

Abstract
The mitochondrial disease often associated with various illnesses in relation to the activity of cells metabolites and the synthesis of adenosine triphosphate (ATP), including alteration in the mitochondrial DNA. The mutation of m.9053G>A at the ATP6 gene was found in patients with type 2 diabetes mellitus (DM type 2) and cataract. Therefore, this mutation is predicted to be clinical features of the 2 diseases. ATP6 gene encodes protein subunit of ATPase6, a part of ATP synthase, which is important in the electron transfer and proton translocation in intracellular respiration system. This study aims to investigate the mutation effect of m.9053G>A at the ATP6 gene (S167N) to the structure and function of ATPase6 using bioinformatics method. The structure of ATPase6 was constructed using homology modeling method. The crystal structure of bovine’s ATP synthase (Protein Data Bank ID 5FIL) was used as a template because of high sequence similarity (77%) and coverage (96%) of the input sequence. The effect of mutation was investigated at the proton translocation channel of ATPase6. It is predicted that the channel was disrupted due to changes in electrostatic potential from serine to asparagine. Furthermore, molecular docking suggests that water binding on the proton translocation channel in the S167N mutant was different from the wild type. The result of this study is hoped to be useful in the development of a new genetic marker for DM type 2 and cataract.
Keywords: ATP6, diabetes mellitus, cataract, bioinformatics, proton translocation
Introduction
Mitochondria are organelles of eukaryotic cells which play an important role in producing energy in the form of adenosine triphosphate (ATP) by ATP synthase complex.1 The complex of ATP synthase in mammals is composed of 2 parts, which are F1 that has a catalytic site for ATP synthesis and hydrolysis and F0 that has proton channel to translocate proton across the mitochondrial membrane.2 Two subunits of F0, ie, ATPase6 (subunit a) and ATPase8 (A6L), are coded by mitochondrial DNA (mtDNA), whereas the rest of subunits are coded by nuclear DNA.3 Therefore, mutations in mtDNA, especially that of subunits a and A6L, would affect the cell metabolism in producing energy that is crucial for all organs in human body.4 There are 2 mutations of mtDNA which are related to type 2 diabetes mellitus (DM type 2) that complicated with cataract disease, ie., A3243G and C12258A.5 Thus, mitochondrial disorder–related diseases are interesting to be studied. In 2014, World Health Organization reported that Indonesia is the fifth and the second largest country regarding a number of diabetes and cataract cases, respectively.
In 2010, Maksum reported 6 mutations in mtDNA which specifically occurred in patients having type 2 DM and cataract. Among the mutations, m.9053G>A was located at respiration complex protein, ATPase6, and was found to be unrelated to neuromuscular diseases, eg, myopathy and deafness.6 ATPase6 plays a role as proton translocation channel in the mitochondrial matrix through the rotation of F0 ring and hence triggers the change of catalytic site of F1 for ATP synthesis.7 Maassen et al8 suggested that mitochondrial disorder was related to diabetes due to the decreasing of insulin secretion as a result of the low concentration of ATP.
This study aims to investigate the changes of structural properties of ATPase6 due to m.9053G>A mutation (S167N) using bioinformatics methods. The structure of ATPase6 was built using homology modeling technique. Furthermore, the electrostatic properties of protein were calculated using Adaptive Poisson-Boltzmann Solver (APBS) program. The water affinity on the proton translocation channel was predicted using molecular docking by AutoDock Vina.
Methods
Homology modeling of human ATPase6
A structure of bovine mitochondrial ATP synthase, resolved by electron microscopy, was used a template for homology modeling (Protein Data Bank [PDB] ID 5FIL).9 The sequence of the human ATP6 gene was retrieved from MITOMAP (https://www.mitomap.org/MITOMAP). Furthermore, the structure of human ATPase6 and its mutant was modeled using MODELLER.10 At the amino acid level, the mutation of m.9053G>A was the substitution of serine by asparagine at the position 167 (S167N). The Discrete Optimized Protein Energy (DOPE) score, a statistical potential used to assess homology models in protein structure prediction, was calculated for the structure model. The structure with the lowest DOPE score was selected as the best model. The quality of both models was assessed using Ramachandran plot by PROCHECK in PDBsum server (Laskowski et al, 1997).
Atomic electrostatic calculations
The atomic electrostatic charge of both models was calculated using APBS method in PDB2PQR Web server (http://nbcr-222.ucsd.edu/pdb2pqr_2.1.1/).11 The PARSE force field was selected, and the internal naming scheme of PDB2PQR was used. The resulted structure was downloaded, and its atomic charge was visualized using Discovery Studio Visualizer.12
Molecular docking of water on the proton translocation channel
AutoDock Vina13 was employed to predict the water binding at the proton translocation channel in both wild-type and mutant models of human ATPase6. The structure coordinate was first transformed, parallel to the z-axis using Discovery Studio Visualizer. AutoDockTools 1.5.614 was used to determine the position and the size of grid box, which is required for docking. The grid box was centered at x = 1.317, y = −3.018, and z = 8.242 with size of 45 Å × 18.75 Å × 18.75 Å in x-y-z dimension. The grid box was adjusted to cover the surface of the second proton translocation channel from R150 to the mutation point (S/N167). The docking was repeated until 25 times to generate 500 of possible water binding poses on the proton translocation channel. The maximum number of binding modes of each docking was 20.
Results and Discussions
Structural model of ATPase6
Sequence alignment of human ATPase6 showed 77% of similarity to that of bovine’s ATP synthase, and 96% of this sequence was covered without any gaps. For this reason, the structure of bovine’s ATP synthase (PDB ID 5FIL) was used as a template for homology modeling. Five models were generated using MODELLER. A model with the lowest DOPE score was selected for further analysis. The quality of the model was assessed with Ramachandran plot (Figure 1). It is shown that 94.3% of residues were in most favored regions. As many as 4.1% and 0.5% of residues fell into the additional allowed regions and generously allowed regions, respectively. In general, a protein structure with more than 90% residues in allowed region is considered as a good model.15 It is noted that the mutant model shows similar quality with the wild type, in which more than 90% residues in the allowed region.
Figure 1.
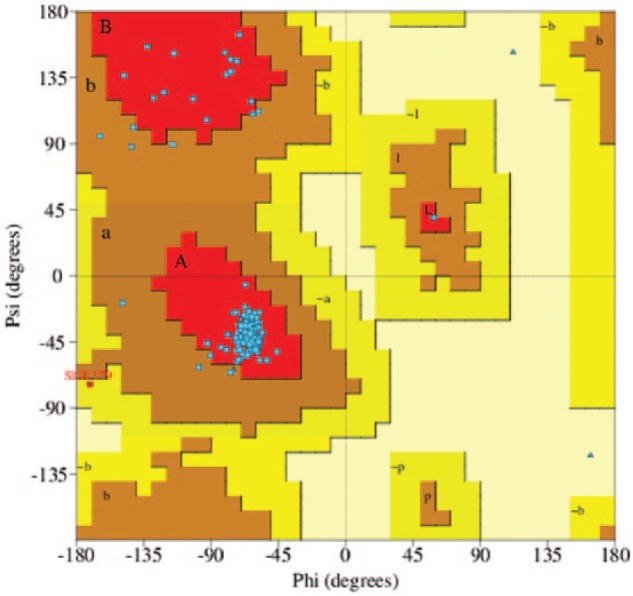
Ramachandran plot of the best ATPase6 model.
The effects of S167N mutation to the structure of ATPase6 and proton translocation
The mutation of S167N is located at the proton translocation channel (Figure 2), more specifically at the second channel from rotor protein (subunit c) to the cytoplasm, starting from residue R150 toward the direction of position 167. For this reason, S167N mutation is predicted to disrupt the translocation of proton to cytoplasm. In general, the overall structure of ATPase6 in wild type and mutant was similar. Further analysis was conducted to the region around the mutation point. Asparagine has a bulkier side chain as compared with serine in wild type. Therefore, a possible sterical hindrance was predicted. However, due to the positively charged nature of proton, there might be another factor that can influence the proton movement, ie, atomic charge throughout the proton translocation channel. Both the wild-type and mutant structures were submitted to PDB2PQR server to calculate the atomic charge using the continuous implicit solvent model by APBS method.
Figure 2.
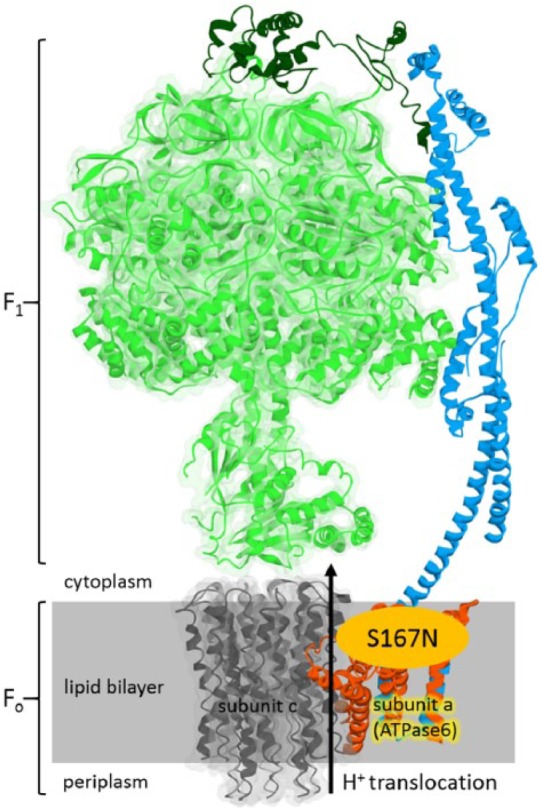
The structure of ATP synthase complex (adapted from Protein Data Bank ID 5FIL). The S167N mutation is located at the subunit a (ATPase6), at the proton translocation channel. This figure was generated using Discovery Studio Visualizer.
Interestingly, a patch of partial negative charge on the surface of proton translocation channel was observed. It was composed of carbonyl oxygen of the backbones of I142, A146, R150, A153, A157, L161, H163, L164, A168 and also oxygen atoms of the side chains of Q143, N154, S167, and E194. Therefore, it is predicted that the H+ is conducted through a proton hopping mechanism, ie, the proton hopped from a lone pair of electrons on oxygen to the next acceptor oxygen atom.16 Because the side chain of S167 was part of the patch of partial negative charge, a possible disruption due to S167N mutation was further studied. In the S167N model, the 2 hydrogen atoms of NH2 group of N167 side chain were exposed on the surface. The charges of these 2 hydrogen atoms were partially positive (+0.2) as compared with the partial negative charge of oxygen in S167 (−0.4) (Figure 3).
Figure 3.

Schematic of proton translocation across ATPase6. The partial positive charge of N167 is exposed on the surface of proton channel, instead of the partial negative charge of S167.
The mechanism of voltage-gated proton channel has been reviewed by DeCoursey.17 There are several mechanisms of proton selectivity through the membrane proteins, eg, hydrogen-bonded chain, histidine as a selective filter, and the frozen water hypothesis. In an original hydrogen-bonded chain model, a proton is passed through the channel by hopping from one serine to another serine group. However, the other amino acids containing an oxygen atom as a proton acceptor, either on the backbone or on the side chain, are also able to transfer the proton at a particular condition. Based on the current findings, it is predicted that the exposure of partial positive charged atoms (N167) on the surface of proton translocation channel would disrupt the proton translocation process. As a result, the flow of proton which is needed for ATP production might be compromised by S167N mutation.
Besides the hydrogen-bonded chain hypothesis, the involvement of water molecules in proton hopping mechanism was also investigated in this study using molecular docking method. AutoDock Vina was used to predicting the possible binding of water molecules on the surface of proton translocation channel of the human ATPase6 model. A total of 500 docked poses was generated using different random seeds. Interestingly, a difference in water binding between R150 and S/N167 in both models was observed (Figure 4). In the wild-type model, the docked water molecules between R150 and S167 were arranged in the shorter intermolecular distance (ranged from 2.4 to 4.7 Å) as compared with S167N mutant (ranged from 2.5 to 6.5 Å). Due to the longer intermolecular distance between waters, it might be indicated that the proton hopping in S167N mutant would be less effective than in the wild type. The docking result suggested that water was more attracted to the asparagine than the serine. Asparagine has a higher number of polar atoms than serine and hence providing more possibility to form hydrogen bonds with the water. Out of 500 docking poses, the numbers of water molecules that are situated within 3.00 Å from S167 (wild type) and N167 (mutant) were 39 and 54, respectively. As a result, the probability of water to bind to the surface in between R150 and N167 in S167N mutant was decreased.
Figure 4.

The docking result of water binding on the proton translocation channel of ATPase6 in wild type (top) and S167N mutant (bottom). The water molecule in wild type and mutant are presented in red and green colored oxygen sphere, respectively. R150 and S/N167 are visualized in space-filling model. The unit of intermolecular distance of water is Å.
The possible effects of S167N mutation to the proton translocation, based on the hydrogen-bonded chain and water binding hypothesis, have been explained in this study. Nevertheless, further experiments such as ATP-based assay are required to strengthen the hypothesis of S167N mutation effect in ATPase6 to the disruption of ATP synthesis, which might lead to DM type 2 and cataract diseases. The relationship between ATP and DM type 2 is associated with its function to stimulate insulin secretion by exocytosis, through the membrane depolarization involving ion channels (K+ and Ca2+) in pancreatic β cells.18 In 2016, it has been reviewed that the antioxidant targeting mitochondria is a powerful therapeutic platform to treat cataract disease.19 The known role of mitochondria in energy metabolism is well established. Mitochondrial DNA mutation which leads to ATP depletion might affect the ATP-dependent chaperone content of the eye lens. The failure in protein renaturation in the eye lens would result in the aggregation of insoluble and light-scattering protein, which caused cataract development.20
The mutation of Mm.9053G>A was found in patients with DM type 2 and cataract, together with Mm.3243A>G mutation.21 The mutant template of Mm.3243A>G has been successfully produced as a positive control in PASA-mismatch three bases of mtDNA mutation analysis.22 Therefore, the result of this study is hoped to be useful to the development of a new specific genetic marker for the 2 diseases.
Conclusions
Previously, an S167N mutation on the ATPase6 was found specifically in patients carrying DM type 2 and cataract. Therefore, this study was conducted to investigate the possible mutation effect of the structure of ATPase6, as part of the ATP synthase complex, using bioinformatics approach. The result showed that S167N mutation point was located at the proton translocation channel, which composed of a continuous patch of negative charges. Instead of a negatively charged OH group serine in wild-type protein, a positively charged of NH2 group of N167 was exposed to the surface of adenosine triphosphatase in mutant type. Therefore, this might lead to the disruption of proton translocation process during ATP synthesis. Furthermore, the possible effect of S167N mutation to the water binding in proton translocation channel was observed using molecular docking study. For the first time, the effect of S167N mutation to the structure of proton translocation channel in ATPase6, from the point of view of surface charges and water binding, was presented in this study.
Footnotes
Peer review:Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 297 words, excluding any confidential comments to the academic editor.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Academic Leadership Grant (ALG) from Universitas Padjadjaran, which has been used to purchase computer and software for the computational experiment.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: IPM and MY conceived and designed the experiments. IPM, SRS, and MY analyzed the data. IPM, SRS, NI, and MY wrote the first draft of the manuscript. IPM, MY, NI, and TS contributed to the writing of the manuscript. IPM, SRS, NI, MY, and TS agree with manuscript results and conclusions. MY, IPM, and TS jointly developed the structure and arguments for the paper. IPM, MY, and TS made critical revisions and approved final version. All authors reviewed and approved the final manuscript. IPM and MY equally contributed to the article.
Disclosure and Ethics: As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
- 1. Galkin A, Drose S, Brandt V. The proton pumping stoichiometry of purified mitochondrial complex I reconstituted into proteoliposomes. Biochim Biophys Acta. 2006;1757:1575–1581. [DOI] [PubMed] [Google Scholar]
- 2. Houštěk JPK, Hermanska JH, Houstkova C, Bogert V, Zeman J. Altered properties of mitochondrial ATP synthase in patients with a T-G mutation in the ATPase 6 (subunit a) gene at position 8993 of mtDNA. Biochim Biophys Acta. 2004;1658:115–121. [DOI] [PubMed] [Google Scholar]
- 3. Houštěk JPK, Pícková A, Vojtíšková T, Mráček P, Pecina P, Jesina P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 2006;1757:1400–1405. [DOI] [PubMed] [Google Scholar]
- 4. Maksum IP, Alchumaira SF, Kamara DS, Rachman SD, Komalaningsih S. The relation of mitochondrial DNA mutation with mitochondrial diseases in coding region. Procedia Chem. 2015;17:84–92. [Google Scholar]
- 5. Nomiyama T, Tanaka Y, Hattori N, et al. Accumulation of somatic mutation in mitochondrial DNA extracted from peripheral blood cells in diabetic patients. Diabetologia. 2002;45:1577–1583. [DOI] [PubMed] [Google Scholar]
- 6. Maksum IP. Varian Genom Mitokondria Pada Pasien Katarak dan Diabetes Melitus Tipe 2 [scientific research dissertation]. Bandung, Indonesia: Universitas Padjadjaran; 2010. [Google Scholar]
- 7. Schon EA, Santra S, Pallotti F, Girvin ME. Pathogenesis of primary defects in mitochondrial ATP synthesis. Semin Cell Dev Biol. 2001;12:441–448. [DOI] [PubMed] [Google Scholar]
- 8. Maassen JA, t’Hart LM, van Essen E, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;52:S103–S109. [DOI] [PubMed] [Google Scholar]
- 9. Zhou A, Rohou A, Schep DG, et al. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. eLife. 2015;10:e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 11. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dassault Systemes. BIOVIA Discovery Studio Visualizer 2016. San Diego, CA: Dassault Systemes; 2016. [Google Scholar]
- 13. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;16:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J App Crystallogr. 1997;26:283–291. [Google Scholar]
- 16. DeCoursey TE, Hosler J. Philosophy of voltage-gated proton channels. J R Soc Interface. 2014;11(92). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeCoursey TE. Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of the HV family. Physiol Rev. 2013;93:599–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fridlyand LE, Jacobson DA, Philipson LH. Ion channels and regulation of insulin secretion in human β-cells: a computational systems analysis. Islets. 2013;5:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Babizhayev MA. Generation of reactive oxygen species in the anterior eye segment. Synergistic codrugs of N-acetylcarnosine lubricant eye drops and mitochondria-targeted antioxidant act as a powerful therapeutic platform for the treatment of cataracts and primary open-angle glaucoma. BBA Clin. 2016;6:49–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moreau KL, King JA. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med. 2012;18:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maksum IP, Natradisastra G, Nuswantara S, Ngili Y. The effect of A3243G mutation of mitochondrial DNA to the clinical features of type-2 diabetes mellitus and cataract. Eur J Sci Res. 2013;96:591–599. [Google Scholar]
- 22. Maksum IP, Farhani A, Rachman SD, Ngili Y. Making of the A3243g mutant template through site directed mutagenesis as positive control in PASA-mismatch three bases. Int J PharmTech Res. 2013;5:441–450. [Google Scholar]