

Abstract
Synthetic proteins bearing site-specific posttranslational modifications have revolutionized our understanding of their biological functions in vitro and in vivo. One such modification, O-GlcNAcylation, is the dynamic addition of β-N-acetyl glucosamine to the side chains of serine and threonine residues of proteins, yet our understanding of the site-specific impact of O-GlcNAcylation remains difficult to evaluate in vivo because of the potential for enzymatic removal by endogenous O-GlcNAcase (OGA). Thioglycosides are generally perceived to be enzymatically stable structural mimics of O-GlcNAc; however, in vitro experiments with small-molecule GlcNAc thioglycosides have demonstrated that OGA can hydrolyze these linkages, indicating that S-linked β-N-acetyl glucosamine (S-GlcNAc) on peptides or proteins may not be completely stable. Here, we first develop a robust synthetic route to access an S-GlcNAcylated cysteine building block for peptide and protein synthesis. Using this modified amino acid, we establish that S-GlcNAc is an enzymatically stable surrogate for O-GlcNAcylation in its native protein setting. We also applied nuclear magnetic resonance spectroscopy and computational modeling to find that S-GlcNAc is an good structural mimic of O-GlcNAc. Finally, we demonstrate that site-specific S-GlcNAcylation results in biophysical characteristics that are the same as those of O-GlcNAc within the context of the protein α-synuclein. While this study is limited in focus to two model systems, these data suggest that S-GlcNAc broadly resembles O-GlcNAc and that it is indeed a stable analogue in the context of peptides and proteins.
Graphical abstract
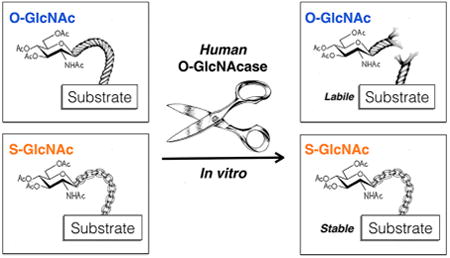
The addition of the single monosaccharide β-N-acetyl glucosamine (O-GlcNAc modification) to the hydroxyl side chains of serine and threonine residues of nuclear, cytosolic, and mitochondrial proteins is an abundant intra-cellular posttranslational modification (PTM) in plants and animals (Figure 1A).1 The installation of this PTM is catalyzed by O-GlcNAc transferase (OGT) and can be subsequently removed by the glycosidase O-GlcNAcase (OGA).2 The proper maintenance of O-GlcNAcylation is required for development in both mice and Drosophila, as genetic knockouts of OGT are lethal and conditional knockouts at the cellular level result in cell-cycle arrest and death.3–6 Moreover, misregulation of O-GlcNAcylation is an important feature of human disease.7–9 For example, all types of cancer that have been examined have overall levels of O-GlcNAcylation higher than those of healthy tissue, and this increase has been shown to be critical for cancer cell survival and tumorigenesis.9 In direct contrast, the global amounts of O-GlcNAcylation are reduced in the brains of Alzheimer's disease patients,10 and a small-molecule inhibitor of OGA that elevates the levels of the modification reduces the rate of progression of neurodegeneration in an Alzheimer's disease mouse model.11 Despite the identification of more than 1000 potentially O-GlcNAcylated proteins,12 the biochemical consequences of most of these events are unknown, and the exact molecular roles for O-GlcNAcylation in both basic biology and disease are far from being completely characterized.
Figure 1.
O-GlcNAcylation and the corresponding S-GlcNAc analogue. (A) O-GlcNAcylation is the addition of N-acetyl glucos-amine to serine and threonine residues of intracellular proteins. It is added by the enzyme O-GlcNAc transferase (OGT) and removed by O-GlcNAcase (OGA). (B) S-GlcNAc analogues of O-GlcNAc could be used as enzymatically stable analogues in synthetic proteins, but whether human OGA can remove these modifications was an open question.
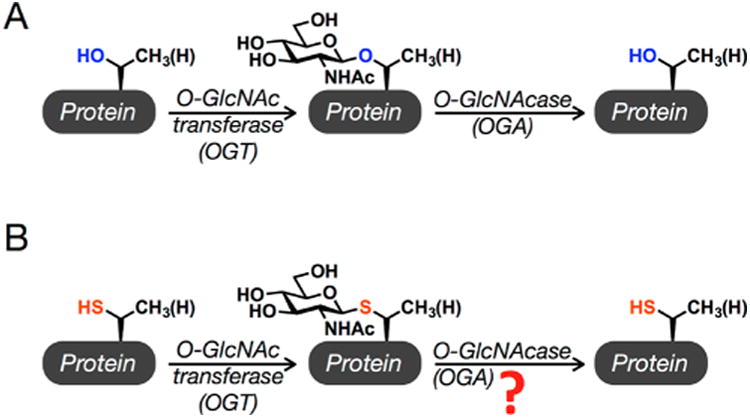
The preparation of site-specifically modified proteins with chemistry has allowed the direct characterization of the effects of several PTMs in an in vitro setting. For example, we previously utilized the technique of expressed protein ligation (EPL) to prepare the Parkinson's disease-associated protein α-synuclein with an O-GlcNAc modification at two different sites. Biochemical and biophysical analysis then revealed that O-GlcNAcylation inhibits α-synuclein aggregation without having a pronounced effect on its ability to bind membranes,13,14 a key feature of its physiological roles. However, extension of these synthetic proteins to the context of cell culture or an in vivo model is limited by the lability of the O-GlcNAc modification to the enzymatic activity of endogenous OGA. This barrier could be overcome by the use of enzymatically stable analogues of the modification, as long as they faithfully mimic its biophysical characteristics. For example, stable analogues of ubiquitin can be made by inserting mutations at its C-terminus that render it resistant to deubiquitinases, which recently permitted the chemical installation of ubiquitin onto histones in nucleo.15 Similarly, the thioglycoside analogue of O-GlcNAc, S-linked β-N-acetyl glucosamine [S-GlcNAc (Figure 1B)] attached to a cysteine residue instead of serine, was used to examine the effect of O-GlcNAcylation on casein kinase II using microinjection into living cells,16 as many thioglycosides have been shown to be stable against removal by glycosidases.17 However, Vocadlo and co-workers18 demonstrated that human OGA (hOGA) is a proficient bifunctional catalyst capable of cleaving both oxygen and small-molecule thioglycosides. While these thioglycosides were aryl and therefore more activated as leaving groups, this result still raises the possibility that it can remove S-GlcNAc from peptides and proteins. Notably, S-GlcNAcylation on cysteine residues of proteins in mice and humans was recently identified using electron transfer dissociation mass spectroscopy (ETD), and the same study biochemically confirmed that recombinant OGT can transfer GlcNAc to cysteine residues to generate S-GlcNAcylation.19 Taken together, these previous results highlight the need to determine both the structural effects of S-GlcNAcylation and its stability against hOGA, as they are critical to both its use as an O-GlcNAc surrogate in synthetic proteins and its investigation as an endogenous cysteine PTM.
Here, we developed a new synthetic route to a cysteine S-GlcNAc amino acid for solid-phase peptide synthesis, which complements other methods like disulfide20 and dehydroalanine tagging.21 We then generated S-GlcNAcylated and O-GlcNAcylated peptides corresponding to the N-terminal region of the mouse β-estrogen receptor, as the O-GlcNAc-modified peptide had been used previously to demonstrate that this modification stabilizes a β-turn in the peptide.22 We demonstrate that both the S and O modifications induce similar structures in these model peptides by using a combination of two-dimensional (2D) nuclear magnetic resonance (NMR) and computational modeling. We then use EPL to generate α-synuclein with S-GlcNAcylation at physiologically relevant residue 87 and use recombinant hOGA to show that this modification is completely enzymatically stable at the peptide and protein level. Finally, we show that S-GlcNAcylation has identical effects on α-synuclein aggregation and membrane binding when compared to the effects of the site-specifically O-GlcNAcylated protein. Together, these data indicate that S-GlcNAc will have similar structural consequences on substrate proteins and demonstrate that it is indeed enzymatically stable and, therefore, a good surrogate for future experiments with synthetic proteins and peptides, including α-synuclein.
Materials and Experimental Details
General
All solvents and reagents were purchased from commercial sources (Sigma-Aldrich, Fluka, EMD, Novagen, etc.) and used without any further purification. All aqueous solutions were prepared using ultrapure laboratory grade water (deionized, filtered, and sterilized) obtained from an in-house ELGA water purification system and filter sterilized with 0.45 μm syringe filters (VWR) before use. Growth media (LB broth, Miller, Novagen, and TB broth, Sigma) were prepared, sterilized, stored, and used according to the instructions of the manufacturer. Antibiotics were prepared as stock solutions at a working concentration of 1000× [100 mg mL−1 ampicillin sodium salt (EMD) and 50 mg mL−1 kanamycin sulfate (EMD)] and stored at −20 °C. All bacterial growth media and cultures were handled using sterile conditions under an open flame. All silica gel column chromatography was performed using 60 Å silica gel (EMD) and all thin-layer chromatography performed using 60 Å, F254 silica gel plates (EMD) with detection by ceric ammonium molybdate (CAM) and/or ultraviolet light. Reverse-phase high-performance liquid chromatography (RP-HPLC) was performed using an Agilent Technologies 1200 Series HPLC instrument with a diode array detector. Unless otherwise stated, the following HPLC buffers were used: buffer A, 0.1% TFA in H2O; buffer B, 0.1% TFA and 90% ACN in H2O. Mass spectra were acquired on an API 3000 LC/MS-MS system (Applied Biosystems/MDS SCIEX). 1H NMR spectra were acquired on a Varian Mercury 400 MHz or Varian VNMRS 500 MHz magnetic resonance spectrometer. TOCSY and NOESY spectra were acquired on a Varian VNMRS-600 three-channel NMR spectrometer equipped with a CryoProbe. IR spectra were obtained on a Bruker Vertex 80v.
Synthesis of Amino Acid Building Blocks
N-{[(9H-Fluoren-9-yl)methoxy]carbonyl}-S-((2S,3R,4R,5S,6R)-4,5-diac-etoxy-6-(acetoxymethyl)-3-{[(2,2,2-trichloroethoxy)-carbonyl]amino}tetrahydro-2H-pyran-2-yl)-l-cysteine (2)
N-Troc-Peracetyl β-d-glucosamine (1.45 mmol, 2.5 equiv), InBr3 (41.3 mg, 0.116 mmol, 0.2 equiv), and N-Fmoc-l-Cys-OH (200 mg, 0.582 mmol, 1 equiv) were suspended in 1,2-dichloroethane (5.8 mL). The reaction mixture was heated to reflux. Reaction progress was monitored by mass spectroscopy and TLC (7:2:1 EtOAc/MeOH/H2O). The reaction mixture was concentrated in vacuo and azeotroped with toluene multiple times to remove the acetic acid generated in situ. The residue was purified by flash chromatography (4:96:0.1 MeOH/CH2Cl2/AcOH) and concentrated in vacuo to afford an off-white solid (422 mg, 90%): 1H NMR (600 MHz, chloroformd) δ 7.75 (d, J = 7.6 Hz, 2H), 7.61–7.57 (m, 2H), 7.41–7.37 (m, 2H), 7.33–7.29 (m, 2H), 5.99 (d, J = 7.6 Hz, 1H), 5.35 (d, J = 9.3 Hz, 1H), 5.17 (t, J = 9.8 Hz, 1H), 5.05 (t, J = 9.7 Hz, 1H), 4.79 (d, J = 12.1 Hz, 1H), 4.72–4.62 (m, 2H), 4.52–4.35 (m, 3H), 4.21 (d, J = 16.0 Hz, 3H), 3.74 (q, J = 10.0 Hz, 1H), 3.63 (s, 1H), 3.32 (d, J = 14.5 Hz, 1H), 3.09 (d, J = 14.1 Hz, 1H), 2.05 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H); 13C NMR (151 MHz, chloroform-d) δ 168.19, 166.85, 140.89, 138.68, 125.22, 124.56, 122.43, 117.46, 82.08, 74.63, 74.42, 74.21, 73.16, 71.81, 70.72, 66.10, 64.89, 59.88, 52.48, 50.81, 44.45, 30.40, 18.15, 18.03; HRMS calcd for (M + H)+ m/z 805.1004, found m/z 805.1012; IR (KBr pellet) 3344.6, 3067.2, 2955.4, 1754.3, 1535.5, 1478.7, 1450.5, 1370.5, 1222.4, 1048.4, 948.5, 917.5, 819.4 cm−1.
(2R,3S,4R,5R,6S)-6-{[(R)-2-({[(9H-Fluoren-9-yl)methoxy]-carbonyl}amino)-3-oxo-3-(perfluorophenoxy)propyl]thio}-2-(acetoxymethyl)-5-{[(2,2,2-trichloroethoxy)carbonyl]amino}-tetrahydro-2H-pyran-3,4-diyl Diacetate (6)
Anhydrous pyridine (2.23 mmol, 4.5 equiv) was added to a stirring solution of 1 (400 mg, 0.496 mmol, 1 equiv) in anhydrous DMF (5.7 mL) under N2. To this solution was added dropwise pentafluorophenyl trifluoroacetate (1.48 mmol, 3 equiv) under N2. The reaction mixture was allowed to stir at room temperature overnight. Reaction progress was monitored by TLC (35% EtOAc/hexane). Once the reaction was complete, the mixture was concentrated in vacuo and azeotroped with toluene multiple times to remove the TFA generated in situ. The residue was purified by flash chromatography (40:60 acetone/hexane) and concentrated in vacuo to afford an off-white solid (440 mg, 91%): 1H NMR (500 MHz, chloroform-d) δ 7.78 (d, J = 7.5 Hz, 2H), 7.63 (dd, J = 13.8, 7.5 Hz, 2H), 7.41 (t, J = 7.5 Hz, 2H), 7.36–7.31 (m, 2H), 6.21 (d, J = 7.7 Hz, 1H), 5.35 (d, J = 9.3 Hz, 1H), 5.22 (t, J = 9.9 Hz, 1H), 5.07 (t, J = 9.7 Hz, 1H), 4.89 (td, J = 7.8, 3.7 Hz, 1H), 4.78 (d, J = 11.9 Hz, 1H), 4.69 (d, J = 10.3 Hz, 1H), 4.61–4.50 (m, 2H), 4.43 (t, J = 8.9 Hz, 1H), 4.28 (t, J = 6.9 Hz, 1H), 4.16–4.05 (m, 2H), 3.83 (q, J = 10.0 Hz, 1H), 3.72–3.67 (m, 1H), 3.50 (dd, J = 14.7, 4.0 Hz, 1H), 3.08 (dd, J = 14.8, 8.3 Hz, 1H), 2.06 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H); 13C NMR (126 MHz, chloroform-d) δ 170.67, 169.29, 166.95, 156.02, 154.30, 143.74, 143.49, 141.32, 127.80, 127.14, 125.08, 124.94, 120.07, 83.71, 76.29, 74.49, 73.02, 68.27, 67.31, 62.10, 54.88, 53.70, 47.11, 31.58, 20.59, 20.50; HRMS calcd for (M + H)+ m/z 971.0846, found m/z 971.0833; IR (KBr pellet) 3339.8, 3068.4, 2955.5, 2670.0, 2461.8, 1753.4, 1519.4, 1450.9, 1374.5, 1223.3, 994.8, 916.0, 878.0, 817.5 cm−1.
(2R,3S,4R,5R,6S)-6-{[(R)-2-({[(9H-Fluoren-9-yl)methoxy]-carbonyl}amino)-3-oxo-3-(perfluorophenoxy)propyl]thio}-5-acetamido-2-(acetoxymethyl)tetrahydro-2H-pyran-3,4-diyl Diacetate (7)
Pfp ester 6 (810 mg, 0.833 mmol, 1 equiv) was dissolved with 12 mL of a 3:2:1 THF/Ac2O/AcOH mixture under N2. Zinc dust (1.08 g, 16.52 mmol, 36.5 equiv) was added to the reaction flask. The reaction mixture was allowed to stir at room temperature overnight. Upon disappearance of the starting material as determined by TLC, the reaction mixture was filtered through Celite and the filtrant concentrated in vacuo. The residue was azeotroped multiple times with toluene to afford an off-white powder. The product was recrystallized from EtOAc using hexanes (488 mg, 70%): 1H NMR (600 MHz, chloroform-d) δ 7.75 (d, J = 7.5 Hz, 2H), 7.62 (t, J = 8.2 Hz, 2H), 7.38 (td, J = 7.5, 2.7 Hz, 2H), 7.30 (td, J = 7.5, 2.6 Hz, 2H), 6.35 (d, J = 7.8 Hz, 1H), 5.73 (d, J = 9.2 Hz, 1H), 5.17 (t, J = 9.8 Hz, 1H), 5.05 (t, J = 9.7 Hz, 1H), 4.84 (td, J = 8.1, 3.9 Hz, 1H), 4.63 (d, J = 10.3 Hz, 1H), 4.51 (dd, J = 10.6, 6.9 Hz, 1H), 4.41 (dd, J = 10.8, 7.0 Hz, 1H), 4.25 (t, J = 6.9 Hz, 1H), 4.11 (td, J = 7.7, 6.2, 3.3 Hz, 2H), 4.05 (dd, J = 12.4, 5.6 Hz, 1H), 3.70–3.66 (m, 1H), 3.46 (dd, J = 14.4, 3.9 Hz, 1H), 3.09 (dd, J = 14.4, 8.4 Hz, 1H), 2.03 (s, 3H), 2.02 (s, 3H), 1.92 (s, 3H), 190 (s, 3H); 13C NMR (151 MHz, chloroform-d) δ 171.02, 170.58, 170.53, 169.17, 167.02, 156.03, 143.68, 143.59, 141.74, 141.28, 141.25, 140.07, 138.69, 137.01, 127.74, 127.71, 127.11, 127.08, 125.07, 124.97, 120.00, 119.98, 83.81, 76.32, 73.33, 68.21, 67.19, 62.13, 53.77, 52.89, 47.10, 31.21, 23.15, 20.62, 20.61, 20.54, 20.47; HRMS calcd for (M + H)+ m/z 839.1909, found m/z 839.1918; IR (KBr pellet) 3334.7, 3068.7, 2926.23, 2854.6, 2669.3, 2461.4, 1752.9, 1518.2, 1450.7, 1373.4, 1218.5, 994.5, 915.9, 874.0, 815.7 cm−1.
Peptide Synthesis
All peptides were synthesized using standard Fmoc solid-phase chemistry on Rink amide ChemMatrix (PCAS BioMatrix, 0.45 mmol g−1) or Dawson Dbz AM (Novabiochem, 0.49 mmol g−1) resin using HBTU (5 equiv, Novabiochem) and DIEA (10 equiv, Sigma) in DMF for 1 h. For activated glycosylated amino acids, 2 equiv of monomer in 3 mL of DMF was coupled overnight. N-Terminal acetylation was conducted using Ac2O (3 equiv) and pyridine (2 equiv) in DMF for 1 h. When peptides were completed, acetyl groups were deprotected with hydrazine monohydrate [80% (v/v) in MeOH] twice for 30 min with mixing. Following deprotection, Dawson linker peptides were activated with p-nitrophenyl chloroformate (5 equiv in DCM, 1.5 h, mixing) followed by treatment with excess DIEA (0.5 M in DMF) for 30 min to cyclize the Dbz linker. All peptides were then cleaved (95:2.5:2.5 TFA/H2O/triisopropylsilane) for 3.5 h at room temperature, precipitated out of cold ether, and purified by reverse-phase HPLC using preparative chromatography. To generate thioester peptides, crude peptides were resuspended in thiolysis buffer [150 mM NaH2PO4 and 150 mM MESNa (pH 7.5)] and incubated at room temperature for 2 h prior to purification. All peptides were characterized by mass analysis using ESI-MS, and the sequence purity was assessed by analytical HPLC.
Protein Expression and Purification
General expression and purification methods were similar to those previously reported. Briefly, BL21(DE3) chemically competent Escherichia coli (VWR) cells were transformed with plasmid DNA by heat shock and plated on selective LB agar plates containing 50 μg mL−1 kanamycin (C-terminus) or 100 μg mL−1 ampicillin (N-terminus). Single colonies were then inoculated and grown to an OD600 of 0.6 at 37 °C while being shaken at 250 rpm, and then expression was induced with IPTG (final concentration of 0.5 mM) at 25 °C while the sample was being shaken at 250 rpm for 18 h. Pellets were harvested by centrifugation (8000g for 30 min at 4 °C) and lysed. The C-terminus was acidified (pH 3.5 with HCl), centrifuged, dialyzed against 3 × 1 L of 1% acetic acid in water (degassed with N2, 1 h/L), and purified by HPLC. The N-terminus was loaded onto a Ni-NTA purification column (HisTrap FF Crude, GE Healthcare), and eluted fractions were dialyzed against 3 × 1 L [100 mM NaH2PO4, 150 mM NaCl, 1 mM EDTA, and 1 mM TCEP HCl (pH 7.2)] and concentrated. Sodium mercaptoethanesulfonate (MESNa) was added to a final concentration of 200 mM along with fresh TCEP (final concentration of 2 mM) overnight to generate the protein thioester and then purified by HPLC. Pure protein fragments were characterized by analytical RP-HPLC and ESI-MS.
Expressed Protein Ligation
General ligation and purification methods were similar to those previously reported. Briefly, the C-terminal fragment (2 mM) and peptide fragment (4 mM) were dissolved in ligation buffer [300 mM NaH2PO4, 6 M guanidine HCl, 100 mM MESNa, and 1 mM TCEP (pH 7.8)] and allowed to react at room temperature. Following completion, as monitored by HPLC, the pH was adjusted to 4 with HCl and methoxylamine was added (final concentration of 100 mM) to deprotect the N-terminal thiazolidine group. Following HPLC purification, the N-terminal fragment (8 mM) and ligation product fragment (2 mM) were dissolved in ligation buffer and following completion were purified by HPLC. Finally, radical desulfurization was performed using desulfurization buffer [200 mM NaH2PO4, 6 M guanidine HCl, and 300 mM TCEP (pH 7.0)] containing 2% (v/v) ethanethiol, 10% (v/v) tert-butyl-thiol, and the radical initiator VA-061 (as a 0.2 M stock in MeOH). The reaction mixture was heated to 37 °C for 15 h and then purified by RP-HPLC to yield synthetic α-synuclein.
Circular Dichroism (CD)
Circular dichroism spectra were recorded on a Jasco J-815 CD spectrometer. Sample aliquots were diluted to 50 μM for peptides or 7.5 μM for proteins in 10 mM phosphate buffer at 7.4 for proteins. Spectra were recorded from 250 to 190 nm with a 0.1 nm data pitch, a 50 nm min−1 scanning speed, a 4 s data integration time, a 1 nm bandwidth, and a 1 mm path length with three accumulations, at 25 °C.
Peptide NMR Spectroscopy
NMR spectra were recorded using a Varian (Palo Alto, CA) VNMRS-600 three-channel NMR spectrometer equipped with a CryoProbe. All samples were prepared in H2O and D2O that had been purged of dissolved O2 gas. One-dimensional and two-dimensional (2D) 1H NMR spectra were recorded at protein concentrations of 1.0 mM in 90% H2O/10% D2O solutions containing 10 mM phosphate at pH 3.5 (uncorrected). Total correlated spectroscopy (TOCSY) spectra were recorded utilizing a mixing time of 120 ms. 2D 1H–1H nuclear Overhauser effect spectroscopy (NOESY) spectra were recorded utilizing a 250 ms mixing time. All spectra were acquired at 5 °C with 512 points in f1 and 2024 points in f2. Chemical shift deviations from “random coil” values shown in Figure 3 are reported relative to a DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) standard.
Figure 3.
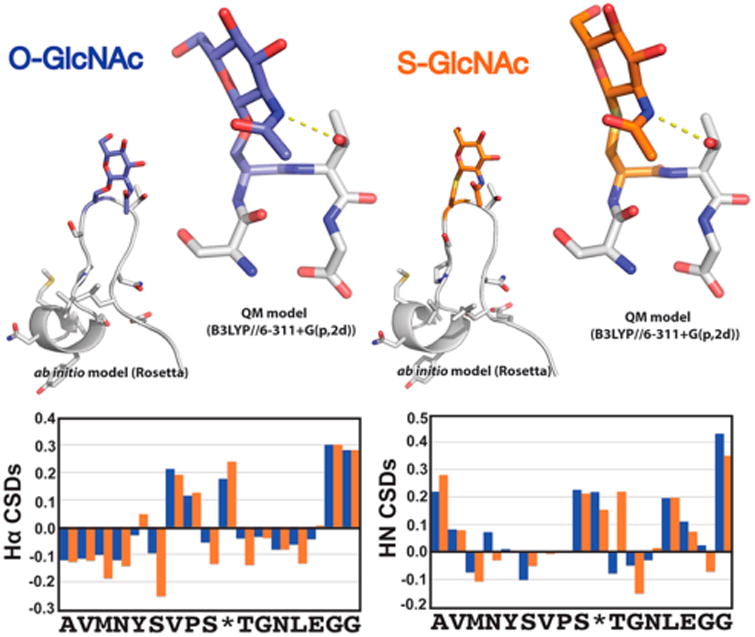
S-GlcNAc is a good structural mimic of O-GlcNAc. Comparison of ab initio folding, QM modeling, and backbone HN and Hα chemical shift deviations (CSDs) for model O/S-GlcNAc-modified peptides. Ab initio folding was conducted within the Rosetta Molecular Design package (see the Supporting Information for “folding funnels” and O/S GlcNAc residue parametrizations). The lowest-energy structure from the ab initio folding is presented. The β-hairpin conformation observed in the lowest-energy ab initio modeling was extracted and geometrically optimized at the B3LYP/6-311+G-(p,2d) level of theory.
Ab Initio Folding and Protein Modeling
Peptides containing both serine O-GlcNAc and cysteine S-GlcNAc modifications were modeled in the Rosetta Molecular Modeling program.23,24 In silico parameters utilized for modeling of the serine and cysteine GlcNAc modifications can be found in the Supporting Information. For ab initio peptide modeling, 25000 decoy structures were initialed with randomized φ and ψ backbone dihedral angles. Side chains, including GlcNAc-modified residues, were repacked and sequences minimized with respect to total molecular energy utilizing a Monte Carlo algorithm. The lowest-energy structure for both serine O-GlcNAc- and cysteine S-GlcNAc-containing model peptides can be seen in Figure 3. The resulting plots of Rosetta-calculated total molecular energy versus RMSD (root-mean-square deviation) to the lowest-energy structure (typically termed the “folding funnel”) can be seen in Figure S2.
Human O-GlcNAcase (hOGA) Assay
Peptides (50 μM, in PBS at pH 7.4) were incubated with hOGA (1 μM) in 200 μL reaction mixtures and allowed to incubate at 37 °C for ≤72 h. Proteins (25 μM, in PBS at pH 7.4) were incubated with hOGA (1 μM) in 125 μL reaction mixtures and allowed to incubate at 37 °C for up to 72 h. Following incubation, hOGA was denatured by being heated at 100 °C for 5 min. Immediately thereafter, reaction mixtures were cooled and analyzed by HPLC.
Circular Dichroism (CD) of α-Synuclein with Lipids
All circular dichroism (CD) spectra were recorded with a Jasco J-815 spectrometer at room temperature. Samples were prepared by mixing a protein and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) in a 1:100 ratio and incubated at room temperature for 20 min. Dried lipid films were solubilized in 10 mM phosphate buffer (pH 7.4) by being vortexed. All spectra (190–250 nm) were recorded with scan rate of 50 nm min−1, a bandwidth of 1 nm, a data integration time of 8 s, and a step resolution of 0.1 nm. Appropriate buffer spectra were subtracted from the final spectra.
Aggregation Reaction
Synthetic or recombinant protein was dissolved with bath sonication in a reaction buffer [10 mM phosphate and 0.05% sodium azide (pH 7.4)] to make its final concentration 25 μM. The solution was centrifuged at 15000 rpm for 15 min at 4 °C to remove any debris, and the supernatant was aliquoted into triplicate reaction mixtures. The samples were incubated at 37 °C with constant agitation (1000 rpm) in a Thermomixer F1.5 (Eppendorf) for 7 days. At each time point, the solution was aliquoted for ThT analysis.
Thioflavin T Fluorescence
Samples from the aggregation assay reaction mixture were diluted in a 96-well plate to a concentration of 1.25 μM with a reaction buffer [10 mM phosphate (pH 7.4) and 0.05% NaN3] containing 10 μM thioflavin T. The plate was read by a Synergy H4 hybrid reader (BioTek). The plate was shaken at 300 rpm for 3 min, followed by data collection (λex = 450 nm, 20 nm band path, λem = 482 nm, 9.5 nm band path, reading from the bottom of a plate, gain = 100, read height = 5.00 mm).
Results and Discussion
Synthesis of S-GlcNAcylated Amino Acids for Peptide and Protein Synthesis
Synthetic routes to generate S-GlcNAc involve the conjugation of β-anomeric thiolates of glucosamine with alanine derivatives typically containing a good leaving group at the β-carbon of the amino acid (Figure 2A).25,26 For example, β-iodoalanine can be employed as the electrophile, but elimination can occur to generate dehydroalanine derivatives, which upon nucleophilic attack by the thiolates led to an undesirable mixture of diastereomers at the α-carbon.25 Although this side reaction can be overcome through the use of a modified Mitsunobu reaction or a cyclic sulfamidate leaving group, they suffer from moderate yields or extensive protecting group chemistry. Moreover, the final glycosylated amino acid was often protected in ways that were incompatible with Fmoc-based solid-phase peptide synthesis. Recently, Polt and co-workers reported the use of minimally competent Lewis acids, in particular In(III)Br3, to promote the conjugation of β-d-glucopyranose peracetates with Fmoc-l-cysteine to afford the corresponding thioglycosides in good yields.27 Notably, the weak Lewis acid was used in catalytic amounts, and the reaction generated exclusively the β-anomer. We envisioned that this same synthetic route could potentially be adapted to glucosamine per-O-acetylated monosaccharides with a carbamate protecting group at the 2-N position.
Figure 2.
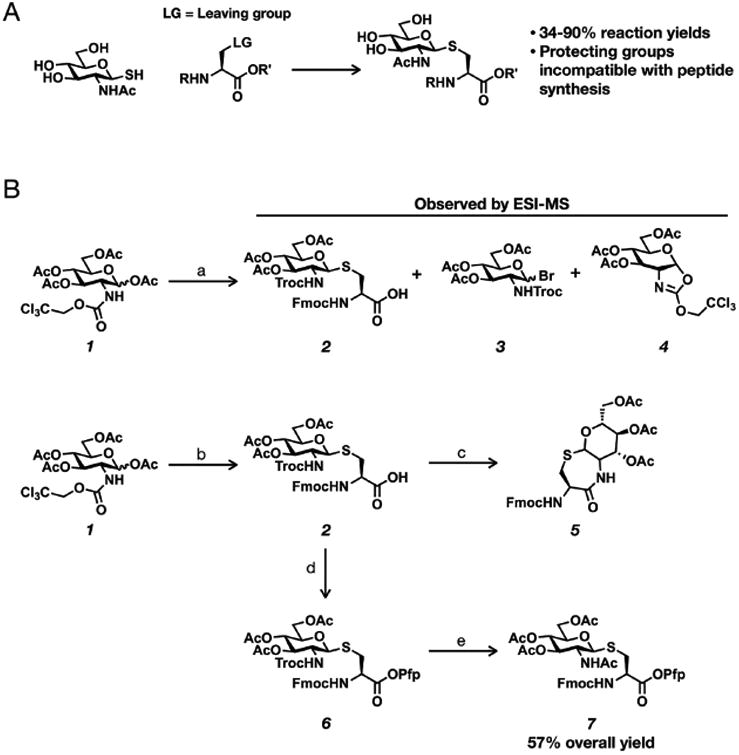
Synthetic routes to S-GlcNAcylated amino acids for solid-phase peptide synthesis. (A) Previous routes involved nucleophilic displacement of leaving groups (LG) on alanine derivatives. (B) The new synthetic route to S-GlcNAcylated cysteine developed here. Reagents: (a) 20 mol % InBr3, Fmoc-Cys-OH, CH2Cl2, reflux, 1–16 h; (b) 20 mol % InBr3, Fmoc-Cys-OH, dichloroethane, reflux, 16 h, 90%; (c) Zn dust, AcOH, Ac2O, 16 h; (d) pentafluorophenyl trifluoroacetate, pyridine, 3 h, 91%; (e) Zn dust, AcOH, Ac2O, 16 h, 70%.
In our initial attempt to use this method to prepare S-GlcNAcylated cysteine (Figure 2B), we chose to use the 2,2,2-trichloroethylcarbamate (Troc) protecting group, as the corresponding per-O-acetyl glucosamine sugar donor (1) can be readily prepared on multigram scales in high yield with no requirement for column chromatography.28,29 Incubation of 1 with N-Fmoc-l-cysteine under conditions similar to those used for glucose monosaccharides (20 mol % InBr3, refluxed in CHCl3, 1 h) resulted in a mixture of products that could be detected by crude ESI-MS. We clearly observed the formation of a small amount of the desired product (2), but also a mixture of the glycosyl bromide of undetermined stereochemistry (3) and the oxazoline generated from the anchimeric assistance of the 2-N-Troc group (4). Interestingly, none of the starting material 1 could be detected; however, continued refluxing for 16 h did not change the distribution of the product and intermediate. These initial results suggested that the amount of Lewis acid (20 mol %) used was sufficient to activate all of the donor sugar and that higher temperatures might be needed to drive the reaction to completion. We therefore exchanged chloroform for 1,2-dichloroethane as the solvent and repeated the reaction while refluxing at 84 °C. Although we were concerned that the higher temperature could potentially decrease the anomeric stereoselectivity of the reaction, we were pleased to see that the increased temperature resulted in the formation of the glycosylated cysteine (2) in 90% yield with complete β-selectivity as determined by the coupling constant (J = 12.1 Hz) in the 1H NMR spectrum. Notably, this excellent yield represents a significant improvement over the previous routes described above, whose yields were modest (∼50%) in the key coupling step or required significant protecting group manipulations before solid-phase peptide synthesis. Following the successful glycosylation to form 2, we attempted to first remove the Troc protecting group and acetylate the resulting free amine in one pot using activated zinc dust in the presence of acetic anhydride. Unfortunately, the major product of the reaction was the cyclic amide formed by the attack of the free amine on the C-terminal mixed anhydride generated in situ, resulting in the lactam (5). This type of side reaction had been observed in the past and was overcome by the protection of the carboxylate as a pentafluorophenyl (Pfp) ester,30 which conveniently activates the amino acid for peptide synthesis. Therefore, we also first protected the C-terminal acid of 2 as a pentafluorophenyl (Pfp) ester 6 in 91% yield. With the C-terminal acid protected, the one-pot deprotection/acetylation of 6 proceeded smoothly to afford compound 7, ready for solid-phase peptide synthesis, in 70% yield and an overall yield of 57%.
S-GlcNAc Is a Good Structural Mimic of O-GlcNAc in the Context of a Model Peptide
After synthesizing the thioglycoside monomer, we next explored whether S-GlcNAc is a good structural mimic for O-GlcNAc. While it is well documented in the literature that O-GlcNAcylation can change the activity or biophysical properties of proteins, the structural details of the O-GlcNAc modifications are mostly unknown. One notable exception to this trend is the analysis of O-GlcNAcylation on a peptide derived from the N-terminus of the murine estrogen receptor (residues 7–23) that is endogenously modified at serine 16.22 More specifically, a combination of NMR spectroscopy and molecular dynamics simulations showed that O-GlcNAcylation promotes a turn in the peptide directly around the glycosylated residue. Following this blueprint, we synthesized the same O-GlcNAcylated peptide (Ac-AVMNYSVPSgSTGNLEGG-NH2) and the corresponding S-GlcNAcylated analogue (Ac-AVMNYSVPSgCTGNLEGG-NH2), which were purified by RP-HPLC and characterized by mass spectrometry (Figure S1). Importantly, we did not observe any major impurities arising from either peptide synthesis, indicating that the S-GlcNAc building block is stable for solid-phase synthesis. With these peptides in hand, we first compared the peptides using CD spectroscopy (Figure S2). The two peptides show very similar CD spectra, with a minimum around 200 nm that is indicative of a largely unfolded conformation and a small negative shoulder around 230 nm that could result from some secondary structure. These results are very consistent with previously published data on the O-GlcNAcylated peptide.22 The spectra from the S-GlcNAcylated peptide do show a slightly more pronounced shoulder at 230 nm, indicating that this modification might induce the peptide turn to a greater extent. We then used a combination of NMR spectroscopy and computational modeling to elucidate the impact of the single-atom substitution. More specifically, we first performed temperature-controlled 2D TOCSY and NOESY experiments to study the effect of S-GlcNAc and O-GlcNAc on the secondary structure of the peptides. Using the sequential assignment strategy, the NMR resonances were then assigned to each amino acid on both peptides. To determine if the glycosylated amino acids induce similar secondary structure, the Hα and HN chemical shifts from both peptides were compared to each other (Table S1) as well as to their deviations from “random coil” Hα and HN chemical shift values (Figure 3).31 It is well established that deviations from random coil shifts are indicators of secondary structure. As is evident from Figure 3, both glycosylated peptides show deviations from random coil and the pattern of deviations looks very similar. These results indicate the possibility that the two motifs are inducing the same effect on secondary structure, consistent with previously published data.22
To computationally model the effects of O- and S-GlcNAcylation on peptide structure, these PTMs were parametrized to facilitate accurate side-chain rotamer repacking within the Rosetta Molecular Design package (see the Supporting Information for .params files). This allowed for ab initio modeling of these peptides, which entailed initiating ∼25000 “decoy” structures and minimizing via a Monte Carlo search of the conformational space of the peptides. This procedure resulted in converged lowest-energy structures for the O- and S-GlcNAcylated peptides prominently featuring a hydrogen bond between the side-chain amide of GlcNAc and the side-chain hydroxyl group of Thr residue i – 1, stabilizing a β-hairpin conformation (Figure 3; see Figure S3 for ab initio “folding funnels”). To gain additional insights into the relative folding energetics of these modifications, we isolated the β-hairpin structures observed in the lowest-energy conformations of the ab initio modeling followed by high-level quantum mechanics-based optimizations and single-point energy calculations at the B3LYP and M06 levels of theory (Figure 3 and Figure S4). A comparison of the differences in energies between a β-hairpin and an extended peptide conformation for each PTM reveals very similar energetics for these two peptide model systems (−3.531 and −2.781 kcal/mol for SgCTG and −3.725 and −2.941 kcal/mol for SgSTG, respectively, at the B3LYP/M06 level of theory). These findings, as well as the extremely minimal differences between the Hα and HN chemical shift deviations from the 2D NMR, strongly support the conclusion that the single-atom substitution has a minimal effect on structure and peptide folding energetics. However, it is important to point out that S-linked oligosaccharides are known to occupy different conformational populations when compared to their O-linked counterparts,17 raising the possibility that S-GlcNAc will occupy different conformations in certain contexts.
S-GlcNAc Is Enzymatically Stable against hOGA in Peptides and Proteins
As noted above, there have been contradictory reports concerning the ability of different OGA enzymes to hydrolyze thioglycoside bonds to GlcNAc.18,19 To evaluate whether hOGA is capable of removing S-GlcNAc on a peptide substrate, purified recombinant hOGA (1 μM) was incubated with either the O-GlcNAc or S-GlcNAc peptides described above (50 μM) in PBS buffer (pH 7.4) at 37 °C for ≤72 h. Following incubation, hOGA was denatured when the samples were heated to 100 °C for 5 min. Immediately thereafter, the reaction mixtures were cooled and analyzed by RP-HPLC and ESI-MS. As expected, hOGA readily removed the O-GlcNAc modification on the peptide (Figure 4 and Figure S5). In contrast, we observed no deprotection with the S-GlcNAc-modified peptide for ≤72 h (Figure 4 and Figure S5), indicating that it is indeed an enzymatically stable surrogate for O-GlcNAcylation at the peptide level.
Figure 4.
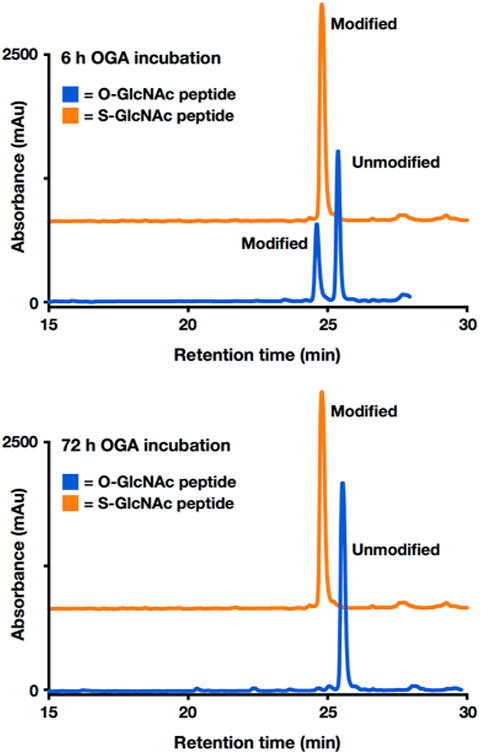
In the context of a peptide, S-GlcNAc is completely stable against human OGA enzymatic deglycosylation. O-GlcNAcylated (blue) or S-GlcNAcylated (orange) peptides (50 μM, in PBS at pH 7.4) were incubated with human OGA (1 μM) at 37 °C for ≤72 h. The S-GlcNAcylated peptide HPLC trace is offset in the y-direction for the sake of clarity. mAU indicates milliabsorbance units. RP-HPLC conditions were 0–70% buffer B over 60 min; buffer A consisted of 0.1% TFA in H2O, and buffer B consisted of 0.1% TFA and 90% ACN in H2O.
Following these results, we wanted to assess the enzymatic stability of S-GlcNAc in a therapeutically relevant protein substrate. Given our previous work on examining the site-specific consequences of O-GlcNAcylation on α-synuclein,13,14 the aggregation-prone protein in Parkinson's disease and other synucleinopathies, we decided to use α-synuclein as our model OGA substrate. More specifically, we chose to examine O- and S-GlcNAcylation at residue 87 (normally serine in α-synuclein), as we have demonstrated that O-GlcNAcylation at this site has interesting effects on α-synuclein,14 particularly in comparison to phosphorylation at the same site.32 To synthesize α-synuclein S-GlcNAcylated at residue 87, termed α-synuclein-(gC87), we used an EPL strategy in combination with one synthetic S-GlcNAcylated peptide and two recombinant α-synuclein fragments (Figure 5A). Accordingly, α-synuclein-(gC87) was retrosynthetically deconstructed (Figure 5A) into a recombinant protein thioester (8, residues 1–75), a synthetic glycopeptide (9, residues 76–90), and a recombinant protein with an N-terminal cysteine residue (10, residues 91–140). Protein fragment 10 was heterogeneously expressed in E. coli. Notably, we found that the initiating methionine residue was conveniently removed during expression by an endogenous methionine aminopeptidase.14 Glycopeptide 9 was prepared using standard Fmoc-based solid-phase peptide synthesis on the Dawson aminobenzyol resin that allows the generation of C-terminal peptide thioesters upon linker activation. On-resin deprotection of the acetyl groups on the S-GlcNAc moiety was accomplished using hydrazine before cleavage and purification of the glycopeptide by RP-HPLC (Figure S6). Importantly, the N-terminal cysteine residue remained protected as a thioproline to prevent autoligation. Incubation of protein 10 and peptide 9 resulted in formation of the ligation product in high yield (Figure S6). The new N-terminal cysteine residue was then deprotected in the same pot by treatment with methoxylamine to give the corresponding protein that was ready for the next ligation reaction (Figure S7). To prepare protein thioester 8, the appropriate α-synuclein fragment (residues 1–75) was recombinantly expressed as an N-terminal fusion with an engineered DnaE intein from Anabaena variabilis. Incubation of this thioester with the S-GlcNAcylated protein fragment gave the full-length α-synuclein product (Figure S8). Finally, radical-based desulfurization was used to convert the two cysteine residues required for the ligations back to the native alanine residues, yielding α-synuclein(gC87) (Figure 5B and Figure S9). To test directly whether hOGA is capable of cleaving S-GlcNAc from α-synuclein(gC87), we again used the same HPLC assay with purified recombinant OGA that was employed for the peptides described above. As a positive control, the O-GlcNAcylated α-synuclein, α-synuclein(gS87), was prepared using EPL as previously described.14 Both α-synuclein(gC87) and α-synuclein(gS87) were separately diluted to a concentration of 25 μM in PBS buffer (pH 7.4) and incubated with hOGA (1 μM) at 37 °C for ≤72 h. Following incubation, hOGA was denatured and the reaction mixtures were centrifuged to remove any possible aggregates. Following centrifugation, the samples were directly injected onto the HPLC instrument and monitored at 214 nm. As expected, hOGA readily hydrolyzed O-GlcNAc-modified α-synuclein, which was confirmed by HPLC and ESI-MS (Figure 6). In contrast, we observed no cleavage of the S-GlcNAc-modified α-synuclein (Figure 6). These results further validate that S-GlcNAc can be used as an enzymatically stable surrogate for O-GlcNAcylation for in vivo and cellular studies.
Figure 5.
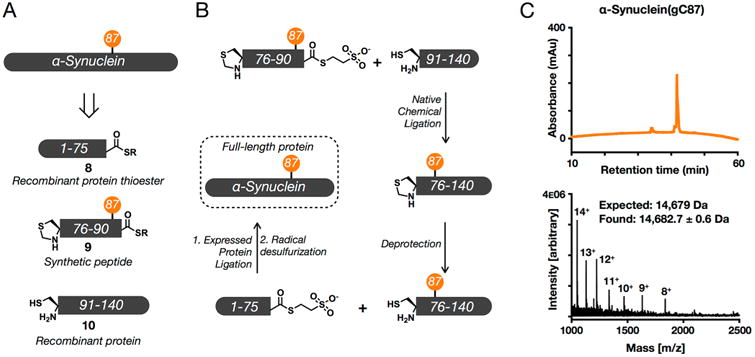
Semisynthesis of S-GlcNAcylated α-synuclein. (A) α-Synuclein was retrosynthetically deconstructed into a recombinant protein thioester (8) obtained using intein chemistry, a synthetic thioester peptide (9), and a recombinant protein (10). (B) These fragments were then combined through iterative ligation reactions. (C) Characterization of synthetic S-GlcNAcylated α-synuclein using RP-HPLC and electrospray ionization mass spectrometry (ESI-MS). Analysis by RP-HPLC showed that synthetic α-synuclein(gC87) was pure, as evidenced by the appearance of only one sharp peak. Characterization by ESI-MS gave a range of charge states that could be deconvoluted to a molecular mass (14682.7 ± 0.6 Da) in excellent agreement with the predicted weight of 14679 Da. RP-HPLC conditions were 0–70% buffer B over 60 min; buffer A consisted of 0.1% TFA in H2O, and buffer B consisted of 0.1% TFA and 90% ACN in H2O.
Figure 6.
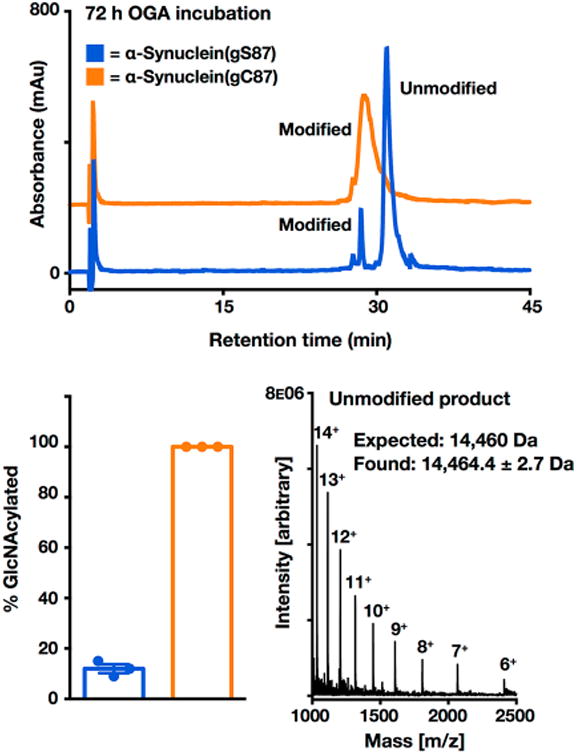
S-GlcNAcylation of α-synuclein is enzymatically stable. α-Synuclein(gS87) or α-synuclein(gC87) (25 μM, in PBS at pH 7.4) was incubated in triplicate with OGA (1 μM) at 37 °C for 72 h. S-GlcNAc is offset in the y-direction for the sake of clarity. mAU indicates milliabsorbance units. Hydrolysis of GlcNAcylated α-synuclein was analyzed by HPLC at the 72 h time point. Deglycosylation was quantitated by area percent using high-performance liquid chromatography (HPLC) at 214 nm, and the deglycosylated product was characterized by ESI-MS. Results are the mean ± the standard error of the mean of three separate biological experiments. RP-HPLC conditions were 35–60% buffer B over 60 min; buffer A consisted of 0.1% TFA in H2O, and buffer B consisted of 0.1% TFA and 90% ACN in H2O.
S-GlcNAc Has the Same Biophysical Effects on α-Synuclein as O-GlcNAcylation Does
To compare the impact of O-GlcNAcylation and S-GlcNAcylation on the gross secondary structure of α-synuclein, we used circular dichroism (CD) spectroscopy using either unmodified α-synuclein, α-synuclein(gC87), or α-synuclein(gS87). Unmodified α-synuclein was recombinantly expressed and purified by RP-HPLC (Figure S10). Unmodified α-synuclein exists as an unstructured random coil in solution, and consistent with our previous results,14 O-GlcNAcylation at serine 87 did not induce any secondary structure in the protein. Not surprisingly on the basis of our structural data given above, the spectra of α-synuclein(gC87) looked identical to those of both the unmodified and O-GlcNAcylated protein controls, demonstrating that this single-atom substitution has essentially no impact on the secondary structure of α-synuclein in solution (Figure S11). Next, we explored the effect of S-GlcNAcylation at residue 87 on the endogenous function of α-synuclein. α-Synuclein is known to interact with negatively charged membranes and vesicles, which induce the protein to become α-helical in structure. This interaction has been shown to be important for both the direct remodeling of membranes33−36 and interactions with other vesicle trafficking proteins.37,38 Notably, this protein–membrane interaction can readily be measured using CD spectroscopy; therefore, we incubated either unmodified α-synuclein, α-synuclein(gS87), or α-synuclein(gC87) with vesicles formed from 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) in a 1:100 protein:lipid ratio. After 20 min, we measured the secondary structure of the α-synuclein proteins by CD and found that neither O-GlcNAcylation nor S-GlcNAcylation at residue 87 had any effect on the membrane binding properties of α-synuclein (Figure 7A). These results are consistent with our previous data on α-synuclein(gS87) and show that S-GlcNAcylation is again a grossly equivalent modification in this context. Next, we examined the effect of S-GlcNAcylation at residue 87 on α-synuclein aggregation using the fluorescence of thioflavin T (ThT), which is a dye that intercalates into the fibers that are formed by amyloid proteins, including α-synuclein. Unmodified α-synuclein, α-synuclein(gS87), or α-synuclein(gC87) was subjected to aggregation by incubation at 37 °C with constant agitation (1000 rpm) at a protein concentration of 25 μM. After 0, 72, 120, and 168 h, aliquots of the reaction mixture were removed and analyzed (Figure 7B). Consistent with our previous results, O-GlcNAcylation at residue 87 did not completely block protein aggregation but did significantly inhibit it. Notably, S-GlcNAcylation at the same position had essentially the same effect, further supporting it as being functionally equivalent to the native modification. Taken together, these data demonstrate that the substitution of oxygen for sulfur to generate S-GlcNAcylation does not dramatically change the biophysical properties of α-synuclein, and we believe that the conservative nature of this modification will also be well tolerated in the context of other proteins.
Figure 7.
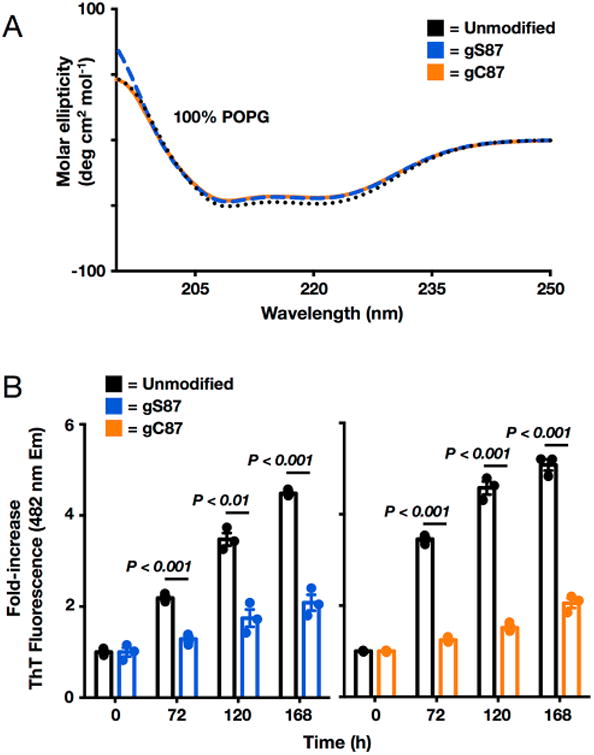
S-GlcNAcylation has effects identical to those of O-GlcNAcylation on the membrane binding and aggregation of α-synuclein. (A) Neither O-GlcNAcylation nor S-GlcNAcylation at residue 87 affects α-synuclein membrane binding. Recombinant α-synuclein, α-synuclein(gS87), or α-synuclein(gC87) was incubated with a 100-fold excess of POPG preformed vesicles and analyzed using circular dichroism (CD). All of the proteins gave essentially indistinguishable CD spectra consistent with the formation of an extended α-helix. POPG denotes 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]. (B) O-GlcNAcylation and S-GlcNAcylation are equally inhibitory toward a-synuclein aggregation. Recombinant α-synuclein, α-synuclein(gS87), or α-synuclein(gC87) was subjected to aggregation conditions (25 μM concentration and agitation at 37 °C) for the indicated lengths of time before analysis of aliquots by ThT fluorescence (λex = 450 nm, and λem = 482 nm). The y-axis shows the fold increase in fluorescence compared with the corresponding protein at time zero. Error bars represent the standard error of the mean from the mean of three biological replicates, and statistical significance was calculated using a two-tailed Student's t test.
Here, we report the facile synthesis and subsequent characterization of S-GlcNAcylated analogues of O-GlcNAcylation and show that this modification is structurally similar and enzymatically stable in a model peptide and protein. First, we developed an improved synthetic route to access a GlcNAc-modified cysteine amino acid by harnessing In(III)Br3 as a catalyst. This reaction is stereospecific, exclusively generating the β-anomer, and does not require the strictly anhydrous conditions of typical O-glycosylation conditions. Furthermore, in the case of S-GlcNAcylated cysteine, the reaction scheme requires only modest protecting group manipulation and provides the monomer protected and activated for solid-phase peptide synthesis. We then experimentally validate, for the first time, that S-GlcNAc is a suitable structural mimic for O-GlcNAc in the context of a model peptide sequence-derived from the β-estrogen receptor. As noted above, this peptide was previously characterized using a combination of NMR spectroscopy and molecular modeling, and the authors found that O-GlcNAcylation at serine 16 stabilizes a β-turn in residues 15–18.22 Consistent with these results, we also observe a β-turn in our O-GlcNAcylated peptide and that the corresponding S-GlcNAcylated peptide has an almost identical structure, as comparison of the chemical shift deviations of the Hα and HN protons revealed a minimal perturbation to the overall fold of the peptide, and we believe the chemical deviation of the threonine residue located just C-terminal to the modified residue could result from the electronic differences between sulfur and oxygen. Importantly, we used the newest version of the Rosetta score function that has been used to model and design glycopeptides.39,40 Substitution of O-GlcNAc with S-GlcNAc in additional synthetic peptides and proteins may present divergent structural features. For example, the linkages will have different bond lengths and may occupy different conformational space due to changes in the exoanomeric effect. However, we expect that this single-atom substitution will be a reasonable structural mimic in the vast majority of scenarios. Unfortunately, there is a dearth of detailed data about the structural effects of O-GlcNAcylation in the context of peptides, so other model systems in which the natural modification has a defined effect are lacking. Therefore, the exact differences between the O and S linkages will need to be further evaluated in different biological and biochemical contexts.
Encouraged by our peptide data, we also used an EPL synthetic strategy to prepare the protein α-synuclein bearing S-GlcNAc at residue 87, α-synuclein(gC87), and compared this protein with the semisynthetic O-GlcNAcylated protein that we previously characterized.14 Separate incubation of these proteins, as well as the O-GlcNAcylated or S-GlcNAcylated β-estrogen receptor peptides, with hOGA showed that while O-GlcNAc can be readily removed the S-GlcNAc modification was completely stable. These data are consistent with previous results obtained by using a bacterial homologue of OGA19 and show that the hydrolysis of GlcNAc thioglycosides by hOGA observed by Vocadlo and co-workers is most likely confined to more activated leaving groups.18 Notably, the activity of OGA for processing several glycosylated proteins has been shown to be independent of the underlying primary sequence, suggesting that the S-GlcNAc analogue should be stable in the context of essentially any protein. Because S-GlcNAc is stable but presumably binds to the OGA active site, it could also function as a substrate-based inhibitor of the enzyme, and this possibility should be considered in future cellular or in vivo experiments. In the case of α-synuclein, we also find that O-GlcNAc and S-GlcNAc similarly have no effect on the secondary structure of α-synuclein in solution or its ability to bind and form an α-helix in the presence of negatively charged membranes. Furthermore, both modifications inhibit the aggregation of α-synuclein, which is a causative factor in the development and progression of Parkinson's disease. These results further support a protective role for O-GlcNAcylation in neurodegenerative diseases. Interestingly, proteomic analysis of mammalian samples recently identified endogenous S-GlcNAcylation of cysteine residues in proteins, and the same authors demonstrated that OGT can indeed modify cysteines of peptides in vitro. This establishes a physiological relevance to studying and synthesizing these modified peptides and proteins for the biological investigation of endogenous site-specific S-GlcNAcylation. In summary, we demonstrate that S-GlcNAc analogues can be used to extend the use of semisynthetic proteins to experiments involving living cells or animals without the complication of O-GlcNAc removal by OGA in the course of the analysis. We anticipate future studies will utilize our general synthetic strategy both to exploit S-GlcNAcylation as a stable O-GlcNAc analogue and to install native cysteine modifications to explore the consequences of both of these important posttranslational modifications in vitro and in vivo.
Supplementary Material
Characterization of the O-GlcNAcylated and S-GlcNA-cylated model peptides (Figure S1), results that show O-GlcNAcylation and S-GlcNAcylation have similar small effects on peptide structure as determined by CD (Figure S2), ab initio “folding funnels” of the O-GlcNAcylated and S-GlcNAcylated model peptides (Figure S3), QM-optimized models of extended and β-hairpin conformations of O- and S-GlcNAcylated tetrapeptides (Figure S4), results that show S-GlcNAcylation in a peptide is resistant to O-GlcNAcase (OGA)-mediated deglycosylation (Figure S5), characterization of S-GlcNAcylated thioester peptide 9 (Figure S6), ligation of peptide 9 and protein 10 and the subsequent deprotection to give the corresponding α-synuclein fragment (Figure S7), ligation with protein thioester 8 to yield full-length S-GlcNAcylated α-synuclein (Figure S8), desulfurization of S-GlcNAcylated α-synuclein (Figure S9), characterization of recombinant, unmodified α-synuclein (Figure S10), evidence that neither O-GlcNAcylation nor S-GlcNAcylation induces secondary structure in α-synuclein (Figure S11), NMR spectra of synthesized small molecules (Figures S12–S14), Hα and HN chemical shift deviations (CSDs) from a random coil of the O-GlcNAcylated and S-GlcNAcylated model peptides (Table S1), residue parameters for the O-GlcNAcylated peptide, and residue parameters for the S-GlcNAcylated peptide (PDF)
Acknowledgments
The authors thank Allan Kershaw (University of Southern California) for NMR assistance and Prof. David Vocadlo (Simon Fraser University, Burnaby, BC) for providing human recombinant O-GlcNAcase.
Funding: C.AD.L. is a fellow of the National Science Foundation Graduate Research Fellowship Program (DGE-0937362). T.W.C. thanks the Washington Research Foundation and the Institute for Protein Design for the Innovation Postdoctoral Fellowship. This research was supported by National Institutes of Health Grant R01GM114537 to M.RP.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00268.
Notes: The authors declare no competing financial interest.
References
- 1.Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vocadlo DJ. O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Curr Opin Chem Biol. 2012;16:488–497. doi: 10.1016/j.cbpa.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Donnell N, Zachara NE, Hart GW, Marth JD. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol. 2004;24:1680–1690. doi: 10.1128/MCB.24.4.1680-1690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sinclair DAR, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, Honda BM. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc) Proc Natl Acad Sci U S A. 2009;106:13427–13432. doi: 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambetta MC, Oktaba K, Muller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009;325:93–96. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- 7.Yuzwa SA, Vocadlo DJ. O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer's disease and beyond. Chem Soc Rev. 2014;43:6839–6858. doi: 10.1039/c4cs00038b. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem. 2014;289:34472–34481. doi: 10.1074/jbc.R114.601351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Z, Vosseller K. O-GlcNAc in cancer biology. Amino Acids. 2013;45:719–733. doi: 10.1007/s00726-013-1543-8. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Iqbal K, Grundke-Iqbal I, Hart G, Gong C. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- 12.Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics. 2014;11:8. doi: 10.1186/1559-0275-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, Arnold DB, Langen R, Pratt MR. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson's disease. Nat Chem. 2015;7:913–920. doi: 10.1038/nchem.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis YE, Galesic A, Levine PM, De Leon CA, Lamiri N, Brennan CK, Pratt MR. O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem Biol. 2017;12:1020–1027. doi: 10.1021/acschembio.7b00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David Y, Vila-Perelló M, Verma S, Muir TW. Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat Chem. 2015;7:394–402. doi: 10.1038/nchem.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarrant MK, Rho HS, Xie Z, Jiang YL, Gross C, Culhane JC, Yan G, Qian J, Ichikawa Y, Matsuoka T, Zachara N, Etzkorn FA, Hart GW, Jeong JS, Blackshaw S, Zhu H, Cole PA. Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat Chem Biol. 2012;8:262–269. doi: 10.1038/nchembio.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Driguez H. Thiooligosaccharides as tools for structural biology. ChemBioChem. 2001;2:311–318. doi: 10.1002/1439-7633(20010504)2:5<311::AID-CBIC311>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 18.Macauley MS, Stubbs KA, Vocadlo DJ. O-GlcNAcase catalyzes cleavage of thioglycosides without general acid catalysis. J Am Chem Soc. 2005;127:17202–17203. doi: 10.1021/ja0567687. [DOI] [PubMed] [Google Scholar]
- 19.Maynard JC, Burlingame AL, Medzihradszky KF. Cysteine S-linked N-acetylglucosamine (S-GlcNAcylation), A New Post-translational Modification in Mammals. Mol Cell Proteomics. 2016;15:3405–3411. doi: 10.1074/mcp.M116.061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernardes GJL, Grayson EJ, Thompson S, Chalker JM, Errey JC, El Oualid F, Claridge TDW, Davis BG. From Disulfide- to Thioether-Linked Glycoproteins. Angew Chem, Int Ed. 2008;47:2244–2247. doi: 10.1002/anie.200704381. [DOI] [PubMed] [Google Scholar]
- 21.Lercher L, Raj R, Patel NA, Price J, Mohammed S, Robinson CV, Schofield CJ, Davis BG. Generation of a synthetic GlcNAcylated nucleosome reveals regulation of stability by H2A-Thr101 GlcNAcylation. Nat Commun. 2015;6:7978. doi: 10.1038/ncomms8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YX, Du JT, Zhou LX, Liu XH, Zhao YF, Nakanishi H, Li YM. Alternative O-GlcNAcylation/O-phosphorylation of Ser16 induce different conformational disturbances to the N terminus of murine estrogen receptor beta. Chem Biol. 2006;13:937–944. doi: 10.1016/j.chembiol.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YEA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drew K, Renfrew PD, Craven TW, Butterfoss GL, Chou FC, Lyskov S, Bullock BN, Watkins A, Labonte JW, Pacella M, Kilambi KP, Leaver-Fay A, Kuhlman B, Gray JJ, Bradley P, Kirshenbaum K, Arora PS, Das R, Bonneau R. Adding Diverse Noncanonical Backbones to Rosetta: Enabling Peptidomimetic Design. PLoS One. 2013;8:e67051. doi: 10.1371/journal.pone.0067051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohnishi Y, Ichikawa M, Ichikawa Y. Facile synthesis of N-Fmoc-serine-S-GlcNAc: a potential molecular probe for the functional study of O-GlcNAc. Bioorg Med Chem Lett. 2000;10:1289–1291. doi: 10.1016/s0960-894x(00)00223-7. [DOI] [PubMed] [Google Scholar]
- 26.Cohen SB, Halcomb RL. Application of Serine-and Threonine-Derived Cyclic Sulfamidates for the Preparation of S-Linked Glycosyl Amino Acids in Solution- and Solid-Phase Peptide Synthesis. J Am Chem Soc. 2002;124:2534–2543. doi: 10.1021/ja011932l. [DOI] [PubMed] [Google Scholar]
- 27.Szabò LZ, Hanrahan DJ, Jones EM, Martin E, Pemberton JE, Polt R. Preparation of S-glycoside surfactants and cysteine thioglycosides using minimally competent Lewis acid catalysis. Carbohydr Res. 2016;422:1–4. doi: 10.1016/j.carres.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitchell SA, Pratt MR, Hruby VJ, Polt R. Solid-phase synthesis of O-linked glycopeptide analogues of enkephalin. J Org Chem. 2001;66:2327–2342. doi: 10.1021/jo005712m. [DOI] [PubMed] [Google Scholar]
- 29.Marotta NP, Cherwien CA, Abeywardana T, Pratt MR. O-GlcNAc Modification Prevents Peptide-Dependent Acceleration of α-Synuclein Aggregation. ChemBioChem. 2012;13:2665–2670. doi: 10.1002/cbic.201200478. [DOI] [PubMed] [Google Scholar]
- 30.Meinjohanns E, Meldal M, Bock K. Efficient synthesis of O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-Ser/Thr building blocks for SPPS of O-GlcNAc glycopeptides. Tetrahedron Lett. 1995;36:9205–9208. [Google Scholar]
- 31.De Simone A, Cavalli A, Hsu STD, Vranken W, Vendruscolo M. Accurate Random Coil Chemical Shifts from an Analysis of Loop Regions in Native States of Proteins. J Am Chem Soc. 2009;131:16332–16333. doi: 10.1021/ja904937a. [DOI] [PubMed] [Google Scholar]
- 32.Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, Chiappe D, Moniatte M, Schneider BL, Aebischer P, Eliezer D, Zweckstetter M, Masliah E, Lashuel HA. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. 2010;30:3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng N, Langen R, Steven AC. Remodeling of lipid vesicles into cylindrical micelles by α-synuclein in an extended α-helical conformation. J Biol Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Middleton ER, Rhoades E. Effects of Curvature and Composition on α-Synuclein Binding to Lipid Vesicles. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burré J, Sharma M, Südhof TC. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–83. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Labonte JW, Adolf-Bryfogle J, Schief WR, Gray JJ. Residue-centric modeling and design of saccharide and glycoconjugate structures. J Comput Chem. 2017;38:276–287. doi: 10.1002/jcc.24679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alford RF, Leaver-Fay A, Jeliazkov JR, O'Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, Labonte JW, Pacella MS, Bonneau R, Bradley P, Dunbrack RL, Das R, Baker D, Kuhlman B, Kortemme T, Gray JJ. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput. 2017;13:ZZZ3031. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of the O-GlcNAcylated and S-GlcNA-cylated model peptides (Figure S1), results that show O-GlcNAcylation and S-GlcNAcylation have similar small effects on peptide structure as determined by CD (Figure S2), ab initio “folding funnels” of the O-GlcNAcylated and S-GlcNAcylated model peptides (Figure S3), QM-optimized models of extended and β-hairpin conformations of O- and S-GlcNAcylated tetrapeptides (Figure S4), results that show S-GlcNAcylation in a peptide is resistant to O-GlcNAcase (OGA)-mediated deglycosylation (Figure S5), characterization of S-GlcNAcylated thioester peptide 9 (Figure S6), ligation of peptide 9 and protein 10 and the subsequent deprotection to give the corresponding α-synuclein fragment (Figure S7), ligation with protein thioester 8 to yield full-length S-GlcNAcylated α-synuclein (Figure S8), desulfurization of S-GlcNAcylated α-synuclein (Figure S9), characterization of recombinant, unmodified α-synuclein (Figure S10), evidence that neither O-GlcNAcylation nor S-GlcNAcylation induces secondary structure in α-synuclein (Figure S11), NMR spectra of synthesized small molecules (Figures S12–S14), Hα and HN chemical shift deviations (CSDs) from a random coil of the O-GlcNAcylated and S-GlcNAcylated model peptides (Table S1), residue parameters for the O-GlcNAcylated peptide, and residue parameters for the S-GlcNAcylated peptide (PDF)