

Abstract
In mammalian cells, cermide-1-phosphate (C1P) is produced via the ATP-dependent mechanism of converting ceramide to C1P by the enzyme, ceramide kinase (CERK). CERK was first described as a calcium-stimulated lipid kinase that co-purified with brain synaptic vesicles, and to date, CERK is the only identified mammalian enzyme known to produce C1P in cells. C1P has steadily emerged as a bioactive sphingolipid involved in cell proliferation, macrophage migration, and inflammatory events. The recent generation of the CERK knockout mouse and the development of CERK inhibitors have furthered our current understanding of CERK-derived C1P in regulating biological processes. In this chapter, the history of C1P as well as the biological functions attributed to C1P are reviewed.
Keywords: Ceramide kinase, Ceramide-1-phosphate, Eicosanoids, Proliferation, Immunity
1 Ceramide-1-Phosphate: The Early Years
Ceramide-1-phosphate (C1P) is synthesized in mammalian cells by the direct phosphorylation of ceramide by ceramide kinase (CERK). To date, CERK is the only known mammalian enzyme to produce C1P (Sugiura et al. 2002), and the enzyme was first described by Bajjalieh and coworkers (1989) as a calcium-stimulated lipid kinase co-purified with brain synaptic vesicles and possessing activity specific for the conversion of ceramide to C1P. Soon after this initial finding, the production of C1P was observed in the human pro-myelocytic leukemia cell line, HL-60 (Dressler and Kolesnick 1990). In this same study, the authors demonstrated that during stimulation, C1P was produced from ceramide derived from sphingomyelin, but not from glycosphingolipids. Follow-up studies by Kolesnick and Hemer (1990) reported a CERK activity distinguishable from diacylglycerol kinase in HL-60 cells verifying the findings of Bajjalieh and coworkers. After these initial studies, over a decade passed before successful cloning of the CERK enzyme was accomplished, and this new molecular “tool” provided researchers with the means to study the role of not only CERK in cellular functions but also C1P.
After the cloning of CERK, the mRNA for the enzyme was found to be expressed in heart, kidney, lung, brain, and hematopoietic cells (Sugiura et al. 2002). Analysis of the CERK mRNA sequence showed that human CERK protein consists of 537 amino acids, which closely resembles the amino acid homology and structure of sphingosine kinase 1 (Sphk1) and 2 (Sphk2). Specifically, CERK was found to contain the five conserved domains (C1–C5) previously identified for Sphk1 and 2. CERK also contains additional conserved regions across several species (M. musculus, D. melanogaster, C. elegans, and O. sativa) that are not homologous to SphK. These include a PH-domain at the N-terminus known to bind the β/γ subunit of heterotrimeric G-proteins, phospoinositol-4,5-bisphophate, and phosphorylated tyrosine residues (Sugiura et al. 2002). These conserved domains have been shown to play a regulatory function for the enzyme. For example, Igarashi and coworkers and Bornancin and coworkers have both demonstrated that the PH-domain is required for the activity of CERK in vitro as well as proper localization of the enzyme in cells (Kim et al. 2005; Carre et al. 2004). Interestingly, expression of the PH-domain alone also demonstrated improper localization suggesting that the catalytic domain also imparts specificity for specific internal membranes of the cell (Kim et al. 2006).
CERK also contains a calcium/calmodulin (CaM)-binding motif of the 1-8-14 type B spanning residues 422–435 [(F/I/L/V/W) XXXXXX (F/A/I/L/V/W) XXXXX (F/I/L/V/W) with a net charge of 2+ to 4+] (Sugiura et al. 2002). The functionality of this calcium/CaM-binding motif was confirmed by Igarashi and coworkers who demonstrated that CaM interacts with CERK and acts as a calcium “sensor” for the enzyme (Mitsutake and Igarashi 2005). Specifically, they showed that the CaM antagonist W-7 decreased both CERK activity and intracellular C1P formation. Additionally, exogenously added CaM enhanced CERK activity in vitro even at low concentrations of Ca2+.
CERK also contains two conserved phosphorylation sites: a casein kinase II phosphorylation site [(S/T) XX (D/E)] at Ser340 and a cAMP-dependent phosphorylation site at Ser424 (Sugiura et al. 2002). There are also many putative protein kinase C (PKC) phosphorylation sites conserved in mammals: Ser72, Thr118, Thr127, Ser230, Ser300, Ser340, and Ser424. At present, the only study investigating the role of phosphorylation in regulating the activity of CERK was carried out by Bornancin and coworkers, which demonstrated that a mutation of Ser340Ala affected the stability of CERK (Chen et al. 2010). The Ser340 residue is located downstream of the catalytic site in a region that has been suggested to possess a regulatory role in CERK activity (Chen et al. 2010). Future identification of the kinases that are involved in CERK phosphorylation may provide additional understanding of how CERK activity is regulated.
Prior to the cloning of CERK, initial studies of the function of C1P utilized exogenous delivery of the sphingolipid followed by the examination of a biological phenotype. In this regard, the first biological activity of C1P was described by the Brindley laboratory. Specifically, Gomez-Munoz et al. (1995) demonstrated that C1P induced DNA synthesis and cell division. Since this initial study, a number of biological activities attributed to C1P have been steadily increasing, further enhancing its recognition as an important lipid-signaling molecule. Currently, C1P has been demonstrated to play a role in DNA synthesis (Gomez-Munoz et al. 1995), macrophage proliferation and migration (Gangoiti et al. 2010; Granado et al. 2009), cPLA2α activation and subsequent production of inflammatory mediators (Pettus et al. 2004; Subramanian et al. 2005; Lamour et al. 2009), as well as inhibition of apoptosis via inhibition of acid sphingomyelinase (A-SMase) (Gomez-Munoz et al. 2004). More recent studies have discovered a potential role for C1P in the processing of the pro-inflammatory cytokine tumor necrosis-alpha (TNFα) (Lamour et al. 2011). These new advances in our knowledge of C1P biology have been facilitated by the development of accurate and reliable methods for detecting the relatively low cellular levels of C1P (Wijesinghe et al. 2010), as well as the availability of CERK-deficient animals, CERK inhibitors, and C1P agonists. Here, we discuss the major findings that have provided substantial evidence supporting a distinct role for C1P in cell growth and inflammatory processes.
2 C1P in Cell Growth and Survival: A Pro-survival Player
As previously stated, the first biological effect for C1P was reported by Gomez-Munoz and coworkers (1995) in regard to cellular proliferation/growth. For example, these early studies demonstrated that short-chain (not naturally found in cells) C1P induced DNA synthesis in Rat-1 fibroblasts (Gomez-Munoz et al. 1995). Additional studies demonstrated that treatment of T17 fibroblasts with natural C1P induced a potent increase in DNA synthesis and levels of proliferating cell nuclear antigen (PCNA) (Gomez-Munoz et al. 1997). Over the course of two reports Gangoiti and coworkers (2008a, b) further demonstrated that C1P stimulates macrophage proliferation through the downstream activation of the extracellularly regulated kinase 1 and 2 (ERK1/2) and c-Jun N-terminal kinase (JNK) pathways. The mechanisms behind the growth promoting role of C1P have recently become a bit more complex, as accompanying reports also implicate the activation of protein kinase C-alpha (PKCα) in C1P-stimulated macrophage proliferation (Gangoiti et al. 2010). Interestingly, these results suggested that the C1P induces the translocation of PKCα from the cytosol to the cell membrane, an event that was shown to be required for the mitogenic effect of C1P in macrophages (Gangoiti et al. 2010). Most recently, the Gomez-Munoz group has demonstrated that C1P also stimulates proliferation in C2C12 myoblasts, a skeletal muscle cell model (Gangoiti et al. 2012). Overall, one of the best described and characterized biological functions for C1P is the role of this lipid in promoting cellular proliferation and growth, which was also corroborated by Bornancin and coworkers using cells from the CERK ablation model (Graf et al. 2008).
C1P has also been implicated as an anti-apoptotic lipid; specifically, a later report from the Gomez-Munoz laboratory demonstrated that C1P also prevented cell death in bone marrow-derived macrophages (BMDMs) after withdrawal of macrophage colony-stimulating factor (M-CSF) (Gomez-Munoz et al. 2004). Treatment of BMDMs with C1P effectively blocked the activation of caspases and prevented DNA fragmentation upon serum removal. In the same study, this laboratory also demonstrated that C1P treatment inhibited ceramide generation from A-SMase. Furthermore, A-SMase was shown to be a direct target of C1P, consequently inducing inhibition of this enzyme (Gomez-Munoz et al. 2004). A follow-up study by Gomez-Munoz et al. (2005) demonstrated that C1P enhanced DNA binding to transcription factor NF-κB via stimulation of phosphatidylinositol 3-kinase (PI3-K) activity and protein kinase B (PKB)/(AKT) phosphorylation. Additionally, C1P treatment resulted in the upregulation of the anti-apoptotic regulator Bcl-XL (Gomez-Munoz et al. 2005). Hence, C1P can activate a number of pro-survival pathways and antagonize the pro-apoptotic effects of ceramide.
Along these same lines, the Gomez-Munoz group has also presented evidence that supports a role for C1P in macrophage migration. Macrophage recruitment is a key event in mediating the inflammatory response as these cells are necessary for the release of cytokines, prostaglandins, and a variety of additional enzymes involved in the innate immune system. This recruitment process is highly dependent on the rate of macrophage proliferation, as well as the rate of migration and efflux (Pollard 2004). In this regard, the Gomez-Munoz laboratory demonstrated that addition of natural C1P stimulated the migration of macrophages (Granado et al. 2009). Interestingly, this finding of Granado and colleagues strongly suggested that C1P-induced migration was independent of intracellular C1P synthesis via CERK activation implicating the existence of cell-surface receptor for C1P. Furthermore, this study demonstrated that migration effects of exogenous C1P did not act through the currently known S1P receptors (Granado et al. 2009), a closely related sphingolipid known to induce cell survival and migration. Hence, the findings of the Gomez-Munoz laboratory suggest that C1P-stimulated macrophage migration is coupled to an, as of yet, unidentified Gi protein receptor. Indeed, a study by Zor and coworkers corroborated the possible existence of receptors for C1P and C1P analogs. Specifically, this laboratory demonstrated that incubation of RAW 264.7 macrophages with the phospho-ceramide analogue-1 (PCERA-1) reduced TNFα production at the mRNA and protein level in response to LPS-stimulation (Goldsmith et al. 2009). Structure–function studies of PCERA-1 show that the phosphate group and the lipid moiety are mutually required for its activity (Matsui et al. 2002a, b). Likewise, this study by the Zor group showed that the anti-inflammatory effect of PCERA-1 was dependent on the presence of PCERA-1 in the cell media during LPS treatment of RAW264.7 macrophages, which suggests that this C1P analogue acts extracellularly by binding a protein target present at the cell membrane (Goldsmith et al. 2009). Although additional biological and biophysical studies are required to validate the existence of a C1P receptor, the biological implications of a new class of lipid receptors are exciting and should be vehemently explored. Regardless, the culmination of these studies further emphasizes the role of C1P in the regulation of cellular homeostasis in macrophages.
3 C1P in Immunity and Inflammation: “The Missing Link”
Eicosanoids are one of the most important classes of lipids, which include prostaglandins, prostacyclins, leukotrienes, and thromboxanes. These eicosanoids give rise to the classical features of inflammation, which are necessary to defend the organism against infection and injury. In spite of these protective efforts, these processes can produce an overwhelming response, which leads to an excess of these molecules. As a result, unnecessary levels of eicosanoids can promote a wide range of disease states including chronic inflammation, allergy, cardiovascular disease, and cancer (Yedgar et al. 2007). The mechanism of prostaglandin synthesis begins with the rate-limiting step, the formation of arachidonic acid (AA), via the activity of phospholipase A2 (Murakami et al. 1996). In many cases, inflammatory cytokines (e.g., TNFα) induce the activation and translocation of Group IVA cytosolic phospholipase A2 (cPLA2α), which requires the association of cPLA2α with membranes in a Ca2+-dependent manner via a C2/CALB domain (Fig. 1).
Fig. 1.
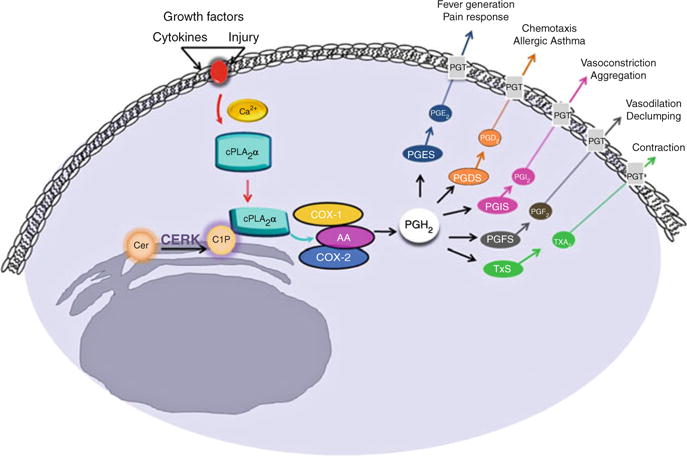
The interaction between C1P and cPLA2α is a crucial link in eicosanoid synthesis. Following an inflammatory stimulus, Ca2+ activated cPLA2α translocates to the golgi membrane where it binds phosphatidylcholine (PC). CERK-derived C1P directly interacts with cPLA2α, thereby enhancing the association of cPLAα to the PC-rich membrane. cPLA2α hydrolyzes PC to produce arachidonic acid (AA), which is further metabolized to several different eicosanoids, one of which is prostaglandin (PGH2). Prostaglandins are involved in various biological processes associated with the inflammatory response
The hypothesis that C1P regulated the activation of a phospholipase A2 (PLA2) and eicosanoid synthesis came from an unlikely source. A link existed between inflammation/eicosanoid synthesis and the venom from Loxosceles recluse (brown recluse spider). Specifically, the main component of this venom is sphingomyelinase D (SMase D), which hydrolyzes sphingomyelin to produce C1P. The pathology of a wound generated from the bite of this spider is that of an intense inflammatory response mediated by AA and prostaglandins. The production of endogenous C1P by the action of SMase D suggested the possibility of C1P acting as a pathophysiologic link in the activation of cPLA2 and the inflammatory response mediated by AA and prostaglandins. Initial evidence for the validation of this hypothesis came from an investigation focused on the regulation of prostanoid synthesis, which showed that the CERK/C1P pathway was required for PLA2 activation in response to calcium ionophore and cytokines (Pettus et al. 2003). Subsequent in vitro studies by our laboratory confirmed these findings, pointing to C1P as a direct activator of cPLA2 through interaction with the C2/CaLB domain (Pettus et al. 2004). Collectively, these findings provided evidence for C1P as the “missing link” in the eicosanoid synthetic pathway (Fig. 1).
The Chalfant group forged forward with mechanistic studies demonstrating that the interaction of C1P and cPLA2α was very specific, as closely related lipids and metabolites were unable to activate cPLA2α in vitro and in cells (Pettus et al. 2004; Subramanian et al. 2005; Wijesinghe et al. 2009). The interaction site for C1P within cPLA2α was characterized in depth over the course of several years, and these investigations provided evidence that C1P interacted with cPLA2α at the C2 domain via a novel and previously undescribed interaction site (Pettus et al. 2004; Subramanian et al. 2005; Stahelin et al. 2007) (Fig. 1). C1P activates cPLA2α by decreasing the dissociation constant of cPLA2α with membranes, and by acting in a manner similar to a positive allosteric activator (Subramanian et al. 2005, 2007). The C1P/cPLA2α paradigm was subsequently tested in cells, which confirmed that this interaction is required for translocation of cPLA2α and subsequent production of eicosanoids in response to several inflammatory agonists (Lamour et al. 2009) (Fig. 1).
In addition to affecting the biochemical pathways through direct interaction with effector proteins like cPLA2α, C1P has also been surmised to induce indirect effects via changes to the structure of resident membranes. Biophysical studies by Kooijman et al. (2008 Kooijman et al. (2009) have demonstrated that C1P has the potential to alter membrane curvature, membrane fluidity, and membrane electrostatics. Specifically, negative curvature of membranes was observed upon incorporation of C1P into glycerophospholipids, which in turn induced the formation of non-lamellar structures (Kooijman et al. 2008). This property was observed even at membrane concentrations of C1P as low as <1 % with large portions of membranes in non-lamellar formations at 5 mol% (Kooijman et al. 2008). Formation of such non-lamellar structures was found to be important in events such as membrane protein insertion (Alonso et al. 2000; Martin et al. 2004) and membrane fusion (Goni and Alonso 2000; Chernomordik et al. 2006), and also has the potential to influence lipid signaling (van Blitterswijk et al. 2003; Kolesnick et al. 2000). Further studies by Koojiman et al. (2009) demonstrated that C1P affected membrane structure in a pH-dependent fashion. Specifically, C1P has a pKa2 of 7.39, and at a slightly acidic pH of around 6 (which is found in many subcellular locations), C1P was observed to form a highly ordered crystalline structure via extensive intermolecular hydrogen bonding (Koojiman et al. 2009). However, at physiologic pH approaching its pKa2, a high proportion of the phosphomonoester moieties of C1P are di-anionic resulting in significant repulsion among C1P molecules leading to a more diffused arrangement (Koojiman et al. 2009). In addition to pH, Ca2+ was also shown to affect the membrane organization of C1P by masking the negative charge, dehydrating the phosphomonoester head group, or linking different C1P molecules together (Koojiman et al. 2009). Additional biophysical studies have demonstrated that C1P formation has the potential to inhibit or reverse the formation of gel-like ceramide domains (Morrow et al. 2009). This finding has implications for a role of C1P in the destabilization of ceramide-rich lipid rafts. Thus C1P, although a relatively simple sphingolipid, has the potential to influence multiple aspects of biological membranes in a pH− and Ca2+-dependent manner, which in turn has the potential to affect a significant array of cellular functions. Indeed, these membrane effects and/or changes in C1P structure may explain the stoichiometry of cPLA2α activation by C1P of >4 molecules per micelle. The possibility of cPLA2α or C1P interacting proteins associating with a specific C1P structure induced by localized pH or Ca2+ changes is an intriguing hypothesis to explore.
The connection between CERK/C1P and cPLA2α activation is also of great interest with regard to disease states involving unnecessary eicosanoid production. In opposition to this link, recent studies published by the Bornancin group using an in vivo disease model for rheumatoid arthritis showed that CERK−/− mice were unaffected and responded similar to wild-type (WT) mice. These studies also suggested that cPLA2α-dependent pathways are unchanged in CERK-deficient mice (Graf et al. 2008). However, this study also found that while the CERK-deficient mice lacked CERK activity, C18:1/16:0C1P was still present at significant levels with minor effects on total C1P. In an independent study utilizing a separate CERK−/− mouse, Igarashi and coworkers confirmed a minor effect on total C1P levels (Mitsutake et al. 2007). These data implicate an alternate mechanism for C1P synthesis and the possibility that the CERK-deficient mice have developed a compensatory system adapting biologically to the lack of CERK.
In contrast to the idea that cPLA2α-dependent pathways are completely functional in the CERK−/− mouse, Niwa et al. (2010) demonstrated that PGE2 levels were reduced in the bronchoalveolar fluid of CERK-deficient mice compared to WT mice, thus providing initial in vivo evidence supporting a role for CERK and its product, C1P, in the regulation of eicosanoid synthesis in vivo. Furthermore, recent studies from our laboratory show that ablation of CERK results in a significant dysregulation/dysfunction in basal eicosanoid synthesis in ex vivo cells (Mietla et al. unpublished observation). In light of these latest findings, the CERK-deficient mouse model may only be partially adapted when cells are removed from the animal model. Why the CERK−/− mouse does not show the same phenotype observed in the cPLA2α−/− mouse is a conundrum, but the relatively unaffected levels of C1P are a likely rationale. Indeed, CERK siRNA-mediated knockout in cultured cell lines results in a significant and major reduction in total C1P levels in contrast to cell from the CERK−/− mouse (Wijesinghe et al. 2010). The phenotypic adaptation of CERK−/− mice in vivo may also be due to the higher levels of C1P found in the serum of these animals, which corresponds to the normal C1P levels found in the liver cells from the CERK−/− mouse. Regardless, the Chalfant laboratory has now created a cPLA2α knock-in mouse, to overcome the controversy surrounding the CERK−/− model and the role of CERK-derived C1P in eicosanoid synthesis/inflammation disorders. This new mouse model expresses a cPLA2α dysfunctional for the C1P interaction and will hopefully determine whether the C1P/cPLA2α interaction plays a role in inflammatory phenotypes in vivo. By examining the C1P/cPLA2α interaction directly in an in vivo model the compensatory mechanisms for C1P production activated in the CERK−/− mouse will be circumvented.
While the C1P/cPLA2α interaction has been a recent major focus in the C1P research field, it is not the only function for C1P in the inflammatory response. Recent work from the Chalfant laboratory has implicated an additional role for C1P as a potential regulator of cytokine secretion, specifically TNFα, as described below (Lamour et al. 2011). TNFα is a major mediator of systemic and acute inflammation, and TNFα secretion is a pro-inflammatory event occurring in response to invading microbes. Unfortunately, dysregulation of this process results in hyper-activation of the immune response accompanied by an unnecessary amount of TNFα production and lethal tissue damage most commonly described as septic shock or sepsis (Lin and Yeh 2005). In addition to sepsis, excessive TNFα production has been linked to other diseases such as rheumatoid arthritis and cancer (Feldmann and Maini 2008; Sethi et al. 2008).
The TNFα protein is synthesized as a membrane-bound pro-peptide (Pro-TNFα) (Kriegler et al. 1988). Proteolytic cleavage of TNFα releases the active C-terminal portion from the cell surface, thus producing the secreted/soluble form of TNFα, which mediates the recruitment of subsequent activation of inflammatory cells to infected tissues or to the site of injury (Old 1988). Several enzymes have been implicated in the processing of TNF, a posttranslational protease-mediated mechanism that has been described as “ectodomain shedding” (Blobel 2000). Specifically, a member of A disintegrin and metalloprotease family (ADAM), ADAM17, has extensively been shown to act as the “sheddase” for TNFα, hence the more common name, TNFα-converting enzyme (TACE) (Moss et al. 1997; reviewed in Black 2002). The sheddase activity of TACE plays a critical role in the regulation of TNFα activation via the direct cleavage of pro-TNFα, thus releasing TNFα (Fig. 2). Indeed, TACE was demonstrated to be the major TNFα sheddase in response to endotoxin stimulation (Horiuchi et al. 2007). Moreover, Blobel and coworkers demonstrated that mice bearing a temporal and conditional inactivation of TACE resulted in significantly decreased serum TNFα levels and were protected from LPS-stimulated endotoxin shock (Horiuchi et al. 2007). Due to the involvement of excessive TNFα release in sepsis and several inflammatory-associated diseases, there is a significant amount of interest in developing therapeutic strategies that can alter TNFα shedding. Thus, TACE sheddase activity has become an attractive target for novel anti-TNFα therapies. Along these lines, the mechanisms regarding the induction or termination of TACE activity following a cellular insult, such as LPS-stimulation, are presently unclear.
Fig. 2.
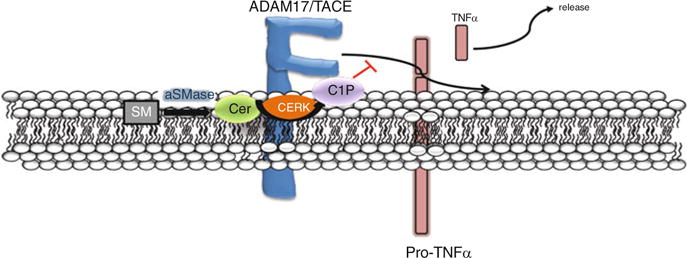
Prospective role of C1P in TNFα production via direct inhibition of TACE. ADAM17/ TACE is the major metalloprotease responsible for cleaving or “shedding” mature TNFα (pro-TNFα) to release the active soluble form. Recent studies have demonstrated a direct interaction between C1P and TACE, which inhibits the sheddase activity and hinders the ability of TACE to release active TNFα. Current investigations are focused on identifying the TACE residues that mediate the interaction of the enzyme with C1P
In regard to C1P playing a role in TNFα secretion, two recent studies have reported findings that suggest a regulatory role for the sphingolipids, ceramide and C1P, in TNFα secretion following LPS stimulation. Initially, studies by Rozenova et al. (2010) demonstrated that A-SMase may act as a regulator of posttranslational processing of TNFα via inhibition of TACE in LPS-stimulated macrophages, providing mechanistic insight for the previous finding that A-SMase-deficient mice were partially protected from the tissue-damaging effects of LPS (Haimovitz-Friedman et al. 1997). The involvement of the sphingolipid pathway in the generation of soluble TNFα was further corroborated in immortalized mouse embryonic fibroblasts (MEFs) and BMDMs (Lamour et al. 2011). Specifically, these recent studies implicated the CERK/C1P pathway in the processing of pro-TNFα to soluble/active TNFα by direct and specific inhibition of TACE (Fig. 2). For example, the Chalfant laboratory demonstrated that genetic ablation of CERK led to a significant increase in TNFα secretion and TACE activity. These findings mirrored those of studies by Nikolova-Karakashian and coworkers in the A-SMase-deficient mice (2010), which also showed increased levels of TACE activity in LPS-treated BMDMs lacking the enzyme. Interestingly, the report by Nikolova-Karakashian and coworkers showed that ceramide did not directly affect the activity of TACE. These findings led to the hypothesis that C1P may directly regulate TACE activity. Following the C1P/cPLA2α model, plausible interaction sites in TACE for C1P were identified, demonstrating that C1P potently and directly inhibited TACE enzyme activity (Lamour et al. 2011). This effect of C1P was specific, as closely related lipids such as ceramide and S1P could not recapitulate this inhibitory effect. While these studies propose a different sphingolipid as an essential component of TNFα production, they clearly depict the same theory, which is the existence of a sphingolipid that regulates the posttranslational processing and secretion of TNFα via the interruption of TACE enzymatic activity. Furthermore, C1P is a direct metabolite of ceramide suggesting that CERK utilizes ceramide derived from A-SMase. Future characterization of the C1P/TACE interaction may explain the anti-inflammatory effects of some C1P analogs. For example, the C1P analogs, pCERA1 and ONO-SM-362, inhibit the production of TNFα in animal models (Avni et al. 2009; Ogata et al. 2008; Goldsmith et al. 2009). Validation of the C1P interaction site within TACE would be a substantial achievement and would allow for testing of the hypothesis that C1P analogs are blocking TNFα production in cells and in vivo via direct inhibition of TACE enzymatic activity. This interaction could perhaps be a future therapeutic target for treating sepsis in addition to chronic inflammatory diseases.
Our current understanding clearly emphasizes the role of the CERK/C1P pathway in the regulation of membrane-bound proteins with a considerable amount of involvement in fundamental inflammatory processes. The establishment and characterization of the C1P/cPLA2α interaction have provided investigators with a representative mechanism that can be used to study additional potential C1P interacting proteins. Furthermore, in vivo relevance of this interaction in inflammatory disease phenotypes is still unclear, but the current subject is under intense investigation by many laboratory groups.
References
- Alonso A, Goni FM, Buckley JT. Lipids favoring inverted phase enhance the ability of aerolysin to permeabilize liposome bilayers. Biochemistry. 2000;39(46):14019–14024. doi: 10.1021/bi001739o. [DOI] [PubMed] [Google Scholar]
- Avni D, Goldsmith M, Ernst O, Mashiach R, Tuntland T, Meijler MM, Gray NS, Rosen H, Zor T. Modulation of TNFalpha, IL-10 and IL-12p40 levels by a ceramide-1-phosphate analog, PCERA-1, in vivo and ex vivo in primary macrophages. Immunol Lett. 2009;123(1):1–8. doi: 10.1016/j.imlet.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Bajjalieh SM, Martin TF, Floor E. Synaptic vesicle ceramide kinase. A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. J Biol Chem. 1989;264(24):14354–14360. [PubMed] [Google Scholar]
- Black RA. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. 2002;34(1):1–5. doi: 10.1016/s1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Blobel CP. Remarkable roles of proteolysis on and beyond the cell surface. Curr Opin Cell Biol. 2000;12(5):606–612. doi: 10.1016/s0955-0674(00)00139-3. [DOI] [PubMed] [Google Scholar]
- Carre A, Graf C, Stora S, Mechtcheriakova D, Csonga R, Urtz N, Billich A, Baumruker T, Bornancin F. Ceramide kinase targeting and activity determined by its N-terminal pleckstrin homology domain. Biochem Biophys Res Commun. 2004;324(4):1215–1219. doi: 10.1016/j.bbrc.2004.09.181. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Zimmerberg J, Kozlov MM. Membranes of the world unite! J Cell Biol. 2006;175(2):201–207. doi: 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WQ, Graf C, Zimmel D, Rovina R, Krapenbauer K, Jaritz M, Parker PJ, Lubec G, Bornancin F. Ceramide kinase profiling by mass spectrometry reveals a conserved phosphorylation pattern downstream of the catalytic site. J Proteome Res. 2010;9(1):420–9. doi: 10.1021/pr900763z. [DOI] [PubMed] [Google Scholar]
- Dressler KA, Kolesnick RN. Ceramide 1-phosphate, a novel phospholipid in human leukemia (HL-60) cells. Synthesis via ceramide from sphingomyelin. J Biol Chem. 1990;265(25):14917–14921. [PubMed] [Google Scholar]
- Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19. doi: 10.1111/j.1600-065X.2008.00626.x. [DOI] [PubMed] [Google Scholar]
- Gangoiti P, Granado MH, Arana L, Ouro A, Gomez-Munoz A. Involvement of nitric oxide in the promotion of cell survival by ceramide 1-phosphate. FEBS Lett. 2008a;582(15):2263–2269. doi: 10.1016/j.febslet.2008.05.027. [DOI] [PubMed] [Google Scholar]
- Gangoiti P, Granado MH, Wang SW, Kong JY, Steinbrecher UP, Gomez-Munoz A. Ceramide 1-phosphate stimulates macrophage proliferation through activation of the PI3-kinase/PKB, JNK and ERK1/2 pathways. Cell Signal. 2008b;20(4):726–736. doi: 10.1016/j.cellsig.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Gangoiti P, Granado MH, Arana L, Ouro A, Gomez-Munoz A. Activation of protein kinase C-alpha is essential for stimulation of cell proliferation by ceramide 1-phosphate. FEBS Lett. 2010;584(3):517–524. doi: 10.1016/j.febslet.2009.11.086. [DOI] [PubMed] [Google Scholar]
- Gangoiti P, Bernacchioni C, Donati C, Cencetti F, Ouro A, Gomez-Munoz A, Bruni P. Ceramide 1-phosphate stimulates proliferation of C2C12 myoblasts. Biochimie. 2012;94(3):597–607. doi: 10.1016/j.biochi.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith M, Avni D, Levy-Rimler G, Mashiach R, Ernst O, Levi M, Webb B, Meijler MM, Gray NS, Rosen H, Zor T. A ceramide-1-phosphate analogue, PCERA-1, simultaneously suppresses tumour necrosis factor-alpha and induces interleukin-10 production in activated macrophages. Immunology. 2009;127(1):103–115. doi: 10.1111/j.1365-2567.2008.02928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Munoz A. Modulation of cell signalling by ceramides. Biochim Biophys Acta. 1998;1391(1):92–109. doi: 10.1016/s0005-2760(97)00201-4. [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A, Duffy PA, Martin A, O’Brien L, Byun HS, Bittman R, Brindley DN. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: antagonism by cell-permeable ceramides. Mol Pharmacol. 1995;47(5):833–839. [PubMed] [Google Scholar]
- Gomez-Munoz A, Frago LM, Alvarez L, Varela-Nieto I. Stimulation of DNA synthesis by natural ceramide 1-phosphate. Biochem J. 1997;325(Pt 2):435–440. doi: 10.1042/bj3250435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Munoz A, Kong JY, Salh B, Steinbrecher UP. Ceramide-1-phosphate blocks apoptosis through inhibition of acid sphingomyelinase in macrophages. J Lipid Res. 2004;45(1):99–105. doi: 10.1194/jlr.M300158-JLR200. [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A, Kong JY, Parhar K, Wang SW, Gangoiti P, Gonzalez M, Eivemark S, Salh B, Duronio V, Steinbrecher UP. Ceramide-1-phosphate promotes cell survival through activation of the phosphatidylinositol 3-kinase/protein kinase B pathway. FEBS Lett. 2005;579(17):3744–3750. doi: 10.1016/j.febslet.2005.05.067. [DOI] [PubMed] [Google Scholar]
- Goni FM, Alonso A. Membrane fusion induced by phospholipase C and sphingomyelinases. Biosci Rep. 2000;20(6):443–463. doi: 10.1023/a:1010450702670. [DOI] [PubMed] [Google Scholar]
- Graf C, Zemann B, Rovina P, Urtz N, Schanzer A, Reuschel R, Mechtcheriakova D, Muller M, Fischer E, Reichel C, Huber S, Dawson J, Meingassner JG, Billich A, Niwa S, Badegruber R, Van Veldhoven PP, Kinzel B, Baumruker T, Bornancin F. Neutropenia with impaired immune response to streptococcus pneumoniae in ceramide kinase-deficient mice. J Immunol. 2008;180(5):3457–66. doi: 10.4049/jimmunol.180.5.3457. [DOI] [PubMed] [Google Scholar]
- Granado MH, Gangoiti P, Ouro A, Arana L, Gonzalez M, Trueba M, Gomez-Munoz A. Ceramide 1-phosphate (C1P) promotes cell migration Involvement of a specific C1P receptor. Cell Signal. 2009;21(3):405–412. doi: 10.1016/j.cellsig.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, Garzotto M, McLoughlin M, Gallily R, Edwards CK, 3rd, Schuchman EH, Fuks Z, Kolesnick R. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186(11):1831–1841. doi: 10.1084/jem.186.11.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179(5):2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- Kim TJ, Mitsutake S, Kato M, Igarashi Y. The leucine 10 residue in the pleckstrin homology domain of ceramide kinase is crucial for its catalytic activity. FEBS Lett. 2005;579(20):4383–4388. doi: 10.1016/j.febslet.2005.06.079. [DOI] [PubMed] [Google Scholar]
- Kim TJ, Mitsutake S, Igarashi Y. The interaction between the pleckstrin homology domain of ceramide kinase and phosphatidylinositol 4,5-bisphosphate regulates the plasma membrane targeting and ceramide 1-phosphate levels. Biochem Biophys Res Commun. 2006;342(2):611–617. doi: 10.1016/j.bbrc.2006.01.170. [DOI] [PubMed] [Google Scholar]
- Kolesnick RN, Hemer MR. Characterization of a ceramide kinase activity from human leukemia (HL-60) cells. Separation from diacylglycerol kinase activity. J Biol Chem. 1990;265(31):18803–18808. [PubMed] [Google Scholar]
- Kolesnick RN, Goni FM, Alonso A. Compartmentalization of ceramide signaling: physical foundations and biological effects. J Cell Physiol. 2000;184(3):285–300. doi: 10.1002/1097-4652(200009)184:3<285::AID-JCP2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Kooijman EE, Sot J, Montes LR, Alonso A, Gericke A, de Kruijff B, Kumara S, Koni FM. Membrane organization and ionization behavior of the minor but crucial lipid ceramide-1-phosphate. Biophys J. 2008;94(11):4320–4330. doi: 10.1529/biophysj.107.121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koojiman EE, Vaknin D, Bu W, Joshi L, Kang SW, Gericke A, Mann EK, Kumar S. Structure of ceramide-1-phosphate at the air-water solution interface in the absence and presence of Ca2+ Biophys J. 2009;96(6):2204–2215. doi: 10.1016/j.bpj.2008.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53(1):45–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- Lamour NF, Subramanian P, Wijesinghe DS, Stahelin RV, Bonventre JV, Chalfant CE. Ceramide 1-phosphate is required for the translocation of group IVA cytosolic phospholipase A2 and prostaglandin synthesis. J Biol Chem. 2009;284(39):26897–26907. doi: 10.1074/jbc.M109.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour NF, Wijesinghe DS, Mietla JA, Ward KE, Stahelin RV, Chalfant CE. Ceramide kinase regulates the production of tumor necrosis factor alpha (TNFalpha) via inhibition of TNFalpha-converting enzyme. J Biol Chem. 2011;286(50):42808–42817. doi: 10.1074/jbc.M111.310169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WJ, Yeh WC. Implication of toll-like receptor and tumor necrosis factor alpha signaling in septic shock. Shock. 2005;24(3):206–209. doi: 10.1097/01.shk.0000180074.69143.77. [DOI] [PubMed] [Google Scholar]
- Martin C, Requero MA, Masin J, Konopasek I, Goni FM, Sebo P, Ostolaza H. Membrane restructuring of Bordetella pertusis adenylatecyclase toxin, a member fo the RTX toxin family. J Bacteriol. 2004;186(12):3760–3765. doi: 10.1128/JB.186.12.3760-3765.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Kondo T, Nishita Y, Itadani S, Nakatani S, Omawari N, Sakai M, Nakazawa S, Ogata A, Mori H, Terai K, Kamoshima W, Ohno H, Obata T, Nakai H, Toda M. Highly potent inhibitors of TNF-alpha production. Part I: discovery of new chemical leads and their structure-activity relationships. Bioorg Med Chem. 2002a;10(12):3757–3786. doi: 10.1016/s0968-0896(02)00381-4. [DOI] [PubMed] [Google Scholar]
- Matsui T, Kondo T, Nishita Y, Itadani S, Tsuruta H, Fujita S, Omawari N, Sakai M, Nakazawa S, Ogata A, Mori H, Ohno H, Obata T, Nakai H, Toda M. Highly potent inhibitors of TNF-alpha production. Part 2: identification of drug candidates. Bioorg Med Chem Lett. 2002b;12(6):907–910. doi: 10.1016/s0960-894x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- Mitsutake S, Igarashi Y. Calmodulin is involved in the Ca2 + -dependent activation of ceramide kinase as a calcium sensor. J Biol Chem. 2005;280(49):40436–40441. doi: 10.1074/jbc.M501962200. [DOI] [PubMed] [Google Scholar]
- Mitsutake S, Yokose U, Kato M, Matsuoka I, Yoo JM, Kim TJ, Yoo HS, Fujimoto K, Ando Y, Sugiura M, Kohama T, Igarashi Y. The generation and behavioral analysis of ceramide kinase-null mice, indicating a function in cerebellar purkinje cells. Biochem Biophys Res Comm. 2007;363(3):519–524. doi: 10.1016/j.bbrc.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Morrow MR, Helle A, Perry J, Vattulainen I, Weidmer SK, Holopainen JM. Ceramide-1-phosphate, in contrast to ceramide, is not segregrated into lateral lipid domains in phosphatidylcholine bilayers. Biophys J. 2009;96(6):2216–2226. doi: 10.1016/j.bpj.2008.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Kudo I. Type II secretory phospholipase A2 associated with cell surfaces via C-terminal heparin-binding lysine residues augments stimulus-initiated delayed prostaglandin generation. J Biol Chem. 1996;271(47):30041–30051. doi: 10.1074/jbc.271.47.30041. [DOI] [PubMed] [Google Scholar]
- Niwa S, Urtz N, Baumruker T, Billich A, Bornancin F. Ovalbumin-induced plasma interleukin-4 levels are reduced in ceramide kinase-deficient DO11.10 RAG1−/− mice. Lipids Health Dis. 2010;9:1. doi: 10.1186/1476-511X-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Yamashita K, Horiuchi H, Okuda K, Todo S. A novel tumor necrosis factor-alpha suppressant, ONO-SM362, prevents liver failure and promotes liver regeneration after extensive hepatectomy. Surgery. 2008;143(4):545–555. doi: 10.1016/j.surg.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Old LJ. Tumor necrosis factor. Sci Am. 1988;258(5):59–60. 69–75. doi: 10.1038/scientificamerican0588-59. [DOI] [PubMed] [Google Scholar]
- Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J Biol Chem. 2003;278(40):38206–38213. doi: 10.1074/jbc.M304816200. [DOI] [PubMed] [Google Scholar]
- Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, Evans JH, Freiberg J, Roddy P, Hannun YA, Chalfant CE. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem. 2004;279(12):11320–11326. doi: 10.1074/jbc.M309262200. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Rozenova KA, Deevska GM, Karakashian AA, Nikolova-Karakashian MN. Studies on the role of acid sphingomyelinase and ceramide in the regulation of tumor necrosis factor alpha (TNFalpha)-converting enzyme activity and TNFalpha secretion in macrophages. J Biol Chem. 2010;285(27):21103–21113. doi: 10.1074/jbc.M109.080671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci. 2008;13:5094–5107. doi: 10.2741/3066. [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Subramanian P, Vora M, Cho W, Chalfant CE. Ceramide-1-phosphate binds group IVA cytosolic phospholipase a2 via a novel site in the C2 domain. J Biol Chem. 2007;282(28):20467–20474. doi: 10.1074/jbc.M701396200. [DOI] [PubMed] [Google Scholar]
- Subramanian P, Stahelin RV, Szulc Z, Bielawska A, Cho W, Chalfant CE. Ceramide 1-phosphate acts as a positive allosteric activator of group IVA cytosolic phospholipase A2 alpha and enhances the interaction of the enzyme with phosphatidylcholine. J Biol Chem. 2005;280(18):17601–17607. doi: 10.1074/jbc.M414173200. [DOI] [PubMed] [Google Scholar]
- Subramanian P, Vora M, Gentile LB, Stahelin RV, Chalfant CE. Anionic lipids activate group IVA cytosolic phospholipase A2 via distinct and separate mechanism. J Lipid Res. 2007;48(12):2701–2708. doi: 10.1194/jlr.M700356-JLR200. [DOI] [PubMed] [Google Scholar]
- Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, Kohama T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem. 2002;277(26):23294–23300. doi: 10.1074/jbc.M201535200. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk WJ, van der Luit AH, Veldman RJ, Verheij M, Borst J. Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem J. 2003;369(Pt 2):199–211. doi: 10.1042/BJ20021528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesinghe DS, Subramanian P, Lamour NF, Gentile LB, Granado MH, Bielawska A, Szulc Z, Gomez-Munoz A, Chalfant CE. Chain length specificity for activation of cPLA2alpha by C1P: use of the dodecane delivery system to determine lipid-specific effect. J Lipid Res. 2009;50(10):1986–1995. doi: 10.1194/jlr.M800367-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesinghe DS, Allegood JC, Gentile LB, Fox TE, Kester M, Chalfant CE. Use of high performance liquid chromatography-electrospray ionization-tandem mass spectrometry for the analysis of ceramide-1-phosphate levels. J Lipid Res. 2010;51(3):641–651. doi: 10.1194/jlr.D000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedgar S, Krimsky M, Cohen Y, Flower RJ. Treatment of inflammatory diseases by selective eicosanoid inhibition: a double-edged sword? Trends Pharmacol Sci. 2007;28(9):459–464. doi: 10.1016/j.tips.2007.07.005. [DOI] [PubMed] [Google Scholar]