantum dots across rat alveolar epithelial cells with decreased TEER values was shown only in the presence of tert-butyl hydroperoxide (known as an oxidative stress inducer) [42]. Exposure of a co-culture of epithelial (NCI-H441) and endothelial (ISO-HAS-1) cells to carboxylated PS nanobeads under mechanical strain induced ROS generation and increased nanobead translocation [43]. In other experiments, we demonstrated that all cell mono-cultures were able to release the superoxide anion after exposure to each nanobead (unpublished data). Therefore, with an increased cell number in tri-cultures compared to bi-cultures, an amplified release of ROS may affect TEER values. Uptake experiments performed on bi- and tri-cultures revealed that NPs were internalized or adsorbed by Calu-3 epithelial cells in bi-cultures, and systematically internalized by macrophages in tri-cultures. Similar results were obtained by Blank et al. who reported in a triple cell co-culture of macrophages and epithelial cells on the apical side, and dendritic cells on the basolateral side, that polystyrene particles (1 μm) were less internalized by macrophages and more by dendritic cells when A549 epithelial cells (exhibiting low TEER values) were employed, and inversely when 16HBE14o-epithelial cells (with higher TEER values) were used [44].
Transwell® membranes are the most used and the only commercially available culture supports allowing transport studies across cellular barriers [4], [16], [19]. Contrary to small molecules, the study of NP translocation requires ideally no interference with membranes. Consequently, we checked whether or not polyester Transwell® membranes (0.4 μm pores) retained PS nanobeads (of different sizes and surface chemistries). We found that PS nanobeads were mainly retained in the apical side of the Transwell® membranes and adsorbed in the micro-porous membrane. Small quantities of PS nanobeads were measured in the basolateral sides, with significantly larger amounts of PS-NF (110 nm) compared to the smallest NPs, indicating a potential effect of particle size. Cartwright et al. recently found that PS nanobeads (50 and 100 nm) have greater adherence to polyester than to polycarbonate Transwell® membranes [10]. Different results were obtained with mono-dispersed nanobeads used in this study, with no differences between PS nanobead translocation across polyester and polycarbonate Transwell® membranes. We also compared 0.4 and 3 μm porosities, but the results remained unchanged (data not shown). We hypothesize that the thickness of the membranes (approximately 10 μm), and the shape and random distribution of the pores, may play more important roles. Consequently, translocation results across cellular models were analysed carefully.
After 24 h of exposure of bi- and tri-cultures, only PS-NF (51 nm) nanobeads were significantly measured in the basolateral sides by spectrofluorometry, while PS-NH2 (52 nm) and PS-NF (110 nm) nanobeads were not. Same experiments were performed on Calu-3 cell monocultures cultivated either on Transwells with 0.4 μm or 3 μm pores, indicating after 14–16 h of incubation with PS-NH2 (46 nm) nanobeads no translocation or 6% translocation respectively [45]. Consequently, we could hypothesize that the retention of PS-NH2 (52 nm) nanobeads in the apical compartment may be due essentially to cell co-culture, and also slightly to the pore size. We also showed that the observed translocation was largely due to active pathways, but also to paracellular pathways, as trafficking decreased significantly but still remained after 24 h exposure to PS-NF (51 nm) at 4 °C compared to 37 °C. Similar results were observed after exposure of rat alveolar epithelial cells to carboxylated PS nanobeads (20 nm) [46]. As we have shown that THP-1 cells internalized PS nanobeads, we could expect a higher translocation in bi-cultures compared to tri-cultures but no difference was seen. It would have been interesting to perform kinetic measurements by stereology of the NPs dose retained by macrophages in tricultures, in order to know if they differentially uptake each type of NPs. Although no significant fluorescence was detected in the basolateral side for PS-NH2 (52 nm) and PS-NF (110 nm) nanobeads, confocal microscopy revealed internalized PS-NH2 (52 nm) nanobeads in the endothelial cells of bi-cultures, but not in tri-cultures. PS-NF (110 nm) nanobeads were also detected in the endothelial cells in bi- and tri-cultures. However, due to the lack of accuracy of translocation studies by fluorescence measurements, we cannot affirm that PS-NH2 (52 nm) and PS-NF (110 nm) nanobeads completely move across co-cultures in basal side. These results emphasize the relevancy of microscopy methods to study nanoparticle uptake across co-cultures, and further research efforts still need to be done to adapt support membranes toward translocation studies. Interestingly, PS-NF (51 nm) nanobeads were not detected in HPMEC-ST1.6R endothelial cells, whereas they were able to significantly translocate in the basolateral sides across bi- and tri-cultures. Further experiments of TJ labelling are necessary to determine if these NPs translocate between cells. As only the time 24 h was tested to observe nanobead translocation, we can hypothesize that PS-NF (51 nm) could be internalized earlier by endothelial cells and then eliminated in the basal side. However, the retention of nanobeads in the Transwell® insert may have affected the accuracy of the measurements (Fig. 10, Fig. 11), and it might therefore be difficult to ascertain precisely the effects of the physico-chemical properties and the differences between active and passive transport using this system. Due to very limited nanoparticle translocation observed in vivo in secondary organs after inhalation and possible adsorption of nanobeads in membranes, it seems difficult to quantitatively compare with translocation rates obtained in this in vitro study [3]. A recent study shows that ultrathin porous Si membranes might be an interesting alternative to Transwell® membranes for the study of nanoparticle translocation [47].
In conclusion, we developed tri-cultures resembling the alveolo-capillary barrier, and exhibiting acceptable barrier properties for the study of NP translocation. Macrophages, which play an important role in NP clearance in vivo, seem also to play an important role in these models by internalizing NPs and probably affecting barrier integrity. Thus, co-cultures with macrophages, epithelial cells, and endothelial cells are appropriate for studying NP translocation. However, the Transwell® membranes commercially available do not allow sensitive assessment of NP translocation, and further research efforts must be made to develop novel adapted culture supports to address this issue more accurately.
Conflict of interest statement
The authors report no conflict of interests.
Acknowledgements
We thank Dr Vera Krump-Konvalinkova and Prof. C.J. Kirkpatrick at The Institute of Pathology, Johannes Gutenberg University, Mainz, Germany, for their kind gift of the HPMEC-ST1.6R cell line used in this study. The authors also thank René Lai-Kuen for his help with TEM analysis at the “Plateforme d’imagerie cellulaire et moléculaire” at the Faculty of Pharmacy, Paris Descartes, and Patrice Delalain for SEM analysis at INERIS. Thanks are also due to Dr Vincent Paget for his valuable assistance.
This work has been supported by a grant of the French Ministry for Ecology, Sustainable Development, and Energy (MEDDE), National Research Program NANOTRANS, and UTC Foundation for Innovation.
Footnotes
Available online 12 May 2014
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.toxrep.2014.03.003.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Tetley T.D. Health effects of nanomaterials. Biochem. Soc. Trans. 2007;35:527–531. doi: 10.1042/BST0350527. [DOI] [PubMed] [Google Scholar]
- 2.Oberdorster G., Oberdorster E., Oberdorster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreyling W.G., Semmler-Behnke M., Takenaka S., Moller W. Differences in the biokinetics of inhaled nano- versus micrometer-sized particles. Acc. Chem. Res. 2013;46:714–722. doi: 10.1021/ar300043r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kreyling W.G., Hirn S., Moller W., Schleh C., Wenk A. Air–blood barrier translocation of tracheally instilled gold nanoparticles inversely depends on particle size. ACS Nano. 2014;8:222–233. doi: 10.1021/nn403256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gehr P., Bachofen M., Weibel E.R. The normal human lung: ultrastructure and morphometric estimation of diffusion capacity. Respir. Physiol. 1978;32:121–140. doi: 10.1016/0034-5687(78)90104-4. [DOI] [PubMed] [Google Scholar]
- 6.Crapo J.D., Barry B.E., Gehr P., Bachofen M., Weibel E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982;126:332–337. doi: 10.1164/arrd.1982.126.2.332. [DOI] [PubMed] [Google Scholar]
- 7.Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res. 2001;2:33–46. doi: 10.1186/rr36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawgood S., Clements J.A. Pulmonary surfactant and its apoproteins. J. Clin. Invest. 1990;86:1–6. doi: 10.1172/JCI114670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clements J.A. Functions of the alveolar lining. Am. Rev. Respir. Dis. 1977;115:67–71. doi: 10.1164/arrd.1977.115.S.67. [DOI] [PubMed] [Google Scholar]
- 10.Lambrecht B.N. Alveolar macrophage in the driver's seat. Immunity. 2006;24:366–368. doi: 10.1016/j.immuni.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Schneeberger-Keeley E.E., Karnovsky M.J. The ultrastructural basis of alveolar-capillary membrane permeability to peroxidase used as a tracer. J. Cell Biol. 1968;37:781–793. doi: 10.1083/jcb.37.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann A.D., Daum N., Bur M., Lehr C.M., Gehr P. An in vitro triple cell co-culture model with primary cells mimicking the human alveolar epithelial barrier. Eur. J. Pharm. Biopharm. 2010;77:398–406. doi: 10.1016/j.ejpb.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Klein S.G., Serchi T., Hoffmann L., Blomeke B., Gutleb A.C. An improved 3D tetraculture system mimicking the cellular organisation at the alveolar barrier to study the potential toxic effects of particles on the lung. Part. Fibre Toxicol. 2013;10:31. doi: 10.1186/1743-8977-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Napierska D., Thomassen L.C., Vanaudenaerde B., Luyts K., Lison D. Cytokine production by co-cultures exposed to monodisperse amorphous silica nanoparticles: the role of size and surface area. Toxicol. Lett. 2012;211:98–104. doi: 10.1016/j.toxlet.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Alfaro-Moreno E., Nawrot T.S., Vanaudenaerde B.M., Hoylaerts M.F., Vanoirbeek J.A. Co-cultures of multiple cell types mimic pulmonary cell communication in response to urban PM10. Eur. Respir. J. 2008;32:1184–1194. doi: 10.1183/09031936.00044008. [DOI] [PubMed] [Google Scholar]
- 16.Rothen-Rutishauser B.M., Kiama S.G., Gehr P. A three-dimensional cellular model of the human respiratory tract to study the interaction with particles. Am. J. Respir. Cell Mol. Biol. 2005;32:281–289. doi: 10.1165/rcmb.2004-0187OC. [DOI] [PubMed] [Google Scholar]
- 17.Rothen-Rutishauser B., Muhlfeld C., Blank F., Musso C., Gehr P. Translocation of particles and inflammatory responses after exposure to fine particles and nanoparticles in an epithelial airway model. Part. Fibre Toxicol. 2007;4:9. doi: 10.1186/1743-8977-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothen-Rutishauser B., Blank F., Muhlfeld C., Gehr P. In vitro models of the human epithelial airway barrier to study the toxic potential of particulate matter. Expert Opin. Drug Metab. Toxicol. 2008;4:1075–1089. doi: 10.1517/17425255.4.8.1075. [DOI] [PubMed] [Google Scholar]
- 19.Hermanns M.I., Unger R.E., Kehe K., Peters K., Kirkpatrick C.J. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: development of an alveolo-capillary barrier in vitro. Lab. Invest. 2004;84:736–752. doi: 10.1038/labinvest.3700081. [DOI] [PubMed] [Google Scholar]
- 20.Gazdar A.F., Linnoila R.I., Kurita Y., Oie H.K., Mulshine J.L. Peripheral airway cell differentiation in human lung cancer cell lines. Cancer Res. 1990;50:5481–5487. [PubMed] [Google Scholar]
- 21.Hermanns M.I., Kasper J., Dubruel P., Pohl C., Uboldi C. An impaired alveolar-capillary barrier in vitro: effect of proinflammatory cytokines and consequences on nanocarrier interaction. J. R. Soc. Interface. 2009;7(Suppl. 1):S41–S54. doi: 10.1098/rsif.2009.0288.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stentebjerg-Andersen A., Notlevsen I.V., Brodin B., Nielsen C.U. Calu-3 cells grown under AIC and LCC conditions: implications for dipeptide uptake and transepithelial transport of substances. Eur. J. Pharm. Biopharm. 2011;78:19–26. doi: 10.1016/j.ejpb.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 23.Krump-Konvalinkova V., Bittinger F., Unger R.E., Peters K., Lehr H.A. Generation of human pulmonary microvascular endothelial cell lines. Lab. Invest. 2001;81:1717–1727. doi: 10.1038/labinvest.3780385. [DOI] [PubMed] [Google Scholar]
- 24.Lanone S., Rogerieux F., Geys J., Dupont A., Maillot-Marechal E. Comparative toxicity of 24 manufactured nanoparticles in human alveolar epithelial and macrophage cell lines. Part. Fibre Toxicol. 2009;6:14. doi: 10.1186/1743-8977-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Legendre A., Froment P., Desmots S., Lecomte A., Habert R. An engineered 3D blood–testis barrier model for the assessment of reproductive toxicity potential. Biomaterials. 2010;31:4492–4505. doi: 10.1016/j.biomaterials.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 26.Lemieux M., Bouchard F., Gosselin P., Paquin J., Mateescu M.A. The NCI-N87 cell line as a gastric epithelial barrier model for drug permeability assay. Biochem. Biophys. Res. Commun. 2011;412:429–434. doi: 10.1016/j.bbrc.2011.07.106. [DOI] [PubMed] [Google Scholar]
- 27.Dekali S., Divetain A., Kortulewski T., Vanbaelinghem J., Gamez C. Cell cooperation and role of the P2X7 receptor in pulmonary inflammation induced by nanoparticles. Nanotoxicology. 2012 doi: 10.3109/17435390.2012.735269. [DOI] [PubMed] [Google Scholar]
- 28.Godfrey R.W. Human airway epithelial tight junctions. Microsc. Res. Tech. 1997;38:488–499. doi: 10.1002/(SICI)1097-0029(19970901)38:5<488::AID-JEMT5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 29.Elbert K.J., Schafer U.F., Schafers H.J., Kim K.J., Lee V.H. Monolayers of human alveolar epithelial cells in primary culture for pulmonary absorption and transport studies. Pharm. Res. 1999;16:601–608. doi: 10.1023/a:1018887501927. [DOI] [PubMed] [Google Scholar]
- 30.Saitou M., Ando-Akatsuka Y., Itoh M., Furuse M., Inazawa J. Mammalian occludin in epithelial cells: its expression and subcellular distribution. Eur. J. Cell Biol. 1997;73:222–231. [PubMed] [Google Scholar]
- 31.Cavanaugh K.J., Jr., Oswari J., Margulies S.S. Role of stretch on tight junction structure in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2001;25:584–591. doi: 10.1165/ajrcmb.25.5.4486. [DOI] [PubMed] [Google Scholar]
- 32.Wan H., Winton H.L., Soeller C., Stewart G.A., Thompson P.J. Tight junction properties of the immortalized human bronchial epithelial cell lines Calu-3 and 16HBE14o. Eur. Respir. J. 2000;15:1058–1068. doi: 10.1034/j.1399-3003.2000.01514.x. [DOI] [PubMed] [Google Scholar]
- 33.Santos M.I., Fuchs S., Gomes M.E., Unger R.E., Reis R.L. Response of micro- and macrovascular endothelial cells to starch-based fiber meshes for bone tissue engineering. Biomaterials. 2007;28:240–248. doi: 10.1016/j.biomaterials.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Sapin C., Baricault L., Trugnan G. PKC-dependent long-term effect of PMA on protein cell surface expression in Caco-2 cells. Exp. Cell Res. 1997;231:308–318. doi: 10.1006/excr.1997.3488. [DOI] [PubMed] [Google Scholar]
- 35.Frohlich E., Meindl C., Roblegg E., Ebner B., Absenger M. Action of polystyrene nanoparticles of different sizes on lysosomal function and integrity. Part. Fibre Toxicol. 2012;9:26. doi: 10.1186/1743-8977-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frohlich E., Samberger C., Kueznik T., Absenger M., Roblegg E. Cytotoxicity of nanoparticles independent from oxidative stress. J. Toxicol. Sci. 2009;34:363–375. doi: 10.2131/jts.34.363. [DOI] [PubMed] [Google Scholar]
- 37.Ruenraroengsak P., Novak P., Berhanu D., Thorley A.J., Valsami-Jones E. Respiratory epithelial cytotoxicity and membrane damage (holes) caused by amine-modified nanoparticles. Nanotoxicology. 2012;6:94–108. doi: 10.3109/17435390.2011.558643. [DOI] [PubMed] [Google Scholar]
- 38.Xia T., Kovochich M., Brant J., Hotze M., Sempf J. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 2006;6:1794–1807. doi: 10.1021/nl061025k. [DOI] [PubMed] [Google Scholar]
- 39.Kasper J., Hermanns M.I., Bantz C., Maskos M., Stauber R. Inflammatory and cytotoxic responses of an alveolar-capillary coculture model to silica nanoparticles: comparison with conventional monocultures. Part. Fibre Toxicol. 2011;8:6. doi: 10.1186/1743-8977-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jantzen K., Roursgaard M., Desler C., Loft S., Rasmussen L.J. Oxidative damage to DNA by diesel exhaust particle exposure in co-cultures of human lung epithelial cells and macrophages. Mutagenesis. 2012 doi: 10.1093/mutage/ges035. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann A.D., Blank F., Baum O., Gehr P., Rothen-Rutishauser B.M. Diesel exhaust particles modulate the tight junction protein occludin in lung cells in vitro. Part. Fibre Toxicol. 2009;6:26. doi: 10.1186/1743-8977-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geys J., De Vos R., Nemery B., Hoet P.H. In vitro translocation of quantum dots and influence of oxidative stress. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297:L903–L911. doi: 10.1152/ajplung.00029.2009. [DOI] [PubMed] [Google Scholar]
- 43.Huh D., Matthews B.D., Mammoto A., Montoya-Zavala M., Hsin H.Y. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blank F., Rothen-Rutishauser B., Gehr P. Dendritic cells and macrophages form a transepithelial network against foreign particulate antigens. Am. J. Respir. Cell Mol. Biol. 2007;36:669–677. doi: 10.1165/rcmb.2006-0234OC. [DOI] [PubMed] [Google Scholar]
- 45.Geys J., Coenegrachts L., Vercammen J., Engelborghs Y., Nemmar A. In vitro study of the pulmonary translocation of nanoparticles: a preliminary study. Toxicol. Lett. 2006;160:218–226. doi: 10.1016/j.toxlet.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Yacobi N.R., Demaio L., Xie J., Hamm-Alvarez S.F., Borok Z. Polystyrene nanoparticle trafficking across alveolar epithelium. Nanomedicine. 2008;4:139–145. doi: 10.1016/j.nano.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Halamoda Kenzaoui B., Angeloni S., Overstolz T., Niedermann P., Chapuis Bernasconi C. Transfer of ultrasmall iron oxide nanoparticles from human brain-derived endothelial cells to human glioblastoma cells. ACS Appl. Mater. Interfaces. 2013;5:3581–3586. doi: 10.1021/am401310s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.