

ABSTRACT
Facilitating the development of alternative targeted therapeutic strategies is urgently required to improve outcome or circumvent chemotherapy resistance in children, adolescents, and adults with recurrent/refractory de novo mature B-cell (CD20) non-Hodgkin lymphoma, including Burkitt lymphoma (BL). Romidepsin, a histone deacetylase inhibitor (HDACi), has been used to treat cutaneous T-cell lymphoma. We have demonstrated the significant anti-tumor effect of anti-CD20 chimeric antigen receptor (CAR) modified expanded peripheral blood natural killer (exPBNK) against rituximab-sensitive and -resistant BL. This study examined the anti-tumor activity of romidepsin alone and in combination with anti-CD20 CAR exPBNKs against rituximab-sensitive and -resistant BL in vitro and in vivo. We found that romidepsin significantly inhibited both rituximab-sensitive and -resistant BL cell proliferation in vitro (P < 0.001) and induced cell death in rituximab-sensitive Raji (P < 0.001) and cell cycle arrest in rituximab-resistant Raji-2R and Raji-4RH (P < 0.001). Consistent with in vitro observations, we also found romidepsin significantly inhibited the growth of rituximab-sensitive and -resistant BL in BL xenografted NSG mice. We also demonstrated that romidpesin significantly induced the expression of Natural Killer Group 2, Member D (NKG2D) ligands MICA/B in both rituximab-sensitive and -resistant BL cells (P < 0.001) resulting in enhancement of exPBNK in vitro cytotoxicity through NKG2D. Finally, we observed the combination of romidepsin and anti-CD20 CAR exPBNK significantly induced cell death in BL cells in vitro, reduced tumor burden and enhanced survival in humanized BL xenografted NSG mice (p < 0.05). Our data suggests that romidepsin is an active HDAC inhibitor that also potentiates expanded NK and anti-CD20 CAR exPBNK activity against rituximab-sensitive and -resistant BL.
KEYWORDS: anti-CD20 chimeric antigen receptor, expanded Natural Killer Cells, rituximab sensitive and resistant Burkitt Lymphoma, romidepsin, targeted immunotherapy
Introduction
The outcome for children, adolescents and adults with de novo mature B-cell (CD20+) non-Hodgkin lymphoma (B-NHL), including Burkitt lymphoma (BL) has improved significantly over the last 2 decades.1-7 Unfortunately, for patients within this group who relapse or progress, the prognosis is dismal due to chemo-radiotherapy resistance.4, 5, 8 Therefore, facilitating the development of alternative targeted therapeutic strategies is required to improve outcome and/or circumvent chemotherapy resistance in these patients. Czuczman et al developed several rituximab-resistant BL cell-lines and found that the rituximab-resistance was also associated with resistance to multiple chemotherapy agents commonly used to treat BL.9, 10 These resistant cell lines provide an excellent model to design alternative novel targeted therapeutic strategies.
Natural killer (NK) cells activities are balanced by signals delivered from both inhibitory and activating receptors.11, 12 NK KIR ligands mismatch following CD34 enriched haploidentical allogeneic stem cell transplantation in patients with poor risk acute myeloid leukemia (AML) has been associated with a significant reduction of the AML relapse rate and a significant increase in overall survival.13 The NKG2D (a NK activating receptor) binding ligands, major histocompatibility complex class I-related chain A and B (MICA/B) and ULBP 1,2,3, enhance NK cell killing against malignant cells.14 However, factors limiting NK therapeutic approaches include small numbers of active NK cells in unexpanded peripheral blood and a lack of specific tumor targeting. Our group and other groups have successfully expanded active NK cells in vitro by short-term culture with cytokines alone, co-culture with irradiated EBV-transformed lymphoblastoid cell lines as feeder cells, or co-culture with K562 cells expressing membrane bound IL-15.15-18
CD20 is a glycosylated phosphoprotein expressed on the surface of B cells in all stages of B cell development except on pro-B cells or plasma cells.19, 20 CD20 is also expressed in more than 99% in BL and over 40% in pre-B-ALL.21, 22 To increase the targeting specificity of expanded NK cells, we previously had investigated the functional activity of K562-mbIL15 -41BBL expanded peripheral blood NK cells (exPBNK) modified to express anti-CD20 CAR following mRNA nucleofection against CD20+ B-NHL in vitro and in xenografted NSG mice.23 We demonstrated that the CAR+ exPBNK significantly induced cell death in CD20+ rituximab-sensitive and -resistant BL in vitro and extended survival time and significantly inhibited tumor cells migration to other organs in NSG mice.23 However, interestingly most of the NSG mice succumbed to their disease and therefore new adjuvant approaches are necessary to enhance this targeted cellular therapeutic approach.
Romidepsin is a structurally unique, potent, bicyclic class I selective HDAC inhibitor.24 It induces apoptosis in vitro by downregulation of the BCL-2 family of proteins, and induces G1 cell cycle arrest by enhancing p21 and p53 in several solid tumor models.25, 26 It was reported that romidepsin was rapidly cleared from the circulation with a short half-life of about 3.5 hours in patients and 5.8 hours in Severe Combine Immune Deficiency (SCID) mice.27-29 The FDA has approved romidepsin for cutaneous lymphoma in patients who have received at least one prior systemic therapy and for peripheral T-cell lymphoma in patients who have received at least one prior therapy.30-33
Skov et al. and our group demonstrated a significant increase in expression of NKG2DL MICA/B in some tumor cells after exposure to romidepsin.34, 35 MICA/B are two stress-inducible ligands that bind the immunoreceptor NKG2D and play an important role in mediating the cyotoxicity of NK and T cells.36 However, the efficacy of anti-CD20 CAR NK cells, in combination with romidepsin against BL has not yet to be investigated. Therefore, in this study, we evaluated the anti-tumor activity of romidepsin as a single agent and in combination with anti-CD20 CAR modified expanded NK cells in rituximab-sensitive or -resistant BL models. Our research demonstrates that romidepsin has distinct anti-BL mechanisms including significantly inducing apoptosis, cell cycle arrest and/or enhancing expression of NKG2D ligands and secondary significantly optimizing the cytotoxicity of exPBNK cells.
Results
Romidepsin significantly inhibits cell proliferation in both rituximab-sensitive and -resistant BL
We previously reported that romidepsin at 10ng/ml significantly enhanced MICA/B expression in acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL).35 To examine if romidepsin has any effect on rituximab-sensitive and -resistant BL growth, CD20+ rituximab-sensitive Raji and -resistant Raji-2R and Raji-4RH cells were treated with or without 10ng/ml romidepsin for 3 days. Raji cells but not Raji-2R and Raji-4RH cells showed characteristic morphological changes of apoptosis such as shrinking of the cytoplasm at day 3 (Fig. 1A). Cell proliferation was inhibited in all 3 cell lines treated with romidepsin (Fig. 1B, p < 0.0001). Consistent with the observed apoptosis related morphological changes, cell death, as monitored by flow cytometry with 7-AAD staining, was significantly enhanced in romidepsin-treated Raji cells as compared with romidepsin-treated Raji-2R and Raji-4RH cells (p < 0.001) (Fig. 1C). We also found that romidepsin has a significantly lower GI50 against Raji (0.40 ± 0.08 ng/ml) compared with GI50 against Raji-2R (8.30 ± 1.87 ng/ml) and Raji-4RH (4.81 ± 2.22 ng/ml) (p < 0.05) (Fig. S1). Consistent with what Roychowdhury et al reported that 270.35ng/ml romidepsin induced apoptosis and caspase 3 activation in Epstein-Barr Virus (EBV) positive rituximab-sensitive BL cells,37 10ng/ml romidepsin increased active caspase 3 by intracellular flow cytometry analysis in rituxiamb sensitive Raji cells (Fig. 1D). However, the cleaved active caspase 3 was not detectable in Raji-2R and Raji-4RH at day 1 and day 2 after romidepsin treatment (Fig. 1D).
Figure 1.
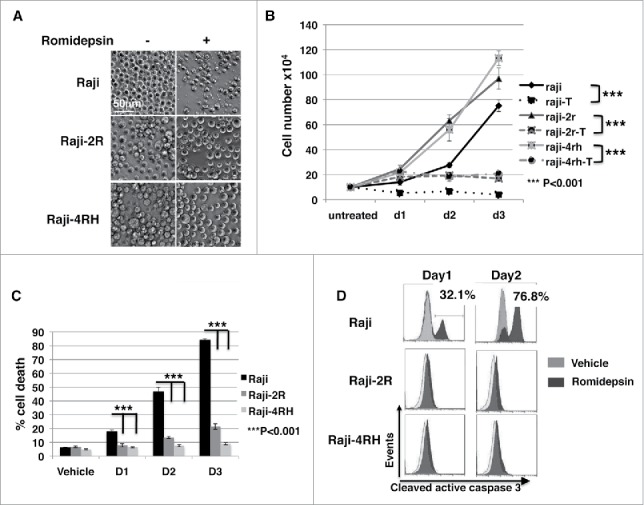
Romidepsin significantly inhibits cell proliferation in both rituximab sensitive and resistant cells and stimulates cell death in rituximab sensitive cells. CD20+ rituximab sensitive Raji and resistant Raji-2R and Raji-4RH cells were treated with or without 10ng/ml romidepsin for 3 days. A, cell phenotypic changes under light microscopy (Carl Zeiss, Thornwood, NY)) are shown at day 3 (original magnification 200x). B, cell proliferation curves were generated with trypan blue staining of living cells and counting living cells with hemocytometer. C, the percentage of the dead cells was gated with 7-AAD+ by flow cytometry analysis. Average values are reported as the mean ± SEM. P values using unpaired student t test were noted in B and C respectively. D, intracellular caspase 3 activation was monitored by flow cytometry analysis at day 1 and day 2. DMSO was added in equal amounts and served as a vehicle control.
We also observed that romidepsin induced significant cell death in other B cell malignancy cell lines such as Ramos (BL), Daudi (BL), Rs4;11 (pre-B-Acute lymphoblastic leukemia), NALM-6 (pre-B-Acute lymphoblastic leukemia), and U-698-M (pre-B- lymphoblastic lymphoma) (Fig. S2).
Romidepsin significantly inhibits rituximab-sensitive and -resistant BL cell growth in xenografted mice
To assess the in vivo anti-tumor activity of romidepsin, 5 × 105 of Raji- Luc or Raji-2R-Luc cells were injected subcutaneously in the right flanks of NSG mice as we previous described.23 Three days after tumor inoculation, mice were randomized to equalize tumor burden and injected intraperitoneally with 4.4mg/kg romidepsin weekly for continuous 3 weeks. The control group was injected with vehicle with the same amount of Dimethyl Sulfoxide (DMSO) diluted in PBS. We demonstrated that the luciferase signals measured in the romidepsin treated Raji-Luc group were significantly reduced compared with the vehicle-treated mice (p < 0.05) (Fig. 2A and B). The tumor size measured in the romidepsin-treated Raji-luc group was also significantly smaller than that in the vehicle-treated mice (p < 0.05) (Fig. 2C). Similarly, the luciferase signals measured in the romidepsin treated Raji-2R-Luc group were significantly reduced compared with the vehicle-treated mice (p < 0.05) (Fig. 2D and E). The tumor size measured in the romidepsin-treated Raji-2R-luc group was also significantly smaller than that in the vehicle-treated mice (p < 0.05) (Fig. 2F). These data demonstrate that romidepsin significantly inhibits rituximab-sensitive and -resistant cells growth in xenografted mice. We further confirmed the conclusion with Daudi xenografts. 1 × 106 Daudi-Luc cells were injected i.v. to NSG mice. After tumor xenografting at day 14, 2.2mg/kg romidepsin was injected i.v. to mice 3 days/week for continuous 2 weeks. We found that tumor burden was significantly reduced with significantly reduced luciferase signals in treated mice compared with the vehicle-treated mice (Fig. 2G and H) (p < 0.001). Consistent with reduced tumor burden, romidepsin treated mice had significantly extended survival time compared with the vehicle-treated mice (p < 0.001) (Fig. 2I).
Figure 2.

Romidepsin significantly inhibits Raji and Raji-2R cells growth in xenografted mice. 5 × 105 of Raji-Luc or Raji-2R-Luc cells were s.c. injected in the right flanks of NSG mice. 3 d after tumor inoculation, mice were randomized to equalize tumor burden and injected i.p. with 4.4mg/kg romidepsin weekly for continuous 3 weeks; mice treated with vehicle containing the same amount of DMSO were served as controls (A-F). A and D, bioluminescence images were taken once weekly. Live imaging demonstrating the extent of Raji-Luc progression is shown. B and E, photons emitted from luciferase-expression cells were measured in regions of interest that encompassed the entire body and quantified using the Living Image software. Signal intensities (total Flux) are shown at the time points detected in untreated, and romidepsin treated mice and plotted as mean ± SEM. C and F, the tumor size was measured with a caliper once a week and plotted as the mean ± SEM for each group. *p < 0.05. G, H and I, 1 × 106 of Daudi-Luc cells were iv injected in the tails of NSG mice. 14 d after tumor inoculation, mice were randomized to equalize tumor burden and injected iv with 2.2mg/kg romidepsin 3 d weekly for continuous 2 weeks; mice treated with PBS were served as controls. G, bioluminescence images were taken once weekly. Live imaging demonstrating the extent of Daudi-Luc progression is shown. H, photons emitted from luciferase-expression cells were measured in regions of interest that encompassed the entire body and quantified using the Living Image software. Signal intensities (total Flux) are shown at the time points detected in untreated, and romidepsin treated mice and plotted as mean ± SEM. I, mice were followed until death or killed if paralysis of hind legs. The Kaplan–Meier survival curves were generated following therapy using animal death/sacrifice as the terminal event.
Romidepsin induces cell cycle arrest in rituximab-resistant cells
We further investigated whether the growth arrest in Raji-2R and Raji-4RH cells treated with romidepsin could be caused by cell cycle arrest. Romidepsin induced G1 arrest in Raji cells (p = 0.001), significant G2 arrest (p = 0.001) and S phase reduction (p < 0.001) in Raji-2R cells, and significant G1 arrest (p < 0.001) and S phase reduction (p < 0.001) in Raji-4RH cells at day 1 (Fig. 3). Cell cycle analysis also indicated romidepsin caused significant increases in pre-G1 fraction (apoptosis) in Raji cells compared with Raji-2R and Raji-4RH (p < 0.001) (Fig. 3). These rituximab-resistant cell lines have been generated through repeated exposure to rituximab.10 Czuczman et al. reported that prolonged rituximab exposure led to downregulation of the pro-apoptotic Bcl-2 family proteins in these rituximab resistant cells and therefore these cells were also resistant to multiple chemotherapeutic agents.38 Probably due to lack of pro-apoptotic pathway proteins in Raji-2R and Raji-4RH, romidepsin does not directly induce cell apoptosis (Fig. 1D) in these cells, but interestingly, it can control the growth of these rituximab-resistant cells by enhancing the cell cycle arrest.
Figure 3.
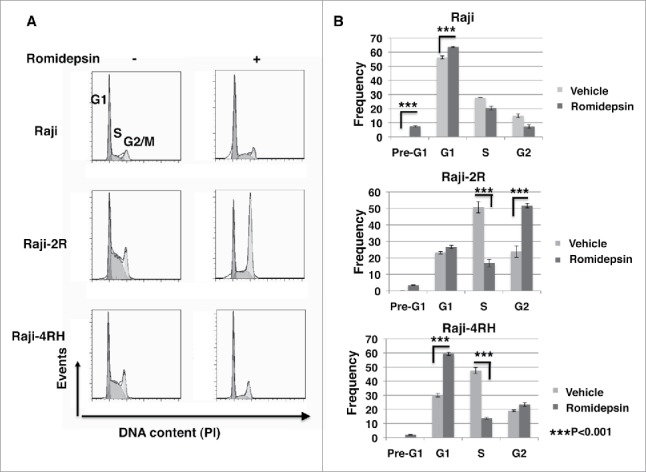
Romidepsin induces cell cycle arrest in rituximab resistant cells. CD20+ rituximab sensitive Raji and resistant Raji-2R and Raji-4RH cells were treated with or without 10ng/ml romidepsin for 24 hours. Propidium iodide (PI)-staining was performed to analyze cell cycle distribution. A, representative histograms illustrate cell cycle profiles of Raji, Raji-2R and Raji-4RH treated with or without romidepsin. B, percentage of Raji, Raji-2R and Raji-4RH. Cells treated with vehicle containing the same amount of DMSO were served as controls.
To explore the potential mechanisms of romidepsin inhibition of cell growth and induction of cell cycle arrest, we performed intracellular flow cytometry and phospho-flow cytometry analysis. We found that romidepsin significantly increased histone H3K9 acetylation (p < 0.05) (Fig. 4A). Consistent with cell cycle arrest, romidepsin increased cell cycle check-point protein p21 in Raji (p = 0.15), Raji-2R (p = 0.06), and Raji-4RH (p = 0.013) compared with the vehicle treated cells (Fig. 4A). Recently, p38 MAPK has been shown to have a central role in resistance to chemotherapeutic agents and inhibition of the p38 MAPK signaling pathway diminished cellular multidrug resistance.40, 41 Therefore, we examined and compared the p38 MAPK phosphorylation level in rituximab-sensitive and resistant cells. We found that p38 MAPK phosphorylation at Thr180/Tyr182 was significantly enhanced in rituximab-resistant Raji-2R (p < 0.01) and Raji-4RH (p < 0.01) cells compared with the sensitive Raji cells (Fig. 4B), indicating p38 MAPK phosphorylation is partially involved in rituximab-resistance. Following, we investigated whether romidepsin could play a role in regulating the level of p38 MAPK phosphorylation. Surprisingly, romidepsin significantly reduced the p38 MAPK phosphorylation in Raji-2R (p < 0.05) and Raji-4RH (p < 0.01) cells but not in Raji cells (Fig. 4B). Considering the p38 MAPK phosphorylation level was still relative high in romidepsin treated rituximab-resistant cells compared with Raji cells, we used a highly selective, cell-permeable inhibitor of p38 MAP kinase on these cells. Our data demonstrate that the combination of romidepsin and a p38 MAPK inhibitor significantly inhibited the growth of Raji-2R and Raji-4RH cells at 24 (Fig. S4) and 72 hours (Fig. 4C). Overall, our results indicate the role of the p38 MAPK signaling pathway in rituximab-resistant BL and the therapeutic potential of the combination of a HDAC inhibitor and targeting p38 MAPK pathway in the treatment of rituximab-resistant BL.
Figure 4.

Romidepsin enhances histone acetylation and cell cycle check point protein p21 expression and reduces phospho-p38MAPK level. Raji, Raji-2R and Raji-4RH cells were treated with or without 10ng/ml romidepsin for 24 hours. A, intracellular protein levels of H3K9 acetylation, p21, phospho-p38 MAPK (Thr180/Tyr182), phospho-p44/42 MAPK (Thr202/Tyr204), phospho-Akt (Ser473) and β-catenin were examined by intracellular flow cytometry or phospho-flow cytometry analysis. B, intracellular phospho-p38 MAPK (Thr180/Tyr182) level was measured by flow cytometry and quantified using the mean fluorescence intensity (MFI) in Raji, Raji-2R and Raji-4RH treated with or without romidepsin. C, Raji, Raji-2R and Raji-4RH cells were treated with 10ng/ml romidepsin, 50uM inhibitor of p38MAPK (SBS202190, Tocris), or combination for 72 hours before relative cell viability/proliferation was measured by MTS assay. Cells treated with vehicle containing the same amount of DMSO were served as controls. The A490 values were normalized to the vehicle control (DMSO).
Ex vivo expanded PBNK has significantly enhanced cytotoxic activity against romidepsin treated rituximab-sensitive and -resistant BL
Recently, we reported that our expanded PBNK cells with K562-mb15 - 41BBL feeder cells were associated with a high expression of the NKG2D receptor.23 To determine if MICA/B expression was increased in rituximab-resistant BL cells exposed to romidepsin and if expanded PBNK K562-mb15 - 41BBL feeder cells were able to induce cell death in the romidepsin treated rituximab-resistant BL cells, we compared MICA/B expression in rituximab sensitive and resistant BL cells treated with or without romidepsin and compared the cytotoxicity of expanded PBNK against rituximab-sensitive and -resistant BL cells treated with or without romidepsin.
Raji, Raji-2R and Raji-4RH cells were treated with or without 10ng/ml romidepsin for 24 hours. MICA/B expression was significantly enhanced in romidepsin treated Raji, Raji-2R and Raji-4RH cells compared with the vehicle untreated cells (p < 0.001) (Fig. 5A). Representative flow cytometry plots of forward scatter (FSC) vs. side scatter (SSC) of the cells treated with or without romidepsin are shown in Fig. S3.
Figure 5.
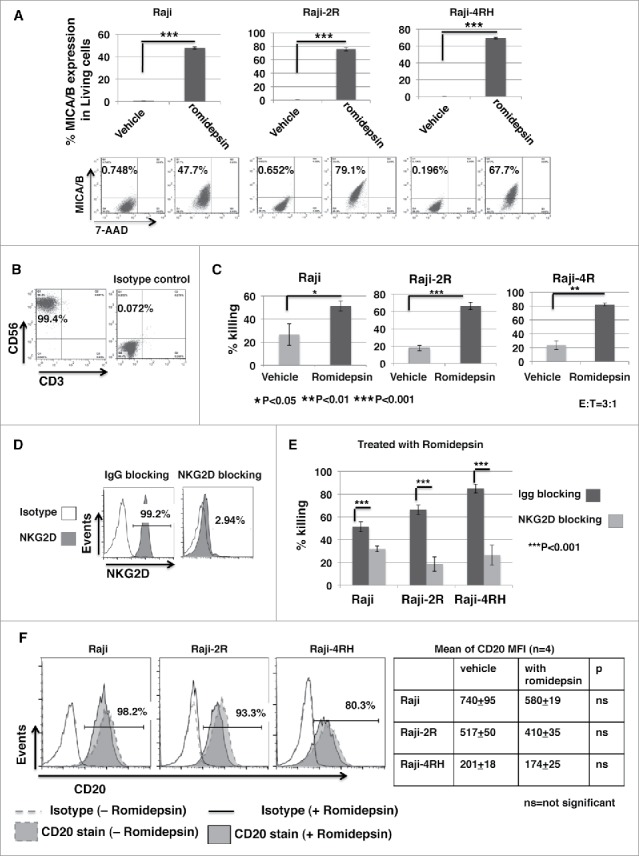
Ex vivo expanded PBNK cells have significantly enhanced cytotoxic activity against romidepsin-treated BL cells compared with the untreated BL. Raji, Raji-2R and Raji-4RH cells were treated with or without 10ng/ml romidepsin for 24 hours. A, MICA/B expression was examined and compared in the cells treated with romidepsin or vehicle containing the same amount of DMSO day 1 with flow cytometry analysis. The top panels show the statistic analysis of % MICA/B expression in gated living cells (7AAD-). ***p < 0.001. The bottom panels are representative dot blots shown from 1 of 3 independent experiments. B, PBNK cells were ex vivo expanded with irradiated K562-mb15 - 41BBL feeder cells for 14 d and purified with NK isolation kits (Miltenyi). Representative dot blots show the purity of final purified expanded PBNK cells with anti-CD56 and anti-CD3 staining. C, in vitro cytotoxicity of ex vivo expanded PBNK cells were measured with europium release assays against Raji, Raji-2R and Raji-4RH treated with or without romidepsin at E:T = 3:1. Cells treated with vehicle containing the same amount of DMSO were served as controls. D, exPBNK cells were incubated with anti-NKG2D antibodies (R & D systems) to block NKG2D receptor. exPBNK cells incubated with IgG isotype were used as controls. Flow cytometry was subsequently performed to quantify surface expression of NKG2D receptors on the exPBNK cell surface. The histograms show the NKG2D expression (left panel). E, cytotoxicity assays were performed after NKG2D receptors were blocked on exPBNK cells against romidepsin treated Raji, Raji-2R and Raji-4RH. exPBNK cells incubated with IgG isotype were used as controls. ***p < 0.001. F, CD20 expression was examined on Raji, Raji-2R and Raji-4RH treated with or without romidepsin by flow cytometry. Left panel show histogram overlays representing isotype controls, CD20 in cells treated with or without romidespin. Right panel show the mean ± SEM of CD20 MFI in cells treated with or without romidespin. n = 4, ns = not significant.
PBNK cells were ex vivo expanded with irradiated K562-mb15 - 41BBL feeder cells for 14 d and purified with NK isolation kits, as we have described previously,23 for more than 99% purity (Fig. 5B). The cytotoxicity of expanded PBNK cells was significantly increased against romidepsin treated Raji cells (p < 0.001), Raji-2R (p < 0.01) and Raji-4RH (p < 0.05) compared with the untreated cells (Fig. 5C).
To further determine whether the enhanced cytotoxicity was in part mediated through the interaction between MICA/B and NKG2D, we performed in vitro NKG2D blocking experiments. ExPBNK cells were incubated with an un-conjugated anti-NKG2D antibody to block NKG2D receptor. After incubation, the free, unblocked NKG2D level was reduced from 99.1 ± 0.03% to 2.97 ± 0.14% detected by a fluorescent conjugated anti-NKG2D antibody (Fig. 5D), demonstrating that the NKG2D receptors were bound by the anti-NKG2D blocking antibody. ExPBNK cells incubated with anti-NKG2D antibodies were used for cytotoxicity assays against romidepsin-treated Raji, Raji-2R and Raji-4RH. ExPBNK cells incubated with Immunoglobulin G (IgG) were used as controls. The cytotoxicity of exPBNK cells was significantly reduced after blocking with anti-NKG2D antibodies compared with exPBNK blocking with IgG against romidepsin treated Raji, Raji-2R and Raji-4RH cells (p < 0.001) (Fig. 5E).
Shimizu et al. have previously reported that CD20 expression on lymphoma cells was enhanced by the HDACi, valproic acid.9 We therefore examined whether romidepsin affects CD20 expression on Raji, Raji-2R and Raji-4RH under our condition. CD20 expression was quantified by mean of fluorescence intensity (MFI). We demonstrated that CD20 MFI was 740 ± 95 in Raji, 517 ± 50 in Raji-2R and 201 ± 18 in Raji-4RH before treatment, and 580 ± 19 in Raji, 410 ± 35 in Raji-2R and 174 ± 25 in Raji-4RH respectively after 10ng/ml romidepsin treatment of 24 hours (Fig. 5F). CD20 expression was not significantly different with 10ng/ml romidepsin treatment of 24 hours in these 3 cell lines, suggesting that CD20 expression on BL is not a major factor on romidepsin-enhanced in vitro cytotoxicity of exPBNK against these 3 BL cell lines.
Anti-CD20 CAR significantly enhanced exPBNK cytotoxic activity against romidepsin treated rituximab-sensitive and -resistant CD20+ BL
To examine whether anti-CD20 chimeric antigen receptor further enhances exPBNK cytotoxocity against romidepsin treated CD20+ BL at a low effector to target ratio, we generated anti-CD20 CAR exPBNK cells by anti-CD20 CAR mRNA nucleofection and the CAR expression was analyzed by flow cytometry at 24 hours after nucleofection (Fig. 6A) as we have described previously.23 The anti-CD20 CAR exPBNK cells at 24 hours after nucleofection were used as effector cells. Mock-nucleofected exPBNK cells, not expressing anti-CD20 CAR were used as control effectors. CD20+ Ramos, Raji, Raji-2R, Raji-4RH and CD20− RS4;11 cells were treated with or without 10ng/ml romidepsin for 24 hours. In vitro cytotoxicity of anti-CD20 CAR exPBNK cells were measured against romidepsin treated or untreated targets at E:T = 3:1. Consistent with our previous report,23 the in vitro cytotoxicity of anti-CD20 CAR exPBNK cells was significantly enhanced against CD20+ Ramos (p < 0.05) (Fig. 6B), Raji (p < 0.01) (Fig. 6C), Raji-2R (p < 0.05) (Fig. 6D), and Raji-4RH (p < 0.01) (Fig. 6E, ) but not CD20− RS4;11 ((Fig. 6F). More over, the in vitro cytotoxicity of anti-CD20 CAR exPBNK cells was significantly enhanced against romidepsin-treated Ramos, Raji Raji-2R, and Raji-4RH compared with the cytotoxicity of the mock CAR exPBNK cells against the treated or untreated tumor cells (Fig. 6B, C and E). In vitro cytotoxicity of anti-CD20 CAR exPBNK cells were significantly enhanced against romidepsin-treated CD20− RS4;11 (p < 0.001) (Fig. 6F) compared with the untreated RS4;11 cells, but not significantly compared with the cytotoxicity of the mock exPBNK cells against the treated RS4;11. Intracellular CD107a expression in exPBNK and anti-CD20 CAR exPBNK was detected by flow cytometry. Consistent with our previous report23 and the enhanced in vitro cytotoxicity (Fig. 6), intracellular CD107a expression was significantly enhanced in anti-CD20 CAR exPBNK compared with mock exPBNK in response to CD20+ Ramos, Raji, Raji-2R and Raji-4RH (p < 0.001) stimulation but not in response to CD20− RS4;11 (Fig. S5). CD107a expression was also significantly enhanced in exPBNK in response to romidepsin treated Raji, Raji-2R, Raji-4RH and RS4;11 (Fig. S5). CD107a expression was unexpectedly reduced in exPBNK in response to romidepsin treated Ramos for unknown reasons (Fig. S5A). More over, CD107a expression was further enhanced in anti-CD20 CAR exPBNK in response to romidepsin treated CD20+ Raji, Raji-2R and Raji-4RH stimulation but not in response to romidepsin treated CD20− RS4;11 (Fig. S5) compared with all other controls (Fig. S5). Overall, these results indicate that the cytotoxicity induced by the interaction of anti-CD20 CAR with CD20 has an additive effect with the cytotoxicity caused by the romidepsin treatment on tumor targets.
Figure 6.

Anti-CD20 CAR significantly enhanced exPBNK cytotoxic activity against rituximab-sensitive and -resistant CD20+ BL after the tumor cells were treated with romidepsin. CD20+ Ramos, Raji, Raji-2R, Raji-4RH and CD20- RS4;11 cells were treated with or without 10ng/ml romidepsin for 24 hours. After washing 3 times with PBS, these cells were used as targets. Anti-CD20 CAR exPBNK cells were generated by anti-CD20 CAR mRNA nucleofection and the CAR expression was analyzed by flow cytometry at 24 hours after nucleofection. Anti-CD20 CAR exPBNK cells at 24 hours after nucleofection were used as effectors. Mock exPBNK cells without anti-CD20 CAR mRNA nucleofection were used as controls. A, one of the representative density plots illustrates the expression of the anti-CD20 CAR in exPBNK cells after mRNA electroporation at 24 hours. In vitro cytotoxicity of anti-CD20 CAR exPBNK cells were measured with europium release assays against rituximab sensitive Ramos and Raji (B-C), rituximab resistant Raji-2R and Raji-4RH (D-E) treated with or without romidepsin at E:T = 3:1. F, CD20- RS4;11 was used as a negative control for anti-CD20 CAR exPBNK cells mediated cytotoxicity. Cells treated with vehicle containing the same amount of DMSO were served as controls. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant, p > 0.05
Anti-CD20 CAR exPBNK cells combined with romidepsin significantly inhibited Raji cells growth and extended the survival of Raji xenografted NSG mice
The in vivo anti-tumor efficacy of anti-CD20 CAR exPBNK combined with romidepsin was next evaluated in therapeutically relevant xenograft models of human BL. 5 × 105 of Raji-Luc cells were i.p. injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 2.2 mg/kg romidepsin or PBS containing DMSO was i.p. injected to each mouse once a week for 3 continuous weeks. 5 × 106 anti-CD20 CAR exPBNK (with anti-CD20 CAR mRNA electroporation) or mock exPBNK (without anti-CD20 CAR mRNA electroporation) cells were i.p. injected following each romidepsin injection after 24 hours. Mice treated with medium were served as controls. Whole mouse bioluminescent imaging (BLI) was used to quantify tumor burden over time (Fig. 7A and B). We demonstrated that after the third NK injection at d28 when the majority of animals were still alive in most of the groups, the luciferase signals were significantly reduced in all of the treated groups including the mock exPBNK group (p < 0.05), the romidepsin group (p < 0.05), the anti-CD20 CAR exPBNK group (p < 0.01), the mock exPBNK+romidepsin group (p < 0.05), and the anti-CD20 CAR exPBNK+romidepsin (p < 0.01) group compared with the untreated tumor only group (Fig. 7A). More importantly, the luciferase signals were significantly reduced in the anti-CD20 CAR exPBNK+romidepsin group compared with the romidepsin group (d28, p < 0.05) and the anti-CD20 CAR exPBNK group (d35, p < 0.05) (Fig. 7A). The representative bioluminescent images are demonstrated in Fig. 7B. Mice were followed until death or killed if tumor size reached 2 cm3. The Kaplan–Meier survival curves for all groups represent the death of mice caused by tumor cells disseminated to the whole body from the injection site or killed if tumor size reached 2 cm3 or larger. The CAR exPBNK-treated Raji-Luc mice (median survival time: 44 days) had significantly extended survival time compared with the untreated mice (median survival time: 32 days, p < 0.001) and the mock exPBNK-treated mice (median survival time: 30 days, p < 0.05) as we previously reported.23 More importantly, we found that the CAR exPBNK+romidepsin treated mice (some mice survived over 85 days) had significantly extended survival time than the CAR exPBNK-treated mice (median survival time: 44 days) or the romidepsin treated mice (median survival time: 30 days) (Fig. 7C). We were able to detect some degree of enhanced MICA/B in the spleen tissue and other tissue with migrated tumor cells (Fig. S6), indicating MICA/B may in part be associated with mediating the enhanced anti-tumor effect of CAR exPBNK cells in vivo.
Figure 7.
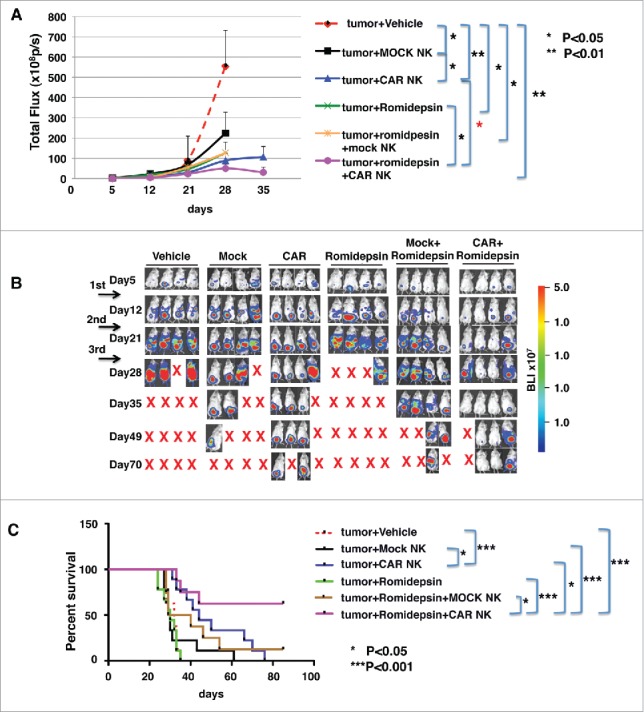
Anti-CD20 CAR exPBNK cells combined with romidepsin significantly inhibited Raji cells growth and extended the survival of xenografted mice. A, 5 × 105 of Raji-Luc cells were i.p. injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 2.2 mg/kg romidepsin or vehicle containing the same amount of DMSO was i.p. injected to each mouse once weekly for continuous 3 weeks. 5 × 106 anti-CD20 CAR exPBNK (with anti-CD20 CAR mRNA electroporation) or mock exPBNK (without anti-CD20 CAR mRNA electroporation) cells were injected i.p. following each romidepsin injection after 24 hours. Mice treated with medium were served as controls. Whole mouse luciferase activity was measured once weekly at various time points. Photons emitted from luciferase-expression cells were measured in regions of interest that encompassed the entire body and quantified using the Living Image software. Signal intensities (total Flux) are shown at the time points plotted as mean ± SEM. B, Live imaging demonstrating the extent of Raji-Luc progression is shown. C, Mice were followed until death or killed if tumor size reached 2 cm3. The Kaplan–Meier survival curves for all groups were generated following therapy initiation using animal sacrifice as the terminal event. Comparison of survival between groups is shown. The romidepsin+CAR exPBNK treated Raji-Luc mice had significantly extended survival time compared with any other listed group. *p < 0.05; ***p < 0.001.
We further confirmed that romidepsin significantly enhanced MICA/B expression in human BL cells in human BL xenografted mice. 1 × 106 of Daudi-Luc or Raji-2R-Luc cells were intravenously injected in NSG mice on day 0. After confirming tumor engraftment at day 14, 2.2mg/kg romidepsin or PBS containing DMSO was intravenously injected to each mouse daily for 2 or 3 consecutive days. We found that MICA/B expression was significantly enhanced in the circulated Daudi in the peripheral blood (Fig. S7A) (p < 0.01) or Raji-2R (Fig. S7B) (p < 0.05) after 2 or 3 consecutive romidepsin injections.
Discussion
In this study, we investigated for the first time the anti-tumor effect of romidepsin; the combined effect of romidepsin with anti-CD20 CAR exPBNK cells against rituximab-sensitive and -resistant CD20+ BL cells; and the enhancement of MICA/B expression by romidepsin in human BL tumor cells in human BL xenografted mice models. We demonstrated that single-agent romidepsin significantly inhibits rituximab-sensitive and -resistant BL growth by inducing cell death and cell cycle arrest in vitro and in vivo. We also demonstrated a significantly synergistic effect between romidepsin and anti-CD20 CAR modified expanded NK cells in vitro and in BL xenografted NSG mice.
Consistent with previous finding many human cancers have alterations in HDACs levels,42, 43 we found that HDAC1, 2, 3, 4 and 6 were highly expressed in most of the BL cells (Fig. S8). Several anti-tumor mechanisms of HDACi have been suggested which include i) upregulation of cell-dependent kinase (CDK) inhibitors like p21/p27 and downregulation of cyclinD1; ii) acetylation of non-histone proteins to regulate cell growth and survival; iii) direct activation of apoptotic pathways; iv) enhanced antitumor immunity through enhancement of tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) or upregulation of antigen expression to facilitate cancer cell recognition.44-46 In our BL pre-clinical models, we found that romidpesin significantly inhibits rituximab sensitive BL growth by inducing cell death and activating apoptotic pathways with the increased active caspase 3 levels (Fig. 1). However, romidepsin did not increase active caspase 3 in rituximab resistant BL cells (Fig. 1), even if the cells had been retreated with romidepsin for a week with a fresh amount of romidepsin after 3 d (Fig. S2), suggesting that romidepsin inhibits rituximab resistant BL growth by other mechanism(s) rather than by activating apoptotic pathways. Resistance to apoptosis is one of the major mechanisms of tumor resistance to conventional therapies and it is selectively and naturally acquired after successive rounds of chemotherapy.47 To explore the mechanisms of the inhibition of rituximab-resistant BL growth, we found that romidepsin induced p21 expression in both rituximab-sensitive and -resistant BL cells (Fig.4A) and G1-phase cell cycle arrest in Raji-4RH, G2-phase cell cycle arrest in Raji-2R rather than apoptosis in rituximab resistant BL at the doses tested (Fig. 3).
Our data demonstrates the in vitro and in vivo effectiveness of romidepsin in inhibiting the growth of the rituximab-sensitive and -resistant BL cells (Fig. 1 and Fig. 2), suggesting that romidepsin can modify the rituximab-resistant phenotype by interfering with signaling pathways to inhibit proliferation. However, the cellular signaling pathways that romidepsin interferes with remains elusive. We performed phosphoflow analysis to examine the changes in p38MAPK, ERK1/2, AKT phosphorylations. Deregulation of these pathways can contribute to the acquired chemoresistance.48, 49 We found that p38MAPK, ERK1/2, AKT were constitutively phosphorylated and activated in Raji, Raji-2R and Raji-4RH. Interestingly, we found that p38MAPK phosphorylation was significantly enhanced in the resistant Raji-2R and Raji-4RH cells compared with the sensitive Raji cells (Fig. 4B). Romidepsin also significantly reduced p38MAPK phosphorylation in Raji-2R and Raji-4RH but not in Raji (Fig. 4B), suggesting that the p38MAPK pathway is involved in the acquired or intrinsic apoptosis-resistance of Raji-2R and Raji-4RH. The combination of romidepsin and p38MAPK inhibitor significantly inhibited the growth of Raji-2R and Raji-4RH (Fig. 4C) compared with each single agent.
In addition to the romidepsin-mediated changes in apoptosis, cell cycle kinetics and the p38MAPK pathway, we also demonstrate that romidespin significantly enhanced NKG2D ligands MICA/B expression in both sensitive and resistant BL cells (Fig. 5A) in vitro and in vivo in BL xenografted mice (Fig. S6 and Fig. S7). MIC A/B binding to NKG2D plays an important role in NK cell activation and tumor immune surveillance .50-52 We previously reported that our ex vivo expanded PBNK cells expressed high levels of NKG2D.23 Previously, it was reported that romidepsin suppressed the NK cytolytic activity of cutaneous T-cell lymphoma patients.53 To reduce the potential toxicity of romidepsin on exPBNK/CAR+ exPBNK cells directly, tumors cells were treated for 24hrs with romidepsin before adding exPBNK/CAR+ exPBNK cells in the in vitro cytotoxicity assays. We demonstrated that the cytotoxicity of exPBNK cells was significantly enhanced against Raji, Raji-2R and Raji-4RH when these cells were treated with romidepsin compared with the cytotoxicity against vehicle treated tumor cells (Fig. 5C). Blocking NKG2D significantly reduced exPBNK cells cytotoxicity even if the tumor cells were treated with romidepsin, suggesting the interaction between NKG2D and MICA/B in cytolysis of tumor cells by NK cells. To reduce the potential in vivo toxicity of romidepsin on exPBNK/CAR+ exPBNK cells, romidepsin was injected into xenografted mice 24 hrs earlier than exPBNK/CAR+ exPBNK injection. With this treatment schedule, we found that CAR+ exPBNK cells combined with romidepsin significantly reduced tumor burden and increased the survival of Raji-Luc xenografted NSG mice compared with the controls (p < 0.01) (Fig. 7).
In conclusion, these results demonstrate the significant effects of romidepsin on rituximab-sensitive and -resistant BL in vitro and in vivo. Our data suggest that romidepsin is an active HDAC inhibitor that potentiates expanded NK and anti-CD20 CAR modified expanded NK activity in vitro and in vivo. Our study offers a rationale for the development of new therapeutic strategies by combining romidepsin with expanded activated NK cells or CAR expanded NK cells for targeting in CD20+ mature BL.
Materials and methods
Cell lines and reagents
Raji, Raji-2R and Raji-4RH cells were generously provided by Matthew Barth, MDand Myron Czuczman, MD from Roswell Park Cancer Institute.10 Romidepsin was generously provided by Celgene Corporation (Summit, NJ). Romidepsin was dissolved in DMSO and further diluted in PBS. As control an equal amount of DMSO was diluted in PBS. See Supplementary Methods for details.
NK cell expansion and anti-CD20-BB- ζ mRNA modified NK generation
Expanded PBNK cells were isolated by negative selection using Miltenyi NK cell isolation kit (Miltenyi Biotec). NK cell purity was confirmed by flow cytometry using anti-CD56 and anti-CD3 antibodies (Becton Dickinson). Anti-CD20–4–1BB- CD3ζ mRNA was transcribed in vitro using the mMESSAGE mMACHINE T7 Ultra kit (Thermofisher) and nucleofected to purified expanded NK cells as described previously.23
In vitro cytotoxicity
NK cytotoxic activity was determined by europium release assays with a standard kit (Perkin Elmer) as we have described previously.23 A detailed description of the experimental procedures is provided in the supplementary methods.
Cell growth assay and GI50 calculation
Cells were treated with 10ng/ml romidepsin. Cell viability was assayed by trypan blue (Thermofisher) exclusion and the cell growth was documented with a hemocytometer by counting the total number of living cells in quadruplicate wells every day for 3 days, or by CellTiter 96 AQueous one solution cell proliferation assay (Pormega) according to the manufacturer's manual. GI50 values for romidepsin on day 1 in BL cells were calculated from nonlinear regression curve fits using GraphPad Prism 5 (Graphpad).
Cell cycle analysis
Romidepsin treated and untreated cells were fixed and stained with propidium iodide (PI) (Acros Organics) in the presence of DNase free RNase (Thermo Scientific). The cells were analyzed via flow cytometry using FACScan (BD), acquiring 10000 events.
NKG2D blocking
NKG2D blocking experiments were performed using anti-NKG2D antibodies (R and D) as described previously with minor modifications.35 Briefly, freshly purified NK cells (1 × 106) were incubated with anti-NKG2D antibodies or IgG control at various concentrations (0–50 µl). Flow cytometry was subsequently performed to quantify surface expression of NKG2D receptors on the NK cell surface with a second incubation with APC conjugated anti-NKG2D antibodies. Cytotoxicity assays were performed after NKG2D receptors were blocked on NK cells.
Flow cytometry analysis of intracellular proteins and phosphoproteins
Fixed and permeabilized cells were stained with mouse anti-human primary antibodies (cellsignaling) H3K9 acetylation (#9649P), p21 (#2947P), β-catenin (#8480p), or phospho-site specific antibodies phospho-p38 MAPK (Thr180/Tyr182) (#4511P), phospho-p44/42 MAPK (Thr202/Tyr204) (#4370P), phospho-Akt (Ser473) (#4060P) followed by anti-rabbit AlexaFluor 647- conjugated secondary antibodies (life technologies). Cells were analyzed using MACSQuant Analyzer (Miltenyi Biotec). No stain, or isotype controls were used for gating.
Animal studies
Six to 8-week old NOD/SCID/γ-chain-/- (NSG) mice (The Jackson Laboratory, Bar Harbor, ME) were bred and maintained under pathogen-free conditions in-house at Comparative Medicine at New York Medical College. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) in New York Medical College.
Xenograft models of human Burkitt lymphoma (BL) and rituximab resistant BL
Luciferase expression tumor cells (Raji-Luc, Daudi-Luc, and Raji-2R-Luc) were generated as we described previously.23 Tumor cells were intraperitoneally or intravenously injected into the NSG mice. Tumor engraftment and progression were evaluated using the Xenogen IVIS-200 system (Caliper Life Sciences) as we have described previously.23 Tumor size was estimated according to the following formula: tumor size (cm3) = length (cm) × width2 (cm) × 0.5. Mice were followed until death or killed if any tumor size reached 2 cm3 or larger.
Statistical analyses
Average values are reported as the mean ± SEM. The one-tailed unpaired Student t-test with p < 0.05 was considered as significant. See Supplementary Methods for details.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Erin Morris, RN, Carmella VanDeVen, MS and Virginia Moore, RN, for their excellent assistance with the preparation of this manuscript. And the authors thank Dr. Dario Campana (St. Jude Children's Research Hospital) and Dr. Terrence Geiger (St. Jude Children's Research Hospital) for kindly providing anti-CD20 scFv, Celgene corporation for providing romidepsin and Dr. Carl Hamby (New York Medical College) for sharing equipments and Bokun Cheng (New York Medical College) for her assistance with western blots and Dr. Matthew Barth (State University of New York at Buffalo) for providing Raji-2R and Raji-4RH cell lines.
Funding
The research for this study was supported by the grant from Pediatric Cancer Research Foundation, St. Baldrick's Foundation, New York Medical College Intramural Research Award, The Touro College and University System Seed Funding Grant and New York Medical College (NYMC) School of Medicine (SOM) Translational Science Institute (TSI) Children Health Translational Research Award.
References
- 1.Pinkerton R, Cairo MS. Childhood Non-Hodgkin Lymphoma In: Cairo MS, Perkins SL, eds. Hematological Malignancies in Children, Adolescents and Young Adults. Singapore: World Scientific, 2012:299-328; ISBN:978-981-4299-60-2; https://doi.org/ 10.1142/9789814299619_0016 [DOI] [Google Scholar]
- 2.Cairo MS, Gerrard M, Sposto R, Auperin A, Pinkerton CR, Michon J, Weston C, Perkins SL, Raphael M, McCarthy K, et al.. Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood 2007; 109:2736-43; PMID:17138821; https://doi.org/ 10.1182/blood-2006-07-036665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerrard M, Cairo MS, Weston C, Auperin A, Pinkerton R, Lambilliote A, Sposto R, McCarthy K, Lacombe MJ, Perkins SL, et al.. Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin's lymphoma: results of the FAB/LMB 96 international study. Br J Haematol 2008; 141:840-7; PMID:18371107; https://doi.org/ 10.1111/j.1365-2141.2008.07144.x [DOI] [PubMed] [Google Scholar]
- 4.Cairo MS, Sposto R, Gerrard M, Auperin A, Goldman SC, Harrison L, Pinkerton R, Raphael M, McCarthy K, Perkins SL, et al.. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (>/ = 15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin's lymphoma: results of the FAB LMB 96 study. J Clin Oncol 2012; 30:387-93; PMID:22215753; https://doi.org/ 10.1200/JCO.2010.33.3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoelzer D, Walewski J, Dohner H, Viardot A, Hiddemann W, Spiekermann K, Serve H, Dührsen U, Hüttmann A, Thiel E, et al.. Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood 2014; 124:3870-9; PMID:25359988; https://doi.org/ 10.1182/blood-2014-03-563627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman S, Smith L, Galardy P, Perkins SL, Frazer JK, Sanger W, Anderson JR, Gross TG, Weinstein H, Harrison L, et al.. Rituximab with chemotherapy in children and adolescents with central nervous system and/or bone marrow-positive Burkitt lymphoma/leukaemia: a Children's Oncology Group Report. Br J Haematol 2014; 167:394-401; PMID:25066629; https://doi.org/ 10.1111/bjh.13040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldman S, Smith L, Anderson JR, Perkins S, Harrison L, Geyer MB, Gross TG, Weinstein H, Bergeron S, Shiramizu B, et al.. Rituximab and FAB/LMB 96 chemotherapy in children with Stage III/IV B-cell non-Hodgkin lymphoma: a Children's Oncology Group report. Leukemia 2013; 27:1174-7; PMID:22940833; https://doi.org/ 10.1038/leu.2012.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miles RR, Arnold S, Cairo MS. Risk factors and treatment of childhood and adolescent Burkitt lymphoma/leukaemia. Br J Haematol 2012; 156:730-43; PMID:22260323; https://doi.org/ 10.1111/j.1365-2141.2011.09024.x [DOI] [PubMed] [Google Scholar]
- 9.Shimizu R, Kikuchi J, Wada T, Ozawa K, Kano Y, Furukawa Y. HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells. Leukemia 2010; 24:1760-8; PMID:20686505; https://doi.org/ 10.1038/leu.2010.157 [DOI] [PubMed] [Google Scholar]
- 10.Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res 2008; 14:1561-70; PMID:18316581; https://doi.org/ 10.1158/1078-0432.CCR-07-1254 [DOI] [PubMed] [Google Scholar]
- 11.Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science 2004; 306:1517-9; PMID:15567854; https://doi.org/ 10.1126/science.1103478 [DOI] [PubMed] [Google Scholar]
- 12.Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol 2007; 7:329-39; PMID:17438573; https://doi.org/ 10.1038/nri2073 [DOI] [PubMed] [Google Scholar]
- 13.Bell E. Mismatch advantages. Nature Reviews Immunology 2002; 2:302-3 [Google Scholar]
- 14.Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol 2007; 7:737-44; PMID:17673918; https://doi.org/ 10.1038/nri2144 [DOI] [PubMed] [Google Scholar]
- 15.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005; 106:376-83; PMID:15755898; https://doi.org/ 10.1182/blood-2004-12-4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayello J, van de Ven C, Fortino W, Wade-Harris C, Satwani P, Baxi L, Simpson LL, Sanger W, Pickering D, Kurtzberg J, et al.. Characterization of cord blood natural killer and lymphokine activated killer lymphocytes following ex vivo cellular engineering. Biol Blood Marrow Transplant 2006; 12:608-22; PMID:16737934; https://doi.org/ 10.1016/j.bbmt.2006.01.009 [DOI] [PubMed] [Google Scholar]
- 17.Robinson KL, Ayello J, Hughes R, van de Ven C, Issitt L, Kurtzberg J, Cairo MS. Ex vivo expansion, maturation, and activation of umbilical cord blood-derived T lymphocytes with IL-2, IL-12, anti-CD3, and IL-7. Potential for adoptive cellular immunotherapy post-umbilical cord blood transplantation. Experimental hematology 2002; 30:245-51; PMID:11882362; https://doi.org/ 10.1016/S0301-472X(01)00781-0 [DOI] [PubMed] [Google Scholar]
- 18.Ayello J, van de Ven C, Cairo E, Hochberg J, Baxi L, Satwani P, Cairo MS. Characterization of natural killer and natural killer-like T cells derived from ex vivo expanded and activated cord blood mononuclear cells: implications for adoptive cellular immunotherapy. Exp Hematol 2009; 37:1216-29; PMID:19638292; https://doi.org/ 10.1016/j.exphem.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 19.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood 2008; 112:1570-80; PMID:18725575; https://doi.org/ 10.1182/blood-2008-02-078071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol 2006; 6:394-403; PMID:16622478; https://doi.org/ 10.1038/nri1838 [DOI] [PubMed] [Google Scholar]
- 21.Pituch-Noworolska A, Hajto B, Mazur B, Sonta-Jakimczyk D, Balwierz W, Janota-Krawczyk E, Kowalska H, Malinowska I, Modzelewska M, Wasik M. Expression of CD20 on acute lymphoblastic leukemia cells in children. Neoplasma 2001; 48:182-7; PMID:11583286 [PubMed] [Google Scholar]
- 22.Perkins SL, Lones MA, Davenport V, Cairo MS. B-Cell non-Hodgkin's lymphoma in children and adolescents: surface antigen expression and clinical implications for future targeted bioimmune therapy: a children's cancer group report. Clin Adv Hematol Oncol 2003; 1:314-7; PMID:16224429 [PubMed] [Google Scholar]
- 23.Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol Res 2015; 3:333-44; PMID:25492700; https://doi.org/ 10.1158/2326-6066.CIR-14-0114 [DOI] [PubMed] [Google Scholar]
- 24.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol 2010; 6:238-43; PMID:20139990; https://doi.org/ 10.1038/nchembio.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolden JE, Shi W, Jankowski K, Kan CY, Cluse L, Martin BP, MacKenzie KL, Smyth GK, Johnstone RW. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis 2013; 4:e519; PMID:23449455; https://doi.org/ 10.1038/cddis.2013.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, Cheng H, Kwan W, Lubieniecka JM, Nielsen TO. Histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in clear cell sarcoma models. Mol Cancer Ther 2008; 7:1751-61; PMID:18566246; https://doi.org/ 10.1158/1535-7163.MCT-07-0560 [DOI] [PubMed] [Google Scholar]
- 27.Woo S, Gardner ER, Chen X, Ockers SB, Baum CE, Sissung TM, Price DK, Frye R, Piekarz RL, Bates SE, et al.. Population pharmacokinetics of romidepsin in patients with cutaneous T-cell lymphoma and relapsed peripheral T-cell lymphoma. Clin Cancer Res 2009; 15:1496-503; PMID:19228751; https://doi.org/ 10.1158/1078-0432.CCR-08-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, et al.. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood 2005; 105:959-67; PMID:15466934; https://doi.org/ 10.1182/blood-2004-05-1693 [DOI] [PubMed] [Google Scholar]
- 29.Kosugi H, Ito M, Yamamoto Y, Towatari M, Ueda R, Saito H, Naoe T. In vivo effects of a histone deacetylase inhibitor, FK228, on human acute promyelocytic leukemia in NOD / Shi-scid/scid mice. Jpn J Cancer Res 2001; 92:529-36; PMID:11376562; https://doi.org/ 10.1111/j.1349-7006.2001.tb01126.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, et al.. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 2009; 27:5410-7; PMID:19826128; https://doi.org/ 10.1200/JCO.2008.21.6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whittaker SJ, Demierre M-F, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, et al.. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol 2010; 28:4485-91; PMID:20697094; https://doi.org/ 10.1200/JCO.2010.28.9066 [DOI] [PubMed] [Google Scholar]
- 32.Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, et al.. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 2012; 30:631-6; PMID:22271479; https://doi.org/ 10.1200/JCO.2011.37.4223 [DOI] [PubMed] [Google Scholar]
- 33.Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, Jaffe ES, Ling A, Turner M, Peer CJ, et al.. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 2011; 117:5827-34; PMID:21355097; https://doi.org/ 10.1182/blood-2010-10-312603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res 2005; 65:11136-45; PMID:16322264; https://doi.org/ 10.1158/0008-5472.CAN-05-0599 [DOI] [PubMed] [Google Scholar]
- 35.Satwani P, Bavishi S, Saha A, Zhao F, Ayello J, van de Ven C, Chu Y, Cairo MS. Upregulation of NKG2D ligands in acute lymphoblastic leukemia and non-Hodgkin lymphoma cells by romidepsin and enhanced in vitro and in vivo natural killer cell cytotoxicity. Cytotherapy 2014; 16:1431-40; PMID:24856896; https://doi.org/ 10.1016/j.jcyt.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 36.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 2003; 3:781-90; PMID:14523385; https://doi.org/ 10.1038/nri1199 [DOI] [PubMed] [Google Scholar]
- 37.Roychowdhury S, Baiocchi RA, Vourganti S, Bhatt D, Blaser BW, Freud AG, Chou J, Chen CS, Xiao JJ, Parthun M, et al.. Selective efficacy of depsipeptide in a xenograft model of Epstein-Barr virus-positive lymphoproliferative disorder. J Natl Cancer Inst 2004; 96:1447-57; PMID:15467034; https://doi.org/ 10.1093/jnci/djh271 [DOI] [PubMed] [Google Scholar]
- 38.Olejniczak SH, Hernandez-Ilizaliturri FJ, Clements JL, Czuczman MS. Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin Cancer Res 2008; 14:1550-60; PMID:18316580; https://doi.org/ 10.1158/1078-0432.CCR-07-1255 [DOI] [PubMed] [Google Scholar]
- 39.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9:537-49; PMID:19629069; https://doi.org/ 10.1038/nrc2694 [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Hong Z, Chen Y. Involvement of p38 MAPK in the Drug Resistance of Refractory Epilepsy Through the Regulation Multidrug Resistance-Associated Protein 1. Neurochem Res 2015; 40:1546-53; PMID:26092535; https://doi.org/ 10.1007/s11064-015-1617-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereira L, Igea A, Canovas B, Dolado I, Nebreda AR. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol Med 2013; 5:1759-74; PMID:24115572; https://doi.org/ 10.1002/emmm.201302732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5:769-84; PMID:16955068; https://doi.org/ 10.1038/nrd2133 [DOI] [PubMed] [Google Scholar]
- 43.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001; 1:194-202; PMID:11902574; https://doi.org/ 10.1038/35106079 [DOI] [PubMed] [Google Scholar]
- 44.Kim H-J, Bae S-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res 2011; 3:166-79; PMID:21416059 [PMC free article] [PubMed] [Google Scholar]
- 45.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res: MCR 2007; 5:981-9; PMID:17951399; https://doi.org/ 10.1158/1541-7786.MCR-07-0324 [DOI] [PubMed] [Google Scholar]
- 46.Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene 2005; 24:4609-23; PMID:15897906; https://doi.org/ 10.1038/sj.onc.1208585 [DOI] [PubMed] [Google Scholar]
- 47.Ng CP, Bonavida B. A new challenge for successful immunotherapy by tumors that are resistant to apoptosis: two complementary signals to overcome cross-resistance. Adv Cancer Res 2002; 85:145-74; PMID:12374285; https://doi.org/ 10.1016/S0065-230X(02)85005-9 [DOI] [PubMed] [Google Scholar]
- 48.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 2004; 23:2934-49; PMID:15077155; https://doi.org/ 10.1038/sj.onc.1207515 [DOI] [PubMed] [Google Scholar]
- 49.Xie Y, Peng Z, Shi M, Ji M, Guo H, Shi H. Metformin combined with p38 MAPK inhibitor improves cisplatin sensitivity in cisplatinresistant ovarian cancer. Mol Med Rep 2014; 10:2346-50; PMID:25118792; https://doi.org/ 10.3892/mmr.2014.2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285:727-9; PMID:10426993; https://doi.org/ 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 51.Obeidy P, Sharland AF. NKG2D and its ligands. Int J Biochem Cell Biol 2009; 41:2364-7; PMID:19631280; https://doi.org/ 10.1016/j.biocel.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 52.Lanier LL. A renaissance for the tumor immunosurveillance hypothesis. Nat Med 2001; 7:1178-80; PMID:11689875; https://doi.org/ 10.1038/nm1101-1178 [DOI] [PubMed] [Google Scholar]
- 53.Kelly-Sell MJ, Kim YH, Straus S, Benoit B, Harrison C, Sutherland K, Armstrong R, Weng WK, Showe LC, Wysocka M, et al.. The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T-cell lymphoma patients. Am J Hematol 2012; 87:354-60; PMID:22367792; https://doi.org/ 10.1002/ajh.23112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.