

Abstract
In the absence of infection, the pathophysiology of endotracheal tube-induced sore throat pain is unclear. Activated neutrophils release elastase, reactive oxygen species, and inflammatory cytokines known to contribute to neuropathic pain. Sterile tissue injury can cause the release of damage-associated molecular patterns such as mitochondrial DNA that promote neutrophil activation. We hypothesized that endotracheal tube-induced sore throat pain is linked to mitochondrial DNA-mediated neutrophil inflammation. A nonrandomized prospective survey for sore throat pain was conducted in 31 patients who required short-term intubation and had no evidence of upper airway infection. Patterns of neutrophil abundance, activation, and mitochondrial DNA levels were analyzed in tracheal lavage fluid following intubation and prior to extubation. Thirteen of 31 patients reported sore throat pain. Sore throat patients had high neutrophilia with elevated adhesion molecule and TLR9 expression and constitutive reactive oxygen species generation. Tracheal lavage fluid from sore throat patients accumulated mitochondrial DNA and stimulated neutrophils to release mediators associated with pain in a TLR9- and DNAse-dependent fashion. Endotracheal tube-induced sore throat is linked to the release of mitochondrial DNA and can drive TLR9-mediated inflammatory responses by neutrophils reported to cause pain. Mitigating the effects of cell-free mitochondrial DNA may prove beneficial for the prevention of endotracheal tube-mediated sore throat pain.
Keywords: Neutrophils, Toll-like receptors, pain, intubation, mitochondrial DNA
Introduction
Endotracheal tube (ETT) placement is commonly associated with postoperative sore throat (POST) pain. The incidence of POST has been reported between 20% and 50% following ETT placement1–3 and is linked with symptoms of upper airway discomfort such as tracheitis, hoarseness, and dysphagia, which may delay patient recovery after surgery.1 Several factors are implicated in POST including ETT materials, cuff pressure, airway instrumentation, and pharmacological agents.1–3 ETT placement is also associated with the development of pneumonia in infectious4,5 and noninfectious settings.5,6 However, in the absence of infection, the underlying mechanisms that contribute to POST remain unclear.
Our previous work has shown that short-term ETT placement results in mechanically mediated tissue injury and induces the influx of neutrophils.7,8 Previous studies have demonstrated the link between neutrophil activation and neuropathic pain.9,10 For example, animal studies have highlighted the importance of neutrophil elastase release in the induction of acute pain.10 Neutrophils also release other proalgesic mediators, including reactive oxygen species (ROS),11 interleukin 1 beta (IL-1β),12 and tumor necrosis factor alpha (TNF-α).13
Neutrophils can be activated by damage-associated molecular patterns released by cells injured during stress such as crush trauma.14 Damage-associated molecular patterns are recognized by pattern recognition receptors (PRRs),15 which trigger inflammatory signaling pathways inducing gene expression. Neutrophils express all major classes of PRRs including Toll-like receptor (TLR), scavenger, and complement receptors.16,17 In particular, TLR9 recognizes hypomethylated CpG DNA motifs within the bacterial genome.18 Consistent with the bacterial ancestry of mitochondria, recent work has demonstrated TLR9-dependent neutrophil activation in response to mitochondrial DNA (mtDNA) released by injured cells.19 Here, we show that tracheal lavage fluid (TLF) neutrophils from patients reporting ETT-mediated POST are activated and contain significantly more mtDNA than TLF from their counterparts who do not report throat pain. In addition, exposure of resting neutrophils to TLF mtDNA triggered inflammatory responses in a TLR9-dependent manner.
Materials and methods
Subjects
After obtaining Washington University School of Medicine in St. Louis’ institutional review board approval #201301037 for human studies and informed written consent, 31 adults aged 18 to 65 years, with American Society of Anesthesiologists health classification I/II requiring endotracheal intubation for same-day laparoscopic gastric, gynecologic, or orthopedic surgeries, were enrolled in this study. Multiple attempts at intubation and any active autoimmune or pulmonary disease, hepatitis, cancer, previous tracheal surgery, surgery or endotracheal intubation within five days prior, smoking history less than six weeks prior, and immunosuppressive medication or azithromycin use excluded patients from participation in this study.
Specimen collection
After the placement of Mallinckrodt™ TaperGuard Evac ETTs (7 mm internal diameter for females and 8 mm for males), two tracheal lavage samples were obtained from each patient: immediately following intubation and prior to extubation. Wall suction was utilized to collect specimens, and neutrophils were immediately isolated using EasySep™ Neutrophil Enrichment Kits (StemCell Technologies, Vancouver, Canada) per manufacturer’s instructions. Two hours postsurgery, the presence or absence of sore throat pain was documented.
Neutrophil TLF phenotyping
Isolated neutrophils were stained with anti-human monoclonal antibodies for CD16 (clone B73.1) and CD66b (clone G10F5) for population identification, and CD11b (clone ICRF44), CD54 (clone HA58), and TLR9 (clone eB72-1665) as adhesion and activity markers, and subsequently characterized by fluorescence-activated cell sorting (FACS; FACScan DxP10, BD Biosciences, San Jose, CA). Respiratory burst was analyzed by priming CD16/CD66b-stained neutrophils with 10 ng/mL phorbol 12-myristate 13-acetate for 10 min at 37℃ followed by the addition of 20 µM dihydrorhodamine 123 for 10 s and characterized by FACS. Thirty thousand events were analyzed with FlowJo® X software (Tree Star, Ashland, OR).
mtDNA quantitation
Primers for human MT-CYB and bacterial 16S rRNA (Integrated DNA Technologies, Coralville, IA) were used to identify extracellular mtDNA concentrations in subject TLF supernatants and exclude bacterial infection of the upper airway, respectively, by quantitative polymerase chain reaction (qPCR). mtDNA purified from human A549 cell mitochondria was used to quantify extracellular mtDNA.
Neutrophil co-culture and TLF enzyme-linked immunosorbent assay
To replicate activation, neutrophils were isolated from 20 mL blood of 51 healthy volunteers and cocultured with TLF from subjects with or without sore throat pain and one of either 3 µg/mL DNAse I, 30 µg/mL DNAse I, or 1 µM inhibitory oligodeoxynucleotide (iODN; TTAGGG A151; Invivogen, San Diego, CA); or with 3 µM proinflammatory 5′-C-phosphate-G-3′ (CpG) ODN and either 3 µg/mL DNAse I, 30 µg/mL DNAse I, or 1 µM iODN as controls; then incubated at 37℃ + 5% CO2 for 6 h and then stained for FACS.
Human neutrophil elastase (HNE) activity was measured by enzymatic hydrolysis of the HNE-specific chromogenic substrate N-methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (MeO-SucAAPVpNA; Sigma Aldrich, St. Louis, MO) into 4-nitroaniline. Supernatants from the experimental cocultures were incubated with 1 mM MeO-SucAAPVpNA in 0.1 M HEPES (pH 7.5) buffer at 37℃ + 5% CO2 for 1 h. Substrate cleavage into 4-nitroaniline by HNE was measured by absorbance at 405 nm via spectrophotometry (NanoDrop 2000, ThermoFisher Scientific, Waltham, MA).
Primers for human IL-1β, IL-8, TNF-α, and TLR9 (ThermoFisher Scientific) were used to evaluate their transcription in these cocultures. All experiments were run on a Bio-Rad CFX 96 thermocycler and analyzed with provided Bio-Rad CFX Manager software (Bio-Rad Laboratories Inc, Hercules, CA). Conditions were 95℃ for 5-min initialization, then 50 cycles of 95℃ for 30-s denaturing, 58℃ for 30-s annealing, and 68℃ for 10-s extension and subsequent plate read; followed by melt curve analysis from 65℃ to 95℃.
TLF IL-1β, IL-8, and TNF-α levels was analyzed with enzyme-linked immunosorbent assays (ProcartaPlex Simplex, ThermoFisher Scientific, Waltham, MA) in accordance with manufacturers recommendations.
Cell reporter assay
A human embryonic kidney (HEK) cell line with transfected human TLR9 and NF-κB/AP-1 alkaline phosphatase transgenes (HEK-Blue™ hTLR9; Invivogen) was grown and cocultured with TLF from subjects with or without sore throat pain and one of either 3 µg/mL DNAse I, 30 µg/mL DNAse I, or 1 µM iODN; or with 3 µM proinflammatory 5′-C-phosphate-G-3′ (CpG) ODN and either 3 µg/mL DNAse I, 30 µg/mL DNAse I, or 1 µM iODN as controls; at 37℃ + 5% CO2 for 6 h. TLR9 activation was analyzed by alkaline phosphatase release, measured using QUANTI-Blue™ calorimetric detection medium (Invivogen) via spectrophotometry.
Statistical analysis
All data were analyzed using SPSS version 17.0 (SPSS Inc, Chicago, IL). Continuous variables obtained for neutrophil counts in the two independent groups “Sore Throat” and “No Sore Throat” were compared using the Student’s t test. Continuous variables for median fluorescence intensity for identifying activation phenotypes, ROS production, and inflammatory marker secretion between both independent groups were tested with the Mann–Whitney U test. Categorical variables—including mean age, gender, body mass index, hypertension, hypercholesterolemia, sleep apnea status, and intubation times in both independent groups—were compared using Fisher’s exact test for contingency tables.
Analyses of qPCR data for determining mtDNA and bacterial DNA concentrations were performed using a Kruskal–Wallis test with Dunn’s post hoc test. Inflammatory qPCR transcript and FACS median fluorescence intensity data from both independent groups treated with DNAse I and CpG ODN or iODN were analyzed with multivariate analysis of variance combined with multiple comparisons post hoc analysis of variance corrections.
Results
Patient demographics
Thirty-one patients admitted for same-day surgery at the Barnes Jewish Hospital without evidence of upper respiratory infection meeting the American Society of Anesthesiologists health classification I/II were asked for a “yes” or “no” response to whether they had sore throat pain 2 h after extubation. As shown in Table 1, nearly half of all patients reported throat pain. Risk of sore throat was unrelated to gender, age, body mass index, hypertension, serum cholesterol, obstructive sleep apnea, or intubation time. However, in line with previous studies,2,3 there was a nonsignificant bias in female patients with 61.5% reporting sore throat.
Table 1.
Patient Demographics.
| Variable | Sore throat | Nonsore throat | P |
|---|---|---|---|
| Number of patients, n | 13 | 18 | – |
| Age, years, mean (range) | 40.92 (26–56) | 40.78 (19–61) | 0.361 |
| Female, n (%) | 8 (61.64%) | 7 (38.89%) | 0.292 |
| Male, n (%) | 5 (38.46%) | 11 (61.11%) | 0.313 |
| Body mass index, n ± SEM | 30.46 ± 7.35 | 33.25 ± 12.45 | 0.442 |
| Hypertension, n (%) | 5 (38.46%) | 4 (22.22%) | 0.433 |
| Hypercholesterolemia, n (%) | 3 (23.08%) | 2 (11.11%) | 0.625 |
| Sleep apnea, n (%) | 2 (15.38%) | 1 (5.55%) | 0.558 |
| Intubation time, min (range) | 213.38 (123–354) | 174.78 (55–363) | 0.395 |
Tracheal neutrophils in sore throat patients have an activation phenotype
Our previous observations that ETT placement in animal models causes tracheal mucosal injury and upper airway neutrophilia8 led us to investigate neutrophil abundance and activation in intubated patients. TLFs from sore throat and nonsore throat patients at the time of intubation had low and comparable numbers of neutrophils (Figure 1). In contrast, sore throat patients prior to extubation had significantly more neutrophils in their TLF when compared to nonsore throat patients. Neutrophils in sore throat TLF were also highly activated as they constitutively produced ROS (Figure 2(a)) and had higher adhesion molecule expression involved in granulocyte trafficking into extracellular spaces as evident by higher plasma membrane levels of CD11b, CD54, and CD66b (Figure 2(b)).
Figure 1.
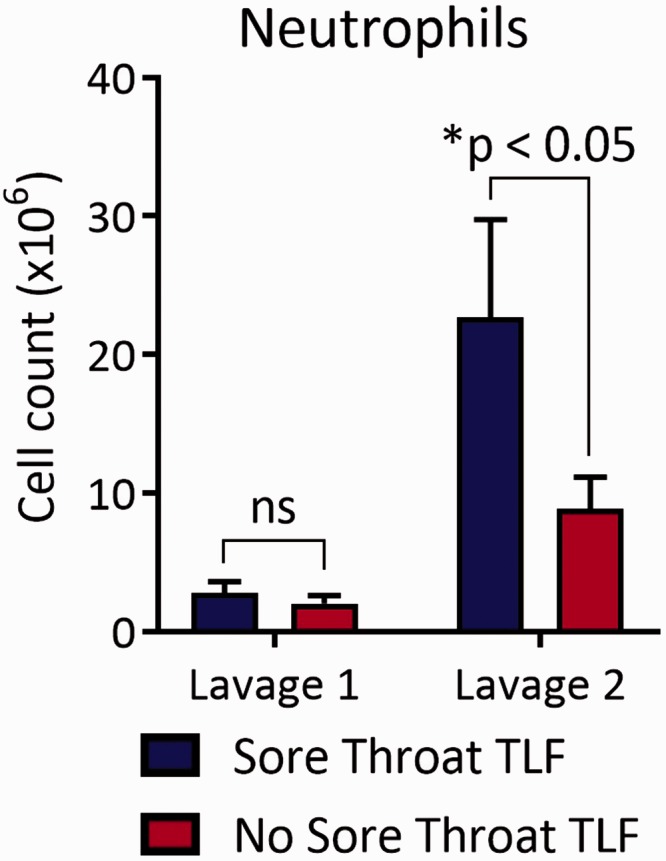
High numbers of TLF neutrophils found in sore throat patients at the time of extubation. Total live cells in TLF were counted by trypan blue exclusion, and the number of neutrophils were calculated by the product of live cells and percent abundance of neutrophils as determined by FACS analysis using a neutrophils Annexin V- SCChi CD66blo-hi CD16lo-hi gate for both tracheal lavages. Results are shown as a mean ± SD derived from sore throat patients (n = 13) and nonsore throat (n = 18) patients. TLF: tracheal lavage fluid.
Figure 2.

Sore throat TLF neutrophils have a distinct activation phenotype. TLF neutrophils were identified by FACS analysis on a SCChi CD66blo-hi CD16lo-hi gate and evaluated for (a) ROS production with DHR 123 dye and plasma membrane expression of (b) CD66b, CD16, CD54, and CD11b using appropriate antibodies. Data (left panel) are representative overlaid histogram result from a sore throat and a nonsore throat patient where solid lines represent indicated antibodies and dotted lines show respective isotype antibody controls. Mean results (right panel) are shown as the MFI ± SD derived from sore throat patients (n = 13) and nonsore throat (n = 18) patients. DHR, dihydrorhodamine; MFI: mean fluorescence intensity; TLF: tracheal lavage fluid.
High mtDNA levels in the TLF of sore throat patients
We next analyzed mtDNA levels in the TLF and circulating blood, as it is a reported mediator of neutrophil activation in lieu of infection.14 Unlike in the circulating blood, TLF mtDNA levels were sharply elevated in sore throat patients when compared to patients who did not report throat pain (Figure 3(a)). Importantly, mtDNA concentrations in the peripheral blood and TLF of nonsore throat patients were also similar suggesting that surgery itself was not a cause of mtDNA release. 16S ribosomal DNA, a commonly used indicator for the presence of bacteria,20 was nearly undetectable in the TLF of both subject groups, indicating the absence of infection in the upper airway (Figure 3(b)). Based on these observations, we asked if TLF neutrophils in sore throat patients express TLR9, a PRR that recognizes hypomethylated CpG DNA motifs found in mitochondria.21 TLR9 expression in TLF neutrophils was significantly elevated in sore throat patients compared to those without sore throat (Figure 3(c)).
Figure 3.

Elevated mitochondrial DNA concentration observed in sore throat patient TLF. (a) Mitochondrial DNA TLF and serum concentrations were determined by real-time quantitative PCR analysis using cytochrome b primers and a cytochrome b standard prepared from human lung mitochondria. (b) Bacterial 16S rRNA DNA levels measured by real-time semiquantiatitive PCR where the quantitation cycle number (Cq) represents DNA levels. Data for (a) and (b) are a representative result of at least three independent experiments. (c) Representative TLF neutrophil TLR9 levels shown as an overlaid histogram from a sore throat and a nonsore throat patient where solid lines represent TLR9 staining and dotted lines represent isotype control staining. Results are shown as indicated mean values ± SD for sore throat patients (n = 13) and nonsore throat (n = 18) patients. TLF: tracheal lavage fluid.
Sore throat TLF stimulates TLR9-dependent neutrophil activation
To address the possibility that mtDNA in TLF drives inflammatory mediator expression, we cocultured sore throat and nonsore throat TLF with a HEK-293 reporter cell line transfected with human TLR9 that measures engagement of CpG DNA through stimulating alkaline phosphatase transgene expression driven by the transcription factors NF-κB and AP-122–24 (Figure 4). Sore throat TLF was nearly five-fold better at stimulating TLR9-mediated reporter activity when compared to nonsore throat TLF. Pretreatment of sore throat TLF with DNAse I or blockade with an iODN that inhibits human TLR925 sharply reduced reporter activity. We further asked if inflammatory mediator accumulation in TLF from sore throat patients differed from nonsore throat patient TLF. Sore throat TLF had a significantly higher concentration of the proinflammatory cytokines IL-1β, TNF-α, and IL-8 when compared to TLF from nonsore throat patients (Figure 5(a)). In line with these observations, sore throat TLF stimulated accumulation of IL-1β, TNF-α, IL-8, and TLR9 mRNA in neutrophils isolated from the peripheral blood of healthy human volunteers (Figure 5(b)–(d)). Several reports have shown that mtDNA triggers neutrophil activation.14,26 To this end, we cocultured sore and nonsore throat TLF with neutrophils isolated from healthy human volunteer peripheral blood. Notably, accumulation of these transcripts could be largely reversed by pretreatment of sore throat TLF with DNAse I or iODN. Moreover, we noted similar patterns of TLR9-mediated neutrophil elastase activity and ROS generation (Figure 6(a) and (b)). Taken together, these data show that extracellular mtDNA in sore throat patients stimulates neutrophil activation in a TLR9-dependent manner.
Figure 4.

Mitochondrial DNA in sore throat TLF stimulates TLR9 signaling. A HEK 293 TLR9 reporter cell line was cultured alone or coincubated with indicated TLFs left untreated or treated with graded amounts of DNAse or TLR9 iODN. Six hours later, supernatant was evaluated for target transgene NF-κB/AP-1 alkaline phosphatase activity by fluorescence spectroscopy. Data are shown as a representative result from three independent experiments where mean alkaline phosphatase activity ± SD is calculated from eight sore throat and eight nonsore throat patients. HEK: human embryonic kidney; ODN: oligodeoxynucleotide; TLF: tracheal lavage fluid.
Figure 5.

Sore throat TLF has higher inflammatory cytokines and triggers neutrophil-mediated inflammatory cytokine expression in a TLR9-dependent manner. TLF analyzed for (a) IL-1β, TNF-α, and chemokine IL-8 levels by enzyme-linked immunosorbent assay. Peripheral blood neutrophils were incubated with indicated TLF, left untreated or treated with grade amounts of DNAse or TLR9 iODN, and 3 h later fractionated for RNA and assessed for (b) IL-1β and TNF-α, (c) IL-8, and (d) TLR9 transcripts by qPCR. Data are shown as a representative result from at least four independent experiments where mean levels ± SD are normalized to uncultured freshly isolated neutrophils and are derived from eight sore throat and eight nonsore throat patients. iODN: inhibitory oligodeoxynucleotide; TLF: tracheal lavage fluid.
Figure 6.

Sore throat TLF drives TLR9-mediated generation of ROS and elastase activity from neutrophils. Peripheral blood neutrophils isolated from healthy human volunteers were incubated with indicated TLF left untreated or treated with graded amounts of DNAse I or TLR9 iODN. One hour later, neutrophils were assessed for (a) human neutrophil elastase (HNE) activity by spectrophotometric assay and (b) ROS by DHR 123 dye staining. Data are shown as a representative result from at least three independent experiments where mean levels ± SD are derived from eight sore throat and eight nonsore throat patients. DHR: dihydrorhodamine; HNE: human neutrophil elastase; iODN: inhibitory oligodeoxynucleotide; TLF: tracheal lavage fluid.
Discussion
Sore throat pain is commonly reported following ETT.1–3 Likewise, nearly half of our 31 patients reported throat pain. Although the underlying mechanisms promoting throat pain following ETT placement are yet to be described, there is evidence that neutrophils may trigger nociception.9–13,27 Previous work has shown that depletion of neutrophils can prevent the induction of hyperalgesia.28 The trachea is highly innervated with a subepithelial network of peripheral nerves that express transient receptor potential vanilloid calcium ion channels (TRPVs),29 which are well-established pain receptors.30 Accordingly, we observed that neutrophilia was significantly greater in patients who reported sore throat when compared to patients without sore throat. Neutrophils of sore throat patients also constitutively produced higher levels of ROS. Several studies have shown that ROS directly promotes hyperalgesia in both acute and inflammatory settings.12 In addition, TLF of sore throat patients induced the release of HNE, a mediator of neuropathic pain.9,10 Recent work has revealed that neutrophil elastase generates pain through the activation of protease-activated TRPV4 receptors on nociceptive neurons.10 Finally, we observed higher levels of IL-1β and TNF-α gene transcription in sore throat TLF-treated neutrophils, and similar secretion of these cytokines, IL-1β and TNF-α, increases the sensitivity of nociceptors by promoting TRPV1 activation.31 Taken together, these data show that TLF from sore throat patients induce neutrophils to release significantly higher amounts of proinflammatory mediators known to trigger peripheral nerve pain.
We also observed that neutrophils from patients with ETT-mediated sore throat have a distinct activation phenotype, which could be distinguished from patients without sore throat by higher mean plasma membrane expression of the adhesion molecules CD66b, CD11b, and CD54. Importantly, high mean neutrophil adhesion molecule expression has been observed in many disease states.32–35 High mean CD66b expression has been reported in the synovium of rheumatoid arthritis patients,33 while elevated CD11b and CD54 levels have been noted in infiltrating neutrophils of infectious or ischemically injured tissue.34,35 In particular, elevated CD11b expression may play a critical role in neutrophil recruitment to the tracheal lumen as it is required for both transendothelial and transepithelial migration.36,37 CD54, otherwise known as I-CAM1, binds to CD11b, suggesting that sore throat neutrophils may additionally promote inflammation by binding to each other or to CD11b expressing myeloid cells such as macrophages.38 Therefore, these data suggest that ETT-mediated sore throat is linked to the generation of a neutrophil phenotype with augmented ability to transmigrate tissue barriers.
Neutrophil activation has primarily been described in the context of infection.39 However, in sore throat patients at the time of extubation, we detected high levels of mtDNA in their TLF suggesting that neutrophil inflammation was generated by sterile injury. As mitochondria encode genes that share considerable homology with their bacterial ancestors, we considered the possibility that bacterial infection could be triggering neutrophil activation in sore throat patients. TLF cultures revealed only normal flora in the throat irrespective of whether they were derived from sore throat patients or not (data not shown). In addition, there were nearly undetectable levels of DNA that encode for the bacterial 16S ribosomal RNA in both sore throat and nonsore throat patient TLF.
The mechanism of mtDNA release was not directly addressed in our study. However, in previous work by our group, we observed in a pig model of ETT-mediated inflammation tracheal histopathology consistent with crush force-mediated epithelial injury.8 Indeed, others have reported high levels of circulating extracellular mtDNA released by blunt force tissue trauma.26 The precise mechanisms by which mtDNA induces neutrophil activation remain unclear but appear to be dependent on several related PRR pathways.40,41 Early studies showed that bacterial-derived CpG DNA induces TLR9-mediated NF-κB activation42 and IL-8 expression.43 Likewise, extracellular mtDNA has been demonstrated to promote IL-8 expression in neutrophils.44 Later reports have linked circulating mtDNA to neutrophil superoxide production45 and the release of neutrophil extracellular traps that may possibly contribute to the additional release of mtDNA.46,47 mtDNA may also augment neutrophilic inflammation in an indirect manner via TLR9-mediated upregulation of adhesion molecules on vascular endothelium, which increase neutrophil adherence and extravasation.48 In line with these previous observations, mtDNA drove the transcription and secretion of the ELR chemokine (CXCL8) IL-8 in neutrophils. IL-8 is likely to promote the trafficking of neutrophils into the trachea. We previously observed the accumulation of IL-8 in TLF following ETT placement.8 Several reports have also shown that the expression of the IL-8 cognate receptor CXCR2 on airway epithelium and endothelium accentuates neutrophil recruitment.49,50 We next determined if TLR9 played a role in triggering inflammatory gene expression in neutrophils from patients with sore throat. To answer this question, we used two independent approaches. First, we tested TLR9 signaling activity in a HEK-293 reporter cell line and demonstrated that sore throat TLF triggered NF-κB and AP-1 driven gene expression. These results were in line with previous observations showing that both transcription factors are activated by CpG DNA stimulation of TLR9 on neutrophils.22–24 DNAse I and iODN treatment of sore throat TLF prevented TLR9 signaling, further confirming these results. Moreover, we observed that this latter method inhibited sore throat TLF-mediated inflammatory gene transcription, ROS, and elastase release.
There are several limitations to this study. Although it is clear that mtDNA in sore throat TLF led to most of the observed neutrophil activation, nuclear DNA may also be contributing to some inflammatory responses as there are hypomethylated CpG motifs on active vertebrate X chromosomes.51 Recent work has also suggested that mtDNA may be generated by neutrophils. Neutrophil extracellular traps, although predominantly made up of nuclear chromatin, may also contain some mtDNA.47
Here, we presented evidence that patients with sore throat release mtDNA into their upper airway, which is sufficient to drive TLR9-dependent neutrophil activation. Our observation that DNAse I inhibited neutrophil activation may advance possible therapeutic interventions aimed at mitigating local inflammation and subsequent pain stemming from sterile tissue injury. Longer term studies of tracheal inflammation mediated by the ETT will be needed to better understand if POST linked to mtDNA also increases the risk of lower airway inflammation or infection.
Acknowledgments
The authors would like to acknowledge Associate Professor William Eades, Director, Siteman Flow Cytometry Core Laboratory, and Jane Blood, RN, Research Coordinator, Department of Anesthesiology and Critical Care.
Authors’ Contributions
CAP participated in experimental design, acquired data, analyzed and interpreted the data, wrote and approved final version of the manuscript and is accountable for all aspects of the study. DP, AE, ER, MK, MHK, ASK, and DK participated in processing specimens, analyzed and interpreted the data, and approved final version of the manuscript. MI participated in processing specimens and approved final version of the manuscript and is accountable for all aspects of the study. AEG participated in experimental design, acquired data, analyzed and interpreted the data, wrote and approved final version of the manuscript and is accountable for all aspects of the study. DP and AE contributed equally to the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CP is supported by Anonymous donors and unrestricted institutional grant from the Department of Anesthesiology Division of Clinical and Translational Research (DoCTR). AEG is supported by NIH grants P01AI116501-01, R01HL113436-01A1, and R01HL121218-01.
References
- 1.McHardy FE, Chung F. Postoperative sore throat: cause, prevention and treatment. Anaesthesia 1999; 54: 444–453. [DOI] [PubMed] [Google Scholar]
- 2.Chandler M. Tracheal intubation and sore throat: a mechanical explanation. Anaesthesia 2002; 57: 155–161. [DOI] [PubMed] [Google Scholar]
- 3.Biro P, Seifert B, Pasch T. Complaints of sore throat after tracheal intubation: a prospective evaluation. Eur J Anaesth 2005; 22: 307–311. [DOI] [PubMed] [Google Scholar]
- 4.Levine SA, Niederman MS. The impact of tracheal intubation on host defenses and risks for nosocomial pneumonia. Clin Chest Med 1991; 12: 523–543. [PubMed] [Google Scholar]
- 5.Divatia JV, Bhowmick K. Complications of endotracheal intubation and other airway management procedures. Indian J Anaesth 2005; 29: 308–318. [Google Scholar]
- 6.Cheung N, Betro G, Luckianow G, et al. Endotracheal intubation: the role of sterility. Surg Infect 2007; 8: 545–552. [DOI] [PubMed] [Google Scholar]
- 7.Puyo CA, Tricomi SM, Dahms TE. Early biochemical markers of inflammation in a swine model of endotracheal intubation. Anesthesiology 2008; 109: 88–94. [DOI] [PubMed] [Google Scholar]
- 8.Puyo CA, Dahms TE. Innate immunity mediating inflammation secondary to endotracheal intubation. Arch Otolaryngol Head Neck Surg 2012; 138: 854–858. [DOI] [PubMed] [Google Scholar]
- 9.Muley MM, Reid AR, Botz B, et al. Neutrophil elastase induces inflammation and pain in mouse knee joints via activation of proteinase-activated receptor-2. Br J Pharmacol 2016; 173: 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao P, Lieu T, Barlow N, et al. Neutrophil elastase activates protease-activated receptor-2 (PAR-2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem 2015; 290: 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salvemini D, Little JW, Doyle T, et al. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med 2011; 51: 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren K, Torres R. Role of interleuking-1β during pain and inflammation. Brain Res Rev 2009; 60: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Empl M, Renaud S, Erne B, et al. TNF-alpha expression in painful and nonpainful neuropathies. Neurology 2001; 56: 1371–1377. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory response to injury. Nature 2010; 464: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas CJ, Schroder K. Pattern recognition receptor function in neutrophils. Trends Immunol 2013; 34: 317–328. [DOI] [PubMed] [Google Scholar]
- 16.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borko A, Cazalet C, Hayes GL, et al. Neutrophil function: from mechanism to disease. Annu Rev Immunol 2012; 30: 459–489. [DOI] [PubMed] [Google Scholar]
- 18.Itagaki K, Adibnia Y, Sun S, et al. Bacterial DNA induces pulmonary damage via TLR-9 through cross-talk with neutrophils. Shock 2011; 36: 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez ME, Fuxman Bass JI, Geffner JR, et al. Neutrophil signaling pathways activated by bacterial DNA stimulation. J Immunol 2006; 177: 4037–4046. [DOI] [PubMed] [Google Scholar]
- 20.Schuurman T, de Boer RF, Kooistra-Smid AMD, et al. Prospective study of use of PCR amplification and sequencing of 16S ribosomal DNA from cerebrospinal fluid for diagnosis of bacterial meningitis in a clinical setting. J Clin Microbiol 2004; 42: 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood 2003; 102: 2660–2669. [DOI] [PubMed] [Google Scholar]
- 22.Miskolci V, Rollins J, Vu HY, et al. NFκB is persistently activated in continuously stimulated human neutrophils. Mol Med 2007; 13: 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang JZ, Liu Z, Liu J, et al. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int J Mol Med 2014; 33: 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Ahmad-Nejad P, Häcker H, Rutz M, et al. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol 2002; 32: 1958–1968. [DOI] [PubMed] [Google Scholar]
- 25.Klinman D, Shirota H, Tross D, et al. Synthetic oligodeoxynucleotides as modulators of inflammation. J Leukoc Biol 2008; 84: 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via P38 MAP kinase. Shock 2010; 34: 55–59. [DOI] [PubMed] [Google Scholar]
- 27.Fiset ME, Gilbert C, Poubelle PE, et al. Human neutrophils as a source of nociception: a novel link between pain and inflammation. Biochemistry 2003; 42: 10498–10505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkins NM, Tracey DJ. Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience 2000; 101: 745–757. [DOI] [PubMed] [Google Scholar]
- 29.Carr MJ, Undem BJ. Ion channels in airway afferent neurons. Resp Physiol 2001; 125: 83–97. [DOI] [PubMed] [Google Scholar]
- 30.Lumpkin EA, Caterina MJ. Mechanisms of sensory transduction in the skin. Nature 2007; 445: 858–865. [DOI] [PubMed] [Google Scholar]
- 31.Binshtok AM, Wang H, Zimmerman K, et al. Nociceptors are interleukin-1β sensors. J Neurosci 2008; 28: 14062–14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt T, Zuöndorf J, Gruöger R, et al. CD66b overexpression and homotypic aggregation of human peripheral blood neutrophils after activation by a gram-positive stimulus. J Leukoc Biol 2012; 91: 791–802. [DOI] [PubMed] [Google Scholar]
- 33.Szekanecz Z, Haines GK, Harlow LA, et al. Increased synovial expression of the adhesion molecules CD66a, CD66b, and CD31 in rheumatoid and osteoarthritis. Clin Immunol Immunopathol 1995; 76: 180–186. [DOI] [PubMed] [Google Scholar]
- 34.Muller Kobold AC, Tulleken JE, Zijlstra JG, et al. Leukocyte activation in sepsis: correlations with disease state and mortality. Intensive Care Med 2000; 26: 883–892. [DOI] [PubMed] [Google Scholar]
- 35.Fortunati E, Kazemir KM, Grutters JC, et al. Human neutrophils switch to an activated phenotype after homing to the lung irrespective of inflammatory disease. Clin Exp Immunol 2008; 155: 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furie MB, Tancinco MC, Smith CW. Monoclonal antibodies to leukocyte integrins CD11a/CD18 and CD11b/CD18 or intercellular adhesion molecule-1 inhibit chemoattractant-stimulated neutrophil transendothelial migration in vitro. Blood 1991; 78: 2089–2097. [PubMed] [Google Scholar]
- 37.Parkos CA, Delp C, Arnaout MA, et al. Neutrophil migration across a cultured intestinal epithelium. J Clin Inv 1991; 88: 1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diamond MS, Staunton DE, Marlin SD, et al. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 1991; 65: 961–971. [DOI] [PubMed] [Google Scholar]
- 39.Pillay J, Ramakers BP, Kamp VM, et al. Functional heterogeneity and differential priming of circulating neutrophil in human experimental endotoxemia. J Leukoc Biol 2010; 88: 211–220. [DOI] [PubMed] [Google Scholar]
- 40.Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012; 485: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012; 36: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeo SJ, Gravis D, Yoon JG, et al. Myeloid differentiation factor 88-dependent transcriptional regulation of cyclooxygenase-2 expression by CpG DNA: role of NF-κB and p38. J Biol Chem 2003; 278: 22563–22573. [DOI] [PubMed] [Google Scholar]
- 43.Bauer S, Kirschning CJ, Häcker H, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA 2001; 98: 9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory response to injury. Nature 2010; 464: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verschoor CP, Loukov D, Naidoo A, et al. Circulating TNF and mitochondrial DNA are major determinants of neutrophil phenotype in the advanced-age, frail elderly. Mol Immunol 2015; 65: 148–156. [DOI] [PubMed] [Google Scholar]
- 46.Itagaki K, Kaczmarek E, Lee YT, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS One 2015; 10: e0120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yousefi S, Mihalache C, Kozlowski E, et al. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Diff 2009; 16: 1438–1444. [DOI] [PubMed] [Google Scholar]
- 48.Kebir DE, József L, Pan W, et al. Bacterial DNA activates endothelial cells and promotes neutrophil adherence through TLR9 signaling. J Immunol 2009; 182: 4386–4394. [DOI] [PubMed] [Google Scholar]
- 49.Hahn CS, Scott DW, Xu X, et al. The matrikine N-α-PGP couples extracellular matrix fragmentation to endothelial permeability. Sci Adv 2015; 1: e1500175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reutershan J, Morris MA, Burcin TL, et al. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Inv 2006; 116: 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alcalay M, Toniolo D. CpG islands of the X chromosome are gene associated. Nucleic Acids Res 1988; 16: 9527–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]