

Summary
DHHC enzymes catalyze palmitoylation, a major post-translational modification that regulates a number of key cellular processes. There are up to 24 DHHCs in mammals and hundreds of substrate proteins that get palmitoylated. However, how DHHC enzymes engage with their substrates is still poorly understood. There is currently no structural information about the interaction between any DHHC enzyme and protein substrates. In this study we have investigated the structural and thermodynamic bases of interaction between the ankyrin repeat domain of Human DHHC17 (ANK17) and Snap25b. We solved a high-resolution crystal structure of the complex between ANK17 and a peptide fragment of Snap25b. Through structure-guided mutagenesis, we discovered key residues in DHHC17 that are critically important for interaction with Snap25b. We further extended our finding by showing that the same residues are also crucial for the interaction of DHHC17 with Huntingtin, one of its most relevant substrates.
Graphical Abstract
Verardi et al. reveal the structural basis of interaction between DHHC17, a neuronal palmitoyltransferase and SNAP25b, a canonical substrate. They show the role of critical residues for substrate-binding and palmitate-transfer using in vitro and cell-based assays and show the involvement of the same residues in binding Huntingtin, another important substrate of DHHC17.
Introduction
Protein palmitoylation refers to post-translational covalent acylation of cysteines, resulting in the formation of a thioester. This modification plays critical roles in a myriad of physiological processes including signaling by oncogenic Ras, localization of synaptic signaling receptors and modulation of protein aggregation in neurons (Fukata and Fukata, 2010; Linder and Deschenes, 2007). More than 500 human proteins are known to be palmitoylated (Tate et al., 2015) and this is likely an underestimate of the cellular palmitoylome (Jones et al., 2012). Protein palmitoylation is carried out by integral membrane enzymes that are characterized by a highly conserved Asp-His-His-Cys motif in a cysteine rich domain (DHHC-CRD) located in a putative intracellular loop. There are several members of the so-called DHHC family of palmitoyltransferases, ranging from 7 in yeast to 24 in mammals (Fukata et al., 2004).
For some DHHC palmitoyltransferases, it has been shown that palmitoylation is catalyzed via a two-step mechanism, whereby the enzymes first get autopalmitoylated at the catalytic cysteine of the DHHC motif and in a subsequent step they transfer the palmitoyl group onto the protein substrate (Jennings and Linder, 2012; Mitchell et al., 2010; 2014). The relatively large number of DHHC enzymes in higher organisms, coupled to the several hundred substrates, indicates a complex enzyme-substrate network. However, there are still no biophysical characterizations and structural insights into how DHHCs engage with protein substrates. For DHHC17 (and the closely related DHHC13) substrate interactions are somewhat better characterized. DHHC17 has two distinct features compared to other DHHCs: 1) it has seven cytoplasmic ankyrin repeats at the N-terminus and 2) it has six predicted transmembrane helices instead of four. DHHC17 is localized to the Golgi membrane and in humans it is highly expressed in the brain (Ohno et al., 2006). Knockout of DHHC17 in mice significantly affects hippocampal memory, synaptic plasticity and produces Huntington-like phenotypes. (Sanders et al., 2015). Recently, Chamberlain and colleagues showed that the cytoplasmic domain of DHHC17 is both necessary and sufficient to bind Snap25b (Synaptosomal-associated protein 25) (Lemonidis et al., 2014). They used alanine scanning mutagenesis and bioinformatics to identify the sequence motif ΨβXXQP (where Ψ = Val, Ile, Ala or Pro β = Val, Ile or Thr X−X = any two residues, Q = Gln, P = Pro) in SNAP25b and other substrates of DHHC17 that is a critical determinant for substrate interactions. Despite the first identification of a sequence motif for any DHHC/substrate interaction, biophysical characterization and atomic insights into how DHHC17 engages with substrates have not been reported in the literature. To answer these questions we solved the crystal structure of the cytoplasmic domain of DHHC17 (ANK17) in complex with a peptide fragment of Snap25b (containing the substrate recognition motif) at 2.1Å resolution. We subsequently showed that mutations of critically important residues identified in the structure impair substrate palmitoylation. In addition, we demonstrate that the same molecular signature is important for interaction with Huntingtin, the most prominent substrate of DHHC17. Our structural data, together with in vitro and in cellulo binding studies, lend the first insights into the chemistry of interactions of protein substrates with DHHC17 palmitoyltransferase.
Results
Characterization of the ANK17/Snap25b complex
DHHC17 regulates the palmitoylation status of several neuronal proteins, among which the synaptosomal Snap25b represents a major target (Huang et al., 2004). The N-terminal cytosolic domain of DHHC17 contains seven ankyrin repeats (Figure 1A), which have been shown to harbor the binding site to Snap25b (Lemonidis et al., 2015). To obtain a more quantitative assessment of the affinity of the ANK17 domain for Snap25b, we performed binding studies using isothermal titration calorimetry (ITC) with purified proteins (Figure 1C–D and Figure S1). In all instances, ANK17 was loaded into the syringe at concentration of around 200–300 μM and Snap25b was loaded into the cell at 10–30 μM. Non-linear regression analysis, assuming a “one-to-one” binding model, shows a relatively high affinity interaction with a Kd of around 0.5 μM (Figure 1D – left panel and Table S1). The cysteine-less mutant (Snap25b-C4A), used in most structural studies of SNARE complex, (Misura et al., 2001a; 2001b; Stein et al., 2009) retains tight-binding to ANK17, albeit with slightly lower affinity (Kd ~ 3 μM) (Figure 1D – middle panel). Finally, we titrated the fragment of Snap25b corresponding to amino acids 111 to 120 in the full-length protein (hereafter referred to as Snap25b111–120) that is necessary and sufficient for the interaction with the ANK17 domain (Lemonidis et al., 2015). The peptide retains binding to ANK17, with ~20 fold lower binding affinity (Kd ~ 11 μM) with respect to the full length Snap25b (Figure 1D-right). For all the species tested, the negative binding Gibbs energy (ΔG) is due to the large negative contribution of the binding enthalpy (ΔH), which compensates for the unfavorable binding entropy (ΔS is negative for all species) (Table S1). This is consistent with the large entropic penalty likely caused by the loss of degrees of freedom upon binding of Snap25 to ANK17, in light of the lack of structure in Snap25b when free in solution (An and Almers, 2004; Misura et al., 2001b). Overall, our results confirm that the ANK17 domain of DHHC17 is sufficient for interaction with Snap25b with sub-micromolar affinity and are consistent with the hypothesis that the region of Snap25b between residues 111 to 120 alone is a major determinant for the binding affinity. To gain further insights into the substrate binding mechanism of DHHC17, we attempted to crystallize ANK17 in complex with full-length Snap25b-C4A. Both ANK17 and Snap25b-C4A show homogenous and monodisperse peaks (Figure S1). Noticeably, although ANK17 and Snap25b-C4A have similar molecular weights, they have distinctly different retention times in size exclusion chromatography. In particular, Snap25b-C4A has a much lower retention time than ANK17, indicative of either the presence of larger molecular weight species or larger effective hydrodynamic radius (Figure S1-B and C). Analytical ultracentrifugation analysis of Snap25b-C4A showed that more than 95% of the protein runs as a monomer at 1.56 S with a best-frictional ratio of 2.1 (Figure S1-B - inset). This frictional ratio is consistent with the fact that Snap25b is largely unstructured in solution compared to the well-folded ANK17. Although the complex between ANK17 and full-length Snap25b-C4A is of relatively high affinity and can be successfully purified by size exclusion chromatography (Figure S1-A), we did not succeed in obtaining crystals.
Figure 1. Initial characterization of ANK17 binding to Snap25b.
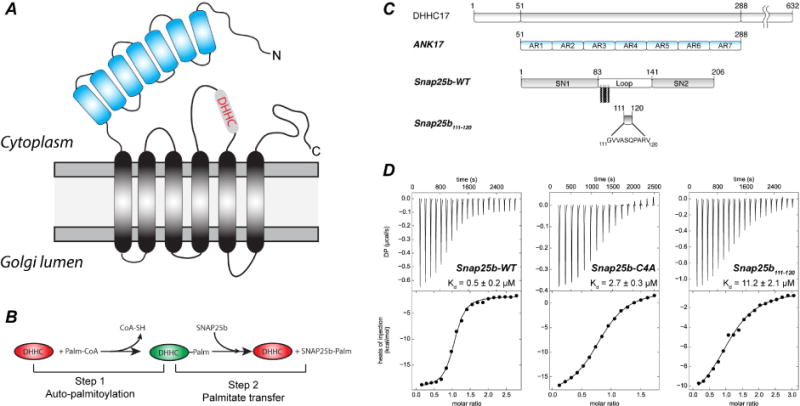
(A) Schematic topology of DHHC17 palmitoyl-transferase. (B) Two-step reaction scheme for a typical DHHC enzyme. (C) List of constructs used for crystallization: ANK17 (corresponding to N-terminal domain of Human DHHC17 - residues 51–288) and full-length Snap25b-WT were produced by recombinant expression in E.coli. Peptide corresponding to amino acids 111 to 120 in Snap25 was produced by solid phase peptide synthesis (D) Representative ITC thermograms and non-linear regression analyses for the interaction between ANK17 and Snap25b (left panel), Snap25b-C4A (central panel) or S25b111–120 (right panel).
Crystal structure of ANK17/Snap25111–120 complex
Since our ITC data showed low micromolar affinity for the binding of ANK17 to Snap25111–120, we decided to attempt crystallization of ANK17 in complex with the Snap25111–120 peptide. Single crystals of ANK17/Snap25111–120 complex diffracted to 2.1 Å resolution and the structure was solved by molecular replacement using the structure of the apo-ANK17 (Gao et al., 2009) (Figure 2 and Table 1).
Figure 2. Crystal structure of the complex between ANK17 and Snap25111–120.
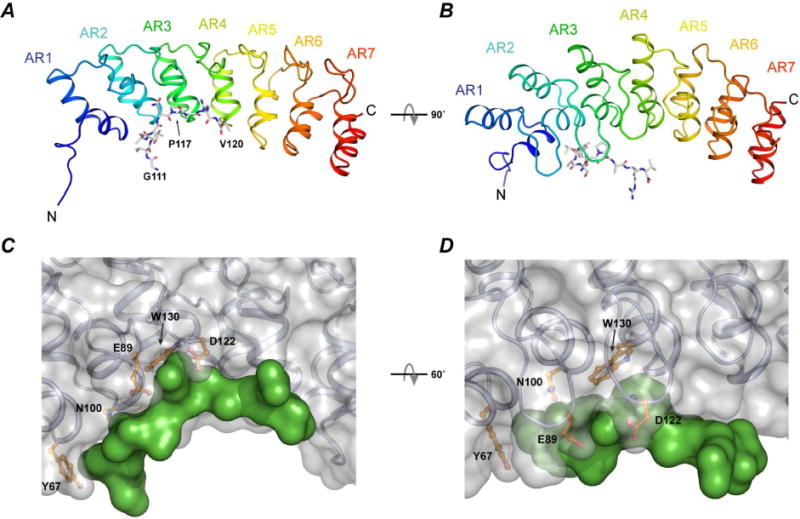
A–B) Cartoon and stick representation of the ANK17 and Snap25b peptide, respectively. C–D) Surface representation of the complex. The peptide (green surface) binds to the concave region of ANK17 between AR1–AR3. Some of the interacting residues in ANK17 are shown in orange.
Table 1.
Statistics for crystallographic data collection and refinement
| ANK17-WT | ANK17-E89A | |
|---|---|---|
| Data collection | ||
| Space group | P64 | P64 |
| Cell constants (Å) | a=b=88.187; c=127.969 α=β=90°; γ=120° |
a=b=87.736; c=128.007 α=β=90°; γ=120° |
| Source | SER-CAT 22BM | SER-CAT 22ID |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Resolution (Å) | 50-2.1 | 50-2.2 |
| Unique reflections | 29948 | 27219 |
| <I>/<σ I> | 17.8 (2.1) | 31.1 (2.7) |
| Redundancy | 3.2 (2.1) | 8.0 (3.2) |
| Completeness (%) | 91.8 (81.4) | 97.0 (85.6) |
| Rmerge (%) | 7.0 (28.8) | 12.1 (33.5) |
| CC1/2 | 0.907 (0.67) | 0.899 (0.47) |
| CC* | 0.973 (0.90) | 0.969 (0.80) |
| Model Refinement | ||
| Resolution (Å) | 20-2.1 | 20-2.2 |
| Reflections | 29948 | 27208 |
| Rwork/Rfree (%) | 17.6/22.2 | 17.0/21.1 |
| RMSD bond lengths (Å) | 0.007 | 0.007 |
| RMSD bond angles (°) | 0.82 | 0.83 |
| Average B-factor (Å2) | ||
| Complex | 35.9 | 43.0 |
| ANK17 | 35.8 | 42.9 |
| Peptide | 38.8 | 44.8 |
| Ramachandran plot | ||
| Favored (%) | 98.3 | 98.1 |
| Allowed (%) | 1.7 | 1.9 |
| Disallowed (%) | 0 | 0 |
| Number of non-hydrogen atoms | ||
| Macromolecules | 3703 | 3732 |
| Solvent | 175 | 97 |
| Buried surface area in complex1 | ||
| Total/apolar/polar Å2 | 528/364/164 | 518/366/152 |
Data collection statistics given in parentheses are for the highest resolution shell.
Calculated with GetArea (Fraczkiewicz and Braun, 1998).
The crystallographic unit contains two ANK17/Snap25b111–120 molecules. The main crystal contacts are located at the N and C-termini of the ANK17 and involve several hydrogen bonds (Figure S3). The N and C-termini of each peptide also make contact to the opposite ANK17 molecule (Figure S3).
In the complex, Snap25111–120 peptide binds between ankyrin repeats 2 and 3 within the concave region of ANK17 (Figure 2). Overall, the backbone structure of the apo-ANK17 and peptide bound-ANK17 are very similar (RMSD ~ 0.4 Å) and only minor differences are visible in finger 2 and finger 3 (Figure S2-B).
Within the complex, only the first half of the peptide (corresponding to residues 112–116 in full-length Snap25b) is close enough to ANK17 to establish inter-molecular contacts, while the C-terminal half of the peptide projects out of the structured region of ANK17 (Figure 2 and Figure S2-A). This is consistent with in vitro and in cellulo data showing the importance of the residues localized between amino acids 112–117 in full-length Snap25b for the binding to DHHC17 and subsequent palmitoylation (Gonzalo et al., 1999; Lemonidis et al., 2015). Overall, ANK17/Snap25111–120 complex formation buries around 530 Å2 of surface, with 70% of the surface consisting of apolar atoms (~360 Å2) and the remaining 30% made of polar atoms (~170 Å2) (Table 1). A closer inspection of the residues involved at the binding interface reveals several close interactions: on the N-terminal side, the side chain of V113 of Snap25b is held in place simultaneously by hydrogen-bonding interactions to the main chain amide of V113 and to the carboxamide of N100 in ANK17 (Figure S4-A). The side chain of V113 also establishes favorable van der Waals (vdW) interactions with the side chain of I99 in ANK17 (Figure S4-B). The second region of interest for the binding of Snap25b to ANK17 is the QP dipeptide (Q116/P117). The carboxamide of Q116 in Snap25b establishes H-bond with both the main chain NH of D122 and the Oδ of E89 in ANK17 (Figure S4-A). The aromatic ring of W130 in ANK17 forms extensive vdW contacts with the Co hydrogens of P117. In addition, the Co hydrogens of R133 pack against the Cβ hydrogens of P117 (Figure S4-C). Finally, following P117, onto the C-terminal side of Snap25b, the main chain A118 packs against L123 of ANK17 possibly involving hydrophobic interactions (Figure S4-D).
Overall, the structure of the putative recognition domain of Snap25b with ANK17 reveals the importance of several residues for both ANK17 as well as Snap25b, with the W130/P117 establishing the most extensive contacts. It also lends structural insight into the existing body of mutagenesis data (Gonzalo et al., 1999; Lemonidis et al., 2015).
Single point mutations of ANK17 residues prevent binding to full-length Snap25b in vitro
The crystal structure of the complex between ANK17 and Snap25b110–120 highlights the possible involvement of several residues from both partners of the binding interface. Analysis of the van der Waals interactions using the small-probe contact dot surfaces technique (Word et al., 1999) indicates that Y67 and W130 of ANK17 make the most extensive contacts with residues in Snap25b peptide (Figure 2 and Figure S4). Additionally, N100 and E89/D122 in ANK17 establish strong H-bonds with the main chain V113 and the side chain of Q116 of Snap25, respectively. We therefore performed alanine mutagenesis of these ANK17 residues and assessed their ability to interact with the full length Snap25b by ITC. Figure 3A shows the results of the ITC titrations of ANK17 mutants into Snap25b-WT. Alanine substitutions at position N100 and W130 in ANK17 completely abolish the interaction with Snap25b, but mutation of D122 to alanine only partially compromises the binding. This is consistent with our crystal structure, which shows D122 interacting with Q116 in Snap25b via the backbone amide and not with the side chain. The role of W130 is also confirmed by the ITC titrations: removal of the aromatic ring prevents Snap25b from binding to ANK17. Intriguingly, mutation of E89 to alanine did not appreciably alters the binding affinity, although in our structure E89 interacts via H-bond with Q116 in Snap25b. In order to shed light on this discrepancy, we crystallized ANK17-E89A mutant in complex with Snap25b111–120 peptide and solved the structure at 2.2 Å resolution (Table 1) The structure reveals that upon mutation of E89 to alanine, the side-chain of Q116 slightly rotates to form hydrogen bond with the side-chain of D122 in ANK17 (Figure S5), essentially compensating for the loss of H-bond to E89. Finally, mutation of Y67 to Alanine reduces the affinity to Snap25b approximately 5-fold.
Figure 3. ITC titration of ANK17 in Snap25b.
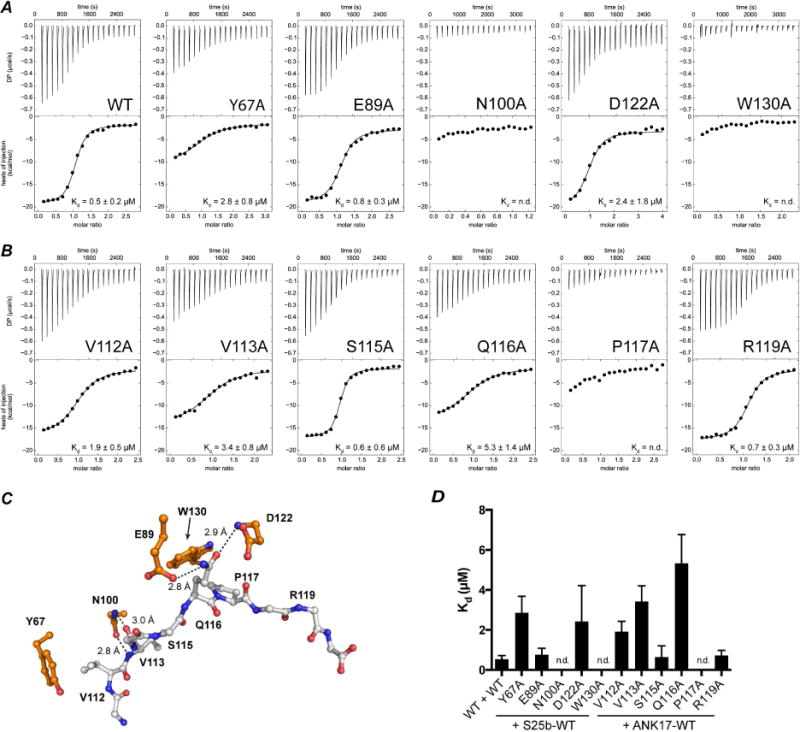
A) ANK17-WT or ANK17-mutants titrated into Snap25b. ANK17-N100A and ANK17-W130A showed only heat of dilution. (B) ANK17-WT titrated into Snap25b mutants. Snap25b-P117A showed only marginal heat of binding and we could not obtain any fitting of the integrated heats. (C) Position of mutated residues in ANK17 and Snap25b in the structure of the complex. (D) Histogram representing binding affinity (Kd) determined by ITC titrations (n.d.: not determined).
We also probed the importance of the residues within Snap25b towards the binding of ANK17. Figure 3B shows the titrations of six different alanine mutants between residues 110–120 in Snap25b. The largest reduction in binding affinity is seen with the P117A mutant, followed by the V113A and Q116A. Partial loss of affinity is also seen with the V112A mutation. Conversely, mutations of S115 or R119 to alanine do not seem to significantly affect the binding to ANK17.
In-cellulo binding assay to validate the interaction surface
We decided to extend our mutational analyses in the context of full length DHHC17 and Snap25b by probing the DHHC17/Snap25b interaction using a cell-based pull-down assay with proteins expressed in mammalian cells. In this assay, GFP-DHHC17 (with a 10xHis tag at the N-terminus) is used to pull-down GFP-Snap25b after co-transfection in HEK-293T cells (Figure 4A). The bottom two panels of Figure 4B show the expression levels of DHHC17 and Snap25b after detergent lysis but prior to Co2+-affinity chromatography. DHHC17 expression levels appear to be equivalent in all cases. The top two panels of Figure 4B show the amount of eluted proteins after IMAC. The intensity of the Snap25b band is proportional to the affinity for DHHC17. Mutation of the Q116 or P117 in Snap25b to alanine severely compromises the ability of DHHC17 to pull-down Snap25b. Decreased interaction is also observed for the Snap25b V113A mutation. These results are remarkably similar to the calorimetric binding studies described above.
Figure 4. Pull-down assay to determine affinity of DHHC17 to Snap25b and effect of single-point mutations.
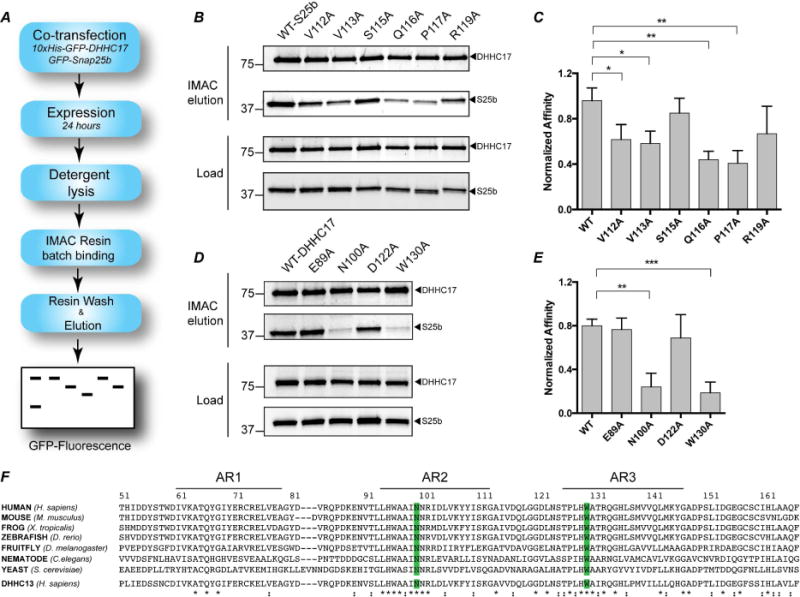
(A) Schematic depiction of the assay (B) 10xHis-GFP-DHHC17 and GFP-Snap25b-WT or mutants after 24 hours co-transfection in HEK-293T cells. Load: expression levels of each protein after lysis but prior to affinity purification. Co2+ affinity pull down: protein eluted from affinity resin after incubation for 16 hours and extensive washing. (C) Quantitation of the experiment in (B) after gel-densitometry analysis. The intensity of the band corresponding to Snap25b was divided the intensity of the corresponding DHHC17 band. Histograms represent average ± standard deviation (SD). One-way ANOVA with Dunnett’s multiple comparison test was used to compare mutants with WT (n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001). (D) and (E) Same as in (B) and (C), except that mutants of DHHC17 were used to pull-down Snap25b-WT. (F) Alignment of the first three ankyrin repeats of DHHC17 in different organisms and Human DHHC13. The conserved N100 and W130 residues are highlighted in blue. Sequence alignment was preformed using Clustal-omega (Sievers et al., 2011).
In order to get a more quantitative estimate of the relative binding affinities between the different constructs, we performed gel-densitometry of the bands and plotted the intensity of the Snap25b relative to the intensity of the DHHC17 band (Figure 4C). This was done to prevent misleading interpretation due to differential expression of DHHC17 across the plate. The three mutants of Snap25b that showed statistically significant reductions in affinity were V113A, Q116A and P117A.
We also corroborated our conclusions by carrying out the experiment with mutants of DHHC17 (Figure 4D–E). We tested two mutations that showed ablation of binding in the ITC experiments, N100A and W130A, as well as two other mutations, E89A and D122A, which did not significantly decrease binding affinity. The N100A and W130A mutants showed the largest decrease in affinity, as evidenced by the very low amount of Snap25b pulled down during affinity chromatography. Consistent with the ITC data, the E89A and D122A mutant did not show as strong an effect compared to wild type. Both N100 and W130 are highly conserved among different organism (Figure 4F), strongly underscoring their potential relevance in DHHC17 function. In summary, results from the pull down assay together with the ITC data with purified proteins are consistent with the structural model of the interaction between DHHC17 and Snap25b, and suggest the importance of residues W130 and N100 for the interaction.
S-acylation of Snap25b by DHHC17
The experiments described thus far investigated the importance of the residues, identified in our structure, for binding of ANK17 to Snap25b. However, substrate binding represents only the initial step in the enzymatic cycle, while the overall reaction results in the transfer of palmitate from palmitoyl-CoA to the target protein (Linder and Deschenes, 2007) (Figure 1B). To investigate the importance of the ankyrin repeat domain in the context of the full catalytic cycle, we performed metabolic labeling experiments in mammalian cells. A well-established method to detect protein palmitoylation is via click-chemistry reaction. In this assay, cells are incubated with a fatty acid containing an alkyne group (Figure 5A). The fatty acid is taken up by the cell and activated to fatty acid Coenzyme A. The latter serves as substrate for palmitoyl transferases. Upon lysis of the cells, acylated proteins can be detected by coupling the alkyne with a fluorescent azide molecule in a click-chemistry reaction (Martin, 2013). In this study, transfected HEK-293T cells were incubated with 17-Octadecynoic Acid (17-ODYA) for four hours and after lysis the alkyne was coupled to Rhodamine azide by click chemistry reaction. As shown in Figure 5B–D, DHHC17-WT readily transfers palmitate to Snap25b, while DHHC17-C467S (mutation of the cysteine of DHHC domain to serine) does not show any appreciable transfer. Mutations in the ankyrin repeat domain of DHHC17 that impair interaction with Snap25b (N100A, W130A) also show severely reduced levels of palmitoylation. In the structure, W130 appears to contact several residues in Snap25b through favorable van der Waals contacts and the interaction with P117 (mediated by the aromatic ring) appears to be the most crucial (Figure 5C). Accordingly, the loss of function of W130A can be recovered by mutation to phenylalanine, underscoring the importance of an aromatic side chain at position 130.
Figure 5. S-acylation of EGFP-Snap25b by DHHC17 and effect of DHHC17 mutations.
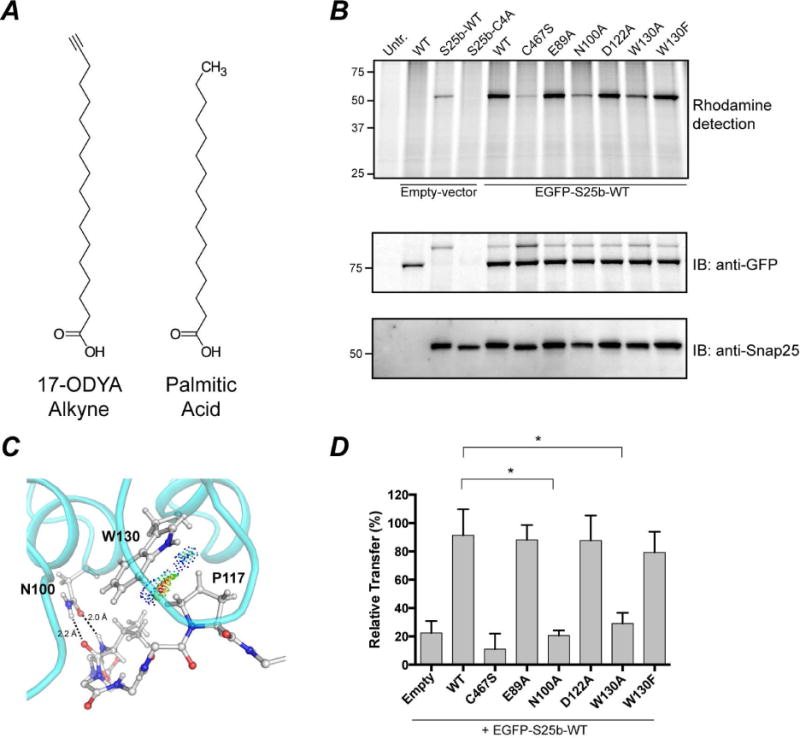
HEK-293T cells were transfected with EGFP-Snap25b, EGFP-Snap25b-C4A (cysteine-less) or empty vector and GFP-DHHC17 or GFP-DHHC17 mutants. Cells were incubated with 17-ODYA fatty acid alkyne for 4 h at 37°C. S-acylation with fatty acid alkyne was detected by coupling the alkyne fatty acid to Rhodamine-azide dye via click chemistry reaction. Isolated proteins were resolved by SDS/PAGE and transferred to PVDF membranes. (A) Chemical structures of 17-ODYA (17-Octadecynoic Acid) and palmitic acid. (B) Representative image of a typical experiment is shown. Click chemistry signal (Top), anti-GFP immunoblot (Middle), and anti-Snap25 immunoblot (Bottom). (C) van der Waals contacts between W130 and P117. Contact dots were obtained using the method of Richardson and co. (Word et al., 1999). (D) Quantification of Snap25b S-acylation. The intensity of the Snap25b band in the immunoblot was divided by the intensity of the Snap25b band in the rhodamine detection gel. Histograms represent average ± standard deviation (n=3). One-way ANOVA with Dunnett’s multiple comparison test was used to compare mutants with WT (n.s., p ≥ 0.05, *p < 0.05).
Taken together this experiment suggests that disruption of substrate interactions prevents the palmitoylation of Snap25b. Interestingly, we did not detect any autoacylation of DHHC17 using this method. This could be due to a fast turnover of palmitate from DHHC17 (not detectable by our end-point assay) or it could be due to different kinetic mechanism for DHHC17 compared to other DHHCs.
An intriguing question arising when studying DHHC17 is whether it follows the same kinetic model proposed for other DHHCs. It has been proposed that some DHHCs (i.e. Erf2/Erf4, DHHC2, DHHC3 and DHHC15) use a two-step Ping-Pong mechanism to transfer palmitate to protein substrates (Jennings and Linder, 2012; Mitchell et al., 2010). In this model, the DHHC enzyme interacts with palmitoyl-CoA and forms a palmitoylated enzyme intermediate. Upon encountering a protein substrate, the enzyme transfer the palmitate group to the protein substrate and becomes available for another round of palmitoylation. The hallmark of this model is a stable palmitoylated-DHHC intermediate.
To test whether DHHC17 interacts with palmitoyl-CoA in a similar fashion as other DHHCs, we expressed DHHC17 and DHHC15 in HEK-293T cells and purified the two enzymes by metal affinity chromatography. Upon incubation of similar amounts of DHHCs with 1 μM of NBD-palmitoyl-CoA (a fluorescent analog of palmitoyl-CoA), a strong band appears for DHHC15, while very low labeling levels of DHHC17 are detected (Figure S6-A). However, when the same reaction is carried out in the presence of purified Snap25b-WT, DHHC17 readily transfers NBD-palmitate to Snap25b (Figure S6B-C). In both instances, DHHC17 does not appear to get autopalmitoylated to appreciable levels. Taken together, these data suggest that DHHC17 might use a different mechanism for the transfer of palmitate to Snap25b compared to DHHC15. We speculate that for DHHC17, the binding of a protein substrate might be necessary before palmitoyl-CoA can interact with the enzyme.
Role of conserved motif for the interaction between ANK17 and Huntingtin
Our structural and functional studies were carried out with Snap25b as a model substrate. However, given the high degree of conservation of W130 in DHHC17 (Figure 4F) and the existence of a consensus motif in protein substrates, we decided to test the importance of the W130 in ANK17 for the interaction with Huntingtin (Htt). Htt is a scaffolding protein whose dysfunction causes Huntington’s disease and is one of the most prominent substrates of DHHC17 (Yanai et al., 2006). The canonical DHHC17 substrate recognition motif in Htt is GHDIITEQPRSQ (Lemonidis et al., 2015). The QP dipeptide is located between residues 493 and 504 in full-length Huntingtin (23 polyQ species). We purified and tested three different constructs of Htt in ITC experiments with wild-type ANK17 domain: 1) wild-type Htt1–548 (23 polyQ), 2) Htt1–492 (a C-terminally truncated version that takes away the canonical sequence motif) and a Q500P501 alanine double mutant (Figure 6). Wild-type Htt binds ANK17 with an affinity of approximately 0.5 μM, but neither the truncated form nor the double mutant show any appreciable binding. We also analyzed binding of ANK17 with the peptide fragment from residues 493–504 of Htt (GHDIITEQPRSQ) to investigate whether interactions with the canonical recognition sequence can recapitulates most of the interactions between ANK17 and Htt1–548. The peptide binds with an affinity of ~2 μM, very similar to that of Htt1–548. In the case of Snap25b, interaction with the QP dipeptide is strongly mediated by W130 of ANK17, as suggested by our structural and functional analyses. When we tested the W130A mutant of ANK17, it showed no discernible signs of binding to wild-type Htt, thus confirming that the same Pro-Trp interaction is likely critical for interactions of ANK17 with Htt. Taken together these results suggest the general importance of the W130 in the interactions of DHHC17 with protein substrates bearing the QP dipeptide in the canonical binding motif.
Figure 6. ITC titrations for the binding of ANK17 to Huntingtin N-terminal region.
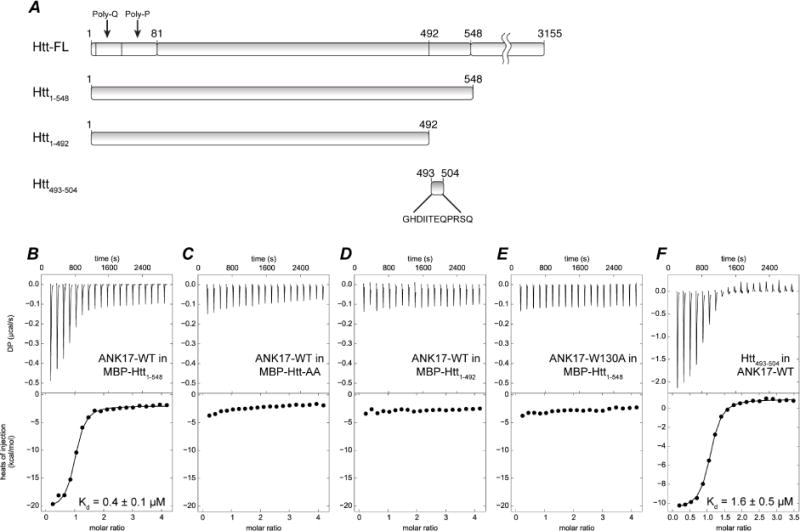
(A) Constructs used in the titrations. ANK17 titrated into (B) MBP-Htt1–548, (C) MBP-Htt1–548-Q500A-P501A, (D) MBP-Htt1–492. (E) ANK17-W130 titrated into MBP-Htt1–548 (F) Peptide corresponding to amino acids 493–504 titrated into ANK17.
Discussion
Since the discovery of the first S-palmitoylating enzymes by the Deschenes and Linder groups (Lobo et al., 2002), there has been an increasing number of protein substrates reported to be enzymatically palmitoylated (Greaves and Chamberlain, 2011). The absence of a distinct palmitoylation motif and lack of structural details for the interaction of DHHC enzymes and their substrates has hampered the understanding of the palmitoylation process as well as slowed down the development of DHHC specific small-molecule regulators. DHHC17 presents an ideal scenario for understanding enzyme/substrate interactions since a clear substrate binding domain (the N-terminal ankyrin repeat domain) is present. The synaptosomal protein Snap25b is a major target of DHHC17 (as well as other DHHCs), with the palmitoylation of the four cysteines being necessary for the proper targeting of Snap25b to the presynaptic membrane and subsequent assembly of the SNARE complex (Gonzalo and Linder, 1998). Moreover, the recently discovered substrate recognition motif of Snap25b is necessary and sufficient for DHHC17-Snap25b interactions (Lemonidis et al., 2014). The objective of the present work was to determine the structural and biophysical underpinnings of the interaction between ANK17 and Snap25b and extend the results to Huntingtin, another substrate of DHHC17.
Although the complex between ANK17 and Snap25b can be purified by size exclusion chromatography, we were not successful in crystallizing such complex. It is likely that the highly dynamic nature of the N- and C-terminal SNARE domains prevents stable packing of the two proteins. Indeed, the crystal structure of ANK17 bound to Snap25b111–120 agrees with this hypothesis. The structure shows that the critical part of the interaction is located between residues 112–117 of Snap25b, while the C-terminus of the peptide projects away from ANK17. We speculate that the engagement of full-length Snap25b with DHHC17 follows a similar arrangement. The interactions described above mediate initial recognition of the Snap25b with the ANK17 domain of DHHC17, thus bringing the four cysteines between residues 85 and 92 in Snap25b proximal to DHHC domain. The cysteines subsequently get palmitoylated and Snap25b is thereby targeted to the presynaptic membrane where it associates with Syntaxin and Synaptobrevin during formation of the SNARE complex. It must be noted however that palmitoylation of the cysteines in Snap25b is a necessary but not sufficient event for proper targeting of Snap25b to the plasma membrane. Recent evidence has pointed to the importance of electrostatic interactions between charged residues in Snap25b and membrane lipids (Weber et al., 2017). In the same report, the authors propose anchoring of Snap25b preceding the palmitoylation by plasma membrane DHHCs. It is however plausible that the same electrostatic interactions might help Snap25b localization to the Golgi membrane prior to palmitoylation by DHHC17.
The major determinants of ANK17/Snap25b interactions appear to be located between ankyrin repeat 2 and 3 and involve residues in both the loops and the a-helices of the concave side of ANK17. Overall, the backbone of ANK17 does not substantially change upon binding of Snap25b. This is consistent with other complexes involving ankyrin repeat proteins such as the tumor suppressor protein p16 binding to the cyclin-dependent kinases 4 and 6 (Russo et al., 1998) or the complex between gankyrin and S6 ATPase C-terminal domain (Nakamura et al., 2007), where binding does not induce any large structural rearrangement of the proteins.
Within the complex, the QP dipeptide of Snap25b establishes the most prominent interactions with ANK17. Specifically, hydrogen bonding interactions to the backbone by the Q116 side chain and hydrophobic interaction between the P117 side chain of Snap25b and the W130 of ANK17. Consistent with this, the QP sequence is absolutely conserved in all the substrates of DHHC17 containing the recognition motif (Lemonidis et al., 2015). The interaction between the pyrrolidine group of proline and the phenyl ring of phenylalanine has been previously described and can have a significant contribution to the binding energy (Riley et al., 2007). The presence of a Proline at position 117 likely serves the purpose of constraining Snap25b in order to wedge into the concave side of ANK17. Mutation of P117 to isoleucine retains some of the binding affinity for ANK17, suggesting that hydrophobicity plays a role in the interaction (Lemonidis et al., 2015). Our mutagenesis data support the central role of W130 in establishing favorable interactions with the P117. Overall, we propose a dual role for P117: 1) it imposes geometrical constraints to Snap25b so that both Q116 and V113 can engage in hydrogen bonding with ANK17 and 2) it forms a multipartite interaction site by favorable van der Waals contacts. Mutation of W130 to Alanine severely hampers the ability of DHHC17 to transfer palmitate to Snap25b, but mutation to a phenylalanine recapitulates the wild type. It is interesting to notice that a crucial Trp/Pro interaction (W188/P500) is present in the complex between the ankyrin-repeat protein ANKRA2 and the peptide from the 3M Syndrome Protein CCDC8, in which mutation of the tryptophan in ANKRA2 leads to complete loss of binding (Nie et al., 2015).
The other part of the Snap25b recognition motif comprises two hydrophobic residues at the N-terminal side. The side chains of V112 and V113 in Snap25b point in opposite directions with respect to the peptide backbone. On one side, the binding site is capped by favorable van der Waals interactions with the aromatic side chain of Y67 of ANK17 and on the other, the side chain of N100 forms hydrogen bonds with the backbone of V113, while the side chain appears to engage in hydrophobic interactions with I99 in ANK17.
In our investigation of the thermodynamics of binding between ANK17 and the N-terminal domain of Huntingtin, we show that a large part of the binding energy is due to the QP dipeptide in the C-terminal region of Htt1–548. Similar to Snap25b, the interaction seems to be mediated by the same residues on ANK17, reinforcing the point that these interactions are generally important for other substrates of DHHC17. A recent study by Hayden and coworkers (Sanders et al., 2014) has suggested the presence of more than one region in DHHC17 that can interact with Huntingtin N-terminal domain. Their study was conducted by co-immunoprecipitation of proteins expressed in mammalian cells. It is possible that the interactions between full-length DHHC17 with Huntingtin might involve other regions of DHHC17 besides the ankyrin repeat domain. In vitro investigation of the full-length DHHC17 and its substrate will likely elucidate this discrepancy. In addition, our in vitro binding data were conducted with Htt fused to maltose binding protein for stability reasons. We cannot currently exclude interference of the fusion tag in the binding process.
Our structural model correctly predicts the effect of mutations on the ability of DHHC17 to transfer palmitate to Snap25b. Using metabolic labeling of HEK-293T cells co-expressing DHHC17 and Snap25b-WT, it was possible to follow both the transfer of palmitate to Snap25b as well as autopalmitoylation of DHHC17. As expected, the presence of mutations that compromise binding of Snap25b to ANK17 also results in low levels of palmitate transfer. The two-step reaction cycle proposed in the literature for several DHHCs (Jennings and Linder, 2012; Mitchell et al., 2012), implies that autopalmitoylation of DHHC enzymes is independent from the subsequent palmitate-transfer step. However, in our experiments we notice that DHHC17 does not become autopalmitoylated to the same extent as DHHC15. This behavior is consistent with what was recently observed comparing the autoacylation of several DHHCs and their ability to transfer palmitate to Snap25b (Greaves et al., 2017). We confirmed this by comparing the reactivity of DHHC17 and DHHC15 to NBD-palmitoyl-CoA. DHHC17 does not become palmitoylated to an appreciable extent whereas DHHC15 rapidly reacts with NBD-palm-CoA. DHHC15 is a typical DHHC palmitoyl transferase that reacts quickly with palmitoyl-CoA to form a palmitoylated enzyme intermediate. This intermediate is capable of transferring palmitate to a protein substrate such as Snap25b (Greaves et al., 2017). Although DHHC17 does not quickly react with palmitoyl-CoA to form the acylated enzyme intermediate, it binds strongly to its substrate and it is capable of transferring palmitate. We therefore propose that the presence of a discrete substrate-binding domain such as the ANK17 in DHHC17 serves a regulatory purpose: only when suitable protein substrates are present, can the enzyme react with palmitoyl-CoA and complete the catalytic cycle.
Although there is no sequence based predictor of palmitoylation, a survey of palmitoylation sites indicates that the majority of them are proximal to the membrane and thus, by spatial constraints, proximal to the active site of DHHC enzymes. For a large number of substrates that are not integral membrane proteins, this initial placement in the vicinity of the membrane is caused by an auxiliary lipidation or a combination of hydrophobicity and charge-based interactions. Several substrates of DHHC17, however, lack another auxiliary lipidation site. Thus the interaction between the ankyrin repeat domain of DHHC17 with the canonical sequence motif (ΨβXXQP) on substrates, might serve to bring the target cysteines close to the active site of DHHC17 for palmitoyl transfer. The structural and biophysical studies presented here provide the first atomic insights into the chemistry of these interactions. Although knockout of single DHHC genes do not have a major effect, knockout of DHHC17 causes profound disruption of synaptic function and a dual knockout of DHHC13 and DHHC17 causes embryonic lethality. It is tempting to speculate that in absence of these two DHHC enzymes that use a distinct mode of substrate engagement, other DHHCs cannot adequately make up for their absence.
STAR Methods text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anirban Banerjee (anirban.banerjee@nih.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK-293T cells (ATCC CRL-3216) were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum, 2mM L-glutamine, 1% Penicillin/Streptomycin at 37°C with 5% CO2 and 70% relative humidity.
METHODS DETAILS
Peptide synthesis
A peptide corresponding to amino acid 111 to 120 of Snap25b (GVVASQPARV) and a peptide corresponding to amino acid 493 to 504 of Huntingtin (GHDIITEQPRSQ) were synthesized by solid phase synthesis (CPC scientific, Sunnyvale, CA). Peptides purity was >95% by HPLC. Lyophilized peptides were stored at −80°C and dissolved in the appropriate buffer prior to use.
Cell Culture and Transfection
HEK-293T cells (ATCC CRL-3216) were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum, 2mM L-glutamine, 1% Penicillin/Streptomycin at 37°C with 5% CO2 and 70% relative humidity. HEK-293T cells were transfected at 80% confluence with Polyethylenimine transfection reagent (25 kDa linear PEI, Polysciences, Inc., cat. No. 23966) at a ratio of PEI:plasmid of 3:1 (w/w).
Constructs used for Isothermal Titration Calorimetry (ITC)
The gene for human DHHC17 was obtained from Open biosystems (GE Lifesciences) and the cytoplasmic domain of human DHHC17 corresponding to residues K50 to D288 was cloned by digestion/ligation using NdeI-XhoI into a pET28b vector (Novagen). The original TEV cleavage site was changed to a Prescission cleavage site. The final vector (pET28-Prx) contained a 6xHis-tag followed by a Ser-Ser-Gly linker, the Prescission cleavage site and His-Met left from NdeI ligation (Nucleotide sequence before K50: CATCATCATCATCATCACAGCAGCGGCCTGGAAGTTCTGTTCCAGGGGCCCCATAT G). A vector containing the sequence for human Snap25b was obtained from Addgene (Plasmid #53235). NdeI was introduced before the first Methionine and the gene was cloned into pET28-Prx by digestion/ligation using NdeI/BamHI. (Linker regions were identical to ANK17 construct). Single point mutations of ANK17 and Snap25b were generated using QuikChange site directed mutagenesis kit (Agilent technology) following the manufacturer’s instructions. All clones were confirmed by DNA sequencing.
The complementary DNA encoding for full-length Htt (with 23 glutamines) was obtained from the Coriell Institute (Catalog ID CH40006). Fragment corresponding to amino acids 1–548 (Htt1–548) was inserted downstream of Maltose Binding Protein in a pET28b vector with N-terminal 6-His tag (Nucleotide sequence between 6xHis tag and first three aa of MBP: CATCACCATCACCATCACGATTACATG). Htt gene was preceded by a TEV cleavage site (ENLYFQG). (Nucleotide sequence between TEV site and first three aa of Htt: GAAAACCTGTATTTTCAGGGCATGGCGACC). Clones were confirmed by DNA sequencing. Double point mutations (Q500A and P501A) were engineered using site directed mutagenesis (Agilent). The C-terminal truncation was obtained by inserting a stop codon after amino acid 492.
Constructs used for pull-down assay
For expression in HEK-293T, all genes were cloned into a pEGFP vector (Addgene) containing a CMV promoter and a N-terminal 10xHis tag followed by eGFP. (Nucleotide sequence between 10xHis tag and eGFP: CATCACCACCACCATCATCATCACCACCATACTGCTGCCGCCGCTGTG). The human DHHC17 gene (accession number: NP_056151) was ligated downstream of eGFP using XhoI/EcoRI. The construct contained a Prescission site between eGFP and DHHC17. (Sequence between Prescission site and DHHC17: TTGGAAGTTTTGTTTCAAGGTCCATCTCGAGCCACCATG). The gene for DHHC15 (accession number: NM_144969) was obtained from Open biosystems (GE Lifesciences) and was inserted using the same restriction sites and contained the same tags and linkers. Snap25b was obtained from Addgene (Plasmid #53235). Snap25b was subcloned into the same pEGFP vector as DHHC17, except that 10xHis-tag was not used. All clones were confirmed by DNA sequencing. Single point mutants of both DHHC17 and ANK17 were generated by site directed mutagenesis, as described above.
Protein expression and purification
ANK17 and Snap25b in pET28-Prx vector were transformed in E.coli BL21(DE3)-Gold and plated onto LB-Kanamycin plates. A single colony was used to inoculate 5mL of LB (with 50 μg/mL of Kanamycin) and cells were grown in shaker incubator at 37°C for 20 hours. One liter of LB-Kan was inoculated with 2 mL of preculture in a 3-L baffled flask and cells were grown in shaker incubator at 37°C and 240 rpm until OD600 was around 1.0. Cells were cooled to 30°C and induced with 0.5mM of IPTG. After 5 hours, cells were harvested by centrifugation at 8000 × g for 10 minutes at 4°C. Cell pellets were flash frozen in liquid nitrogen and stored at −80°C.
For a typical purification, 10 grams of frozen pellet were dissolved by stirring at 4°C (30–40 minutes) in 100mL of lysis buffer containing: 50mM Tris-HCl pH 8.0, 250mM NaCl, 5% glycerol, 5mM β-mercaptoethanol, protease inhibitor cocktail (0.1mg/ml deoxyribonuclease I, 0.1mg/ml pepstatin, 1ug/ml leupeptin, 1ug/ml aprotinin, 1mM benzamidine and 0.1mg/ml soy trypsin inhibitor and 1mM PMSF) and 10mg lysozyme. Resuspended cells were disrupted by sonication with a Q700 Ultrasonic Processor (QSonica) equipped with flat tip using 10 cycles of 5 seconds ON and 30 seconds OFF (50% Amplitude and 4000J total energy). Sonicated slurry was stirred for 20 minutes at 4°C and subsequently centrifuged at 38000 × g for 30 minutes at 4°C. Pellets were discarded and supernatant was batch-bound to 5mL of Ni-NTA resin (previously equilibrated with binding buffer containing 50mM Tris-HCl pH 8.0, 250mM NaCl, 5% glycerol, 5mM β -mercaptoethanol) by rotation at 4°C for 1 hour. Resin slurry was applied to a plastic column and buffer was allowed to flow by gravity. Resin was washed with 10 volumes (around 50 mL) of binding buffer, 10 volumes of binding buffer with 10mM Imidazole, 10 volumes of binding buffer with 30mM imidazole and the protein was finally eluted with binding buffer containing 250mM imidazole. The amount of total protein eluted was quantitated by absorption spectrophotometry at 280nm using extinction coefficients of 48670 M−1cm−1 for ANK17 and 7240 M−1cm−1 for Snap25b. Eluted proteins were incubated at 4°C with rotation for 20 hours in the presence of 1mg/mL of Prescission protease to remove the 6xHis tag. Completeness of cleavage was assessed by gel shift using 12% acrylamide SDS-PAGE after Coomassie staining. Completed cleavage reactions were dialyzed against 4 liters of binding buffer for 15 hours at 4°C to remove imidazole. The dialyzed protein solution was re-applied to 5mL of Ni-NTA resin equilibrated with binding buffer and incubated with rotation at 4°C for 1 hour. Resin was loaded onto a column and flowthrough was collected. At this point the protein was concentrated to around 20 mg/mL using an Amicon concentrator (MWCO 10kDa, 15mL). Concentrated protein was centrifuged at 21000x g for 20min at 4°C to remove any precipitated particles. Around 200μL of concentrated protein was injected onto Superdex-200 equilibrated with SEC buffer (for crystallization: 50mM Tris-HCl pH, 50mM NaCl, 5% glycerol, 1mM DTT, for ITC: 1X PBS pH 7.4 with 1 mM DTT). Fractions corresponding to the protein peak were collected and concentrated using Amicon spin concentrators (MWCO 10kDa). The concentration of purified proteins was determined using the 660nm Assay Kit (Thermo Fisher).
MBP-Htt clones were transformed into E.coli BL21-DE3-Gold cells and plated onto LB-Kanamycin plates. Single colonies were used to inoculated 100mL of LB-Kanamycin media and cells were grown 15 hours at 37°C and 25 0 rpm shaking. Ten ml of preculture were used to inoculate 1 liter of LB-Kanamycin and cells grown to an OD600 of 1. Cultures were cooled on ice, isopropyl β-d-1-thiogalactopyranoside (IPTG, Sigma-Aldrich) was added to a concentration of 1 mM, and cultures were placed in an incubator at 12°C and 250 rpm for 15 h. Bacteria were collected by centrifugation and lysed by sonication after resuspension in PBS with 1% Tween 20 and protease inhibitors (Same inhibitors as for ANK17 expression). Lysates were cleared by centrifugation (30 min, 4°C, 38,000 g) and incubated with 3mL of Ni2+-NTA resin for 1 h at 4°C with rotation. Resin was washed once with 2 0 resin volumes of wash buffer (PBS, 1% Tween 20, 5mM 2-mercaptoethanol), 20 resin volumes of with wash buffer with 10mM imidazole, 20 resin volumes of wash buffer with 50mM imidazole and finally eluted with 5mL of wash buffer with 200mM Imidazole. Eluted MBP-Htt was concentrated and injected onto equilibrated Supredex-200 column as described for the purification of ANK17 and Snap25b (see above for details).
Sedimentation velocity analytical ultracentrifugation
A 90 μM solution of Snap25b-C4A was studied by sedimentation velocity in 50 mM Tris (pH 7.4), 100 mM and 1 mM TCEP. Experiments were conducted at 50,000 rpm and 20°C on a Beckman Coulter ProteomeLab XL-I analytical ultracentrifuge following standard protocols (Zhao et al., 2013a). The sample was loaded into a cell with a 12-mm (400μL) double-sector charcoal-filled Epon centerpiece, placed into an 8-hole An-50 Ti rotor, and thermally equilibrated at zero speed. Absorbance and interference velocity scans were subsequently acquired at speed - absorbance data were collected in a continuous mode as single measurements at 280 nm using a radial spacing of 0.003 cm. Sedimentation data were temporally corrected (Zhao et al., 2013b) with REDATE 1.0.0 (http://biophysics.swmed.edu/MBR/software.html) and analyzed in SEDFIT 15.01c (https://sedfitsedphat.nibib.nih.gov) (Schuck, 2000) in terms of a continuous c(s) distribution of sedimenting species using an uncorrected s range of 0 to 5 with a linear resolution of 100 and a maximum entropy regularization confidence interval of 0.68. Excellent fits were observed with root mean square deviations of 0.0048 absorbance units and 0.012 fringes. The partial specific volume Snap25b was calculated based on the amino acid composition using SEDNTERP (Cole et al., 2008) (http://sednterp.unh.edu/); the solution density and viscosity were also calculated in SEDNTERP based on the buffer composition.
Isothermal Titration Calorimetry
Samples for ITC were prepared as follows: ANK17 purified by size exclusion chromatography in ITC buffer (1X PBS, 1mM DTT) was concentrated to 300μM in an Amicon spin concentrator (10kDa MWCO) and the solution was degassed under vacuum for 20min prior to loading onto calorimeter. Snap25b or MBP-Htt was prepared identically to ANK17, except that the final concentration was less than 30μM. For ITC experiments involving S25b peptide, the peptide was dissolved in size exclusion buffer and degassed for 20 min under vacuum. A MicroCal i200 microcalorimeter (Malvern Instruments) was used for all experiments. Reference temperature was set at 25°C. Syringe contained ANK17 whereas Snap25b was loaded into the sample cell. Each injection was 2μL and the spacing between injections was set at 150 seconds. Twenty total injections were performed for each experiment. Raw data were analyzed using NITPIC v1.2.0 (Brautigam et al., 2016) and the resulting integrated heats were exported to SEDPHAT v12.1b (Brautigam et al., 2016). Non-linear regression analysis was performed assuming one binding site per molecule. Data were plotted using GUSSI v1.1.0 (Brautigam et al., 2016). A minimum of two or three titrations were performed for each construct pair. Thermodynamic parameters are reported as the average and standard deviation.
Pull-down assay by Co2+ affinity chromatography
Prior to co-transfection, HEK-293T cells were split at a density of 0.5 × 106 cells/ml in 6-well plates with 2 mL/well of supplemented DMEM. Cells (at around 80% confluence) were co-transfected with 2 μg of pEGFP-DHHC17-WT (or mutants) and 0.5 μg of pEGFP-Snap25b-WT (or mutants) using three-fold excess of PEI-max by weight, as cotransfection agent. Cells were incubated at 37°C and 5% CO2 and 70% relative humidity for 24 hours. Expression levels were qualitatively assessed by imaging GFP fluorescence using an inverted microscope (EVOS FL Cell Imaging System, ThermoFisher Scientific with Ex. 470nm/Em. 510nm). Prior to lysis, cells were washed twice with 1mL ice-cold PBS and resuspend by pipetting in 1mL of ice-cold PBS and transferred to microfuge tube. Cells were centrifuged 5 minutes at 1000 × g and the supernatant removed. Pellets were resuspended in 400μL of lysis buffer (50mM Hepes pH 7.8, 50mM NaCl, 5% glycerol, 5mM β-mercaptoethanol, protease inhibitors cocktail, 40mM Dodecylmaltoside) and incubated at 4°C for 2 hours with rotation. Tubes were centrifuged at 21000 × g for 30 minutes at 4°C. Samples from the supernatant were used to quantitate the amount of expressed proteins. Supernatants were mixed with 100μL of Talon resin and incubated for 15 hours at 4°C with rotation. Resin was washed with 20 bed volumes of wash buffer (same as lysis buffer but containing 2mM DDM and 10mM or 30mM Imidazole). Proteins were eluted by addition of 100μL of elution buffer (same as wash buffer but with 400mM imidazole. Eluted proteins were separated on a 12% SDS-PAGE (TGX, BioRad) and after rinsing with distilled water, the GFP signal was detected using a ChemiDoc™ MP System (BioRad) with Blue Epi illumination and a 530/25 filter.
Metabolic labeling for palmitate transfer detection
HEK-293T were cultured and co-transfected as described for the pull-down assay above. Typically, 6-well plates were used for the assay. Twenty-four hours post-transfection, growth medium was exchanged with two milliliters of serum-free DMEM containing 100 μM of 17-Octadecynoic Acid (17-ODYA - Sigma). Cells were incubated at 37°C, 5% CO2 and 70% relative humidity for 4 hours and subsequently lysed in 200 μl of detergent-containing buffer (same as described for the pull-down assay). After metabolic labeling, 200 μl of click-chemistry mix containing: 2 mM CuSO4, 2 mM freshly prepared TCEP, 70 μM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA - Sigma), 100 μM tetramethylrhodamine azide (Rhodamine-azide - Lumiprobe) was added to 200 μL of lysis supernatant and incubated for 1 hour at room temperature with rotation. Proteins were precipitated with acetone and the resulting pellet was dissolved in 100 μl of SDS-sample buffer without reducing agent. Samples were denatured by heating for 5 minutes at 95°C. Thirteen μl were of each sample were loaded onto 12% SDS-Page gel. Gels were visualized using a ChemiDoc™ MP System (BioRad). Rhodamine-labeled proteins were detected with Red-epi illumination and a 695/55 filter. The same gel was subsequently transferred to PVDF membrane followed by incubation with blocking buffer (1X TBS, 0.05% Tween-20 and 5% dry milk). Next, the portion of membrane corresponding to DHHC17 was incubated with anti-GFP antibody (Thermo Fisher #A-11122), while the portion of membrane corresponding to Snap25b was incubated with anti-Snap25b antibodies (Thermo Fisher #MA5-17609) overnight at 4°C with gentle agitation (both primary antibodies were diluted 1000-fold in 1X TBS, 0.05% Tween-20 and 5% dry milk). Membranes were washed three times with wash buffer (1X TBS, 0.05% Tween-20) and incubated with fluorescent secondary antibodies (1000-fold dilution in wash buffer) for 1 hour at 4°C with gentle agitation. Alexa-680 conjugated secondary antibodies (Thermo Fisher #A-21057 and #A-21076) were used in both cases and membranes were imaged on a Chemidoc (BioRad) equipped with a Green-epi illumination and a 605/50 filter. Quantitation of protein bands was carried out using ImageJ software.
In vitro autoacylation and palmitate transfer assay
10xHis-GFP-DHHC17, 10xHis-GFP-DHHS17 and 10xHis-DHHC15 were cloned in pEGFP vector (Addgene) under the control of a CMV promoter. HEK-293T cells were grown in 10-cm tissue culture plates and transfected with 10 μg of each plasmid in the presence of 30 μg of PEI. After 48 hours, cells were harvested and lysed in the presence of 40 mM DDM detergent (same buffer as reported for the pull-down assay described above). All steps following harvesting of cells were carried out at 4°C. Clarified lysate was bound to 200 μL of Talon metal affinity resin for two hours with rotation and subsequently. The bound protein was washed with 10 column volumes (CVs) of low-imidazole wash buffer (same as lysis buffer but containing 2 mM DDM and 10 mM imidazole) and 10 CVs of high-imidazole wash buffer (2 mM DDM and 30 mM Imidazole). Finally, protein was eluted in elution buffer (2 mM DDM and 200 mM Imidazole). Eluted proteins were concentrated using 0.5-mL Amicon spin-column with a 10 kDa membrane cutoff. Protein concentration was determined with the 660-nm colorimetric assay (Thermo Fisher). To measure autoacylation of the enzymes, each enzyme was diluted to 0.1 μM in wash buffer containing 1mM DDM and NBD-palmitoyl-CoA (Avanti polar lipids) was added to a concentration of 1μM. Samples were incubated at room temperature (24°C) and aliquots were taken at different time points and mixed with SDS sample buffer in non-reducing conditions.
For in vitro Snap25b acylation assay, purified GFP-DHHC17 (100 nM) and GFP-DHHS17 (100 nM) were incubated with affinity purified Snap25b (1 μM) in reaction buffer (1X PBS, 1mM DDM) for 10 minutes at room temperature (24°C). Reaction was started by addition of 1 μM of NBD palmitoyl-CoA (20 μM stock in reaction buffer). Samples were taken at different time points and reaction quenched with non-reducing SDS sample buffer. Samples were separated in 12% SDS-page gels and fluorescence detected using a Chemidoc using Blue Epi illumination and a 530/25 filter.
Preparation of ANK17/Snap25b complex for crystallization
Individually purified ANK17 and Snap25b in crystallization buffer were mixed at equimolar concentration (750μM each) and incubated for 15 hours with rotation at 4°C. After incubation, the mixture was centrifuged for at 21000 × g for 30min to remove any precipitated particles and concentrated in Amicon spin-concentrators (MWCO 30kDa) to around 30 mg/mL. Concentrated complex was injected onto Superdex-200 equilibrated with crystallization buffer. Fraction corresponding to complex were pooled and concentrated to around 20mg/mL using spin-concentrator (Amicon MWCO 30kDa). Concentrated complex was centrifuged at 21000x g for 30min at 4°C prior to crystallization trials.
For the crystallization of ANK17/Snap25 peptide, purified ANK17 at a concentration of 750 μM was mixed with an equal volume of Snap25b peptide at a concentration of 1.5 mM in identical buffer. Protein mixture was rotate for 15 hours at 4°C. After incubation, the protein/peptide mixture was centrifuged at 21000x g for 30 min at 4°C. Neither size exclusion chromatography step, nor concentration was necessary at this stage.
Crystallization of ANK17/Snap25b111–120 was performed in hanging-drop by vapor diffusion method. Typically, 300nL of reservoir buffer and 300nL of protein solution were spotted in a 96-well cover (Grace) and plates were incubated at 4°C in Rockimager (Formulatrix). Crystals appeared after 24 hours and grew to maximum size of 400um × 60um × 60um, after four days. Best diffracting crystals were obtained in 100 mM Tris-HCl pH 7.5, 12.5% PEG-6000 and soaked in a cryogenic solution (contacting 100mM Tris-HCl pH 7.5, 15% PEG-6000, 30% glycerol) and immediately frozen in liquid nitrogen. Production and crystallization of ANK17-E89A/Snap25b111–120 was performed as described above. Best diffracting crystals of ANK17-E89A/Snap25b111–120 were obtained in 50 mM Glycine pH 9.0, 18.5% PEG-4000, soaked in cryogenic solution (containing 50mM Glycine pH 9.0, 21% PEG-4000, 30% glycerol) and immediately frozen in liquid nitrogen.
X-ray data analysis and model refinement
The diffraction data were collected at 1.0000-Å wavelength and cryogenic temperatures at the Advanced Proton Source, Argonne National Laboratory, beamline SER-CAT 22BM and 22ID. Data were scaled by using HKL2000 (Otwinowski and Minor, 1997) and further processed by using the CCP4 suite (Collaborative Computational Project, Number 4, 1994). Initial phases were determined by the molecular replacement package MOLREP using the coordinates of the ankyrin repeat domain of DHHC17 (Protein Data Bank entry 3EU9) as a search model. The protein model was further refined in Phenix, and the electron-density maps were examined in Coot (Emsley and Cowtan, 2004). Following refinement, 98.5, 1.5 and 0% of the backbone dihedral angles were within the favored, allowed and disallowed regions of the Ramachandran plot, respectively. All structural figures were prepared with PyMol (www.pymol.org/).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of bands for in-gel fluorescence and immunoblots was done by densitometry, using ImageJ (National Institutes of Health, Bethesda, MD) software. All statistical analyses were performed with Prism software (GraphPad).
One-way ANOVA with Dunnett’s multiple comparison test was used for comparisons of averages between wild-type and mutants.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the structures reported in this paper are PDBs: [5W7I] and [5W7J].
Supplementary Material
Highlights.
X-ray structure of ANK17 domain of DHHC17 bound to Snap25b111–120 recognition motif
In vitro and cell-based binding assays validate the structural model
Loss-of-binding mutants affect palmitate transfer in cells
Binding of Huntingtin1–548 to ANK17 share similarity with Snap25b
Acknowledgments
We thank Gisela Storz, Juan Bonifacino and Susan Buchanan for critical reading and helpful comments on the manuscript. Crystals were screened and data were collected at Southeast Regional Collaborative Access Team (SER-CAT) beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. We thank beamline staff SER-CAT for help with data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This work was supported by the Intramural Research Program of the National Institutes of Health, the National Institutes of Child Health and Human Development (R.V., J.-S.K., A.B.), the National Institute of Neurological Disorders and Stroke (R.V. and A.B.) and the National Institute of Diabetes and Digestive and Kidney Diseases (R.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions.
R.V., A.B. conceived the experiments. R.V., performed protein purifications, assays, ITC measurements, crystallizations and X-ray data collection. J-S. K. built and refined the models. R.G performed analytical ultracentrifugation. R.V. and A.B. wrote the manuscript. All authors reviewed the manuscript.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SJ, Almers W. Tracking SNARE complex formation in live endocrine cells. Science. 2004;306:1042–1046. doi: 10.1126/science.1102559. [DOI] [PubMed] [Google Scholar]
- Brautigam CA, Zhao H, Vargas C, Keller S, Schuck P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat Protoc. 2016;11:882–894. doi: 10.1038/nprot.2016.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL, Lary JW, Moody TP, Laue TM. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 2008;84:143–179. doi: 10.1016/S0091-679X(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. Journal of Computational Chemistry. 1998;19:319–333. [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Gao T, Collins RE, Horton JR, Zhang X, Zhang R, Dhayalan A, Tamas R, Jeltsch A, Cheng X. The ankyrin repeat domain of Huntingtin interacting protein 14 contains a surface aromatic cage, a potential site for methyl-lysine binding. Proteins. 2009;76:772–777. doi: 10.1002/prot.22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Linder ME. SNAP-25 palmitoylation and plasma membrane targeting require a functional secretory pathway. Molecular Biology of the Cell. 1998;9:585–597. doi: 10.1091/mbc.9.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Greentree WK, Linder ME. SNAP-25 is targeted to the plasma membrane through a novel membrane-binding domain. J Biol Chem. 1999;274:21313–21318. doi: 10.1074/jbc.274.30.21313. [DOI] [PubMed] [Google Scholar]
- Greaves J, Chamberlain LH. DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends in Biochemical Sciences. 2011;36:245–253. doi: 10.1016/j.tibs.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Greaves J, Munro KR, Davidson SC, Riviere M, Wojno J, Smith TK, Tomkinson NCO, Chamberlain LH. Molecular basis of fatty acid selectivity in the zDHHC family of S-acyltransferases revealed by click chemistry. Proceedings of the National Academy of Sciences. 2017;114:E1365–E1374. doi: 10.1073/pnas.1612254114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, et al. Huntingtin-Interacting Protein HIP14 Is a Palmitoyl Transferase Involved in Palmitoylation and Trafficking of Multiple Neuronal Proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Jennings BC, Linder ME. DHHC Protein S-Acyltransferases Use Similar Ping-Pong Kinetic Mechanisms but Display Different Acyl-CoA Specificities. Journal of Biological Chemistry. 2012;287:7236–7245. doi: 10.1074/jbc.M111.337246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ML, Tay CL, Rayner JC. Getting stuck in: protein palmitoylation in Plasmodium. Trends Parasitol. 2012;28:496–503. doi: 10.1016/j.pt.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Lemonidis K, Gorleku OA, Sanchez-Perez MC, Grefen C, Chamberlain LH. The Golgi S-acylation machinery comprises zDHHC enzymes with major differences in substrate affinity and S-acylation activity. Molecular Biology of the Cell. 2014;25:3870–3883. doi: 10.1091/mbc.E14-06-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonidis K, Sanchez-Perez MC, Chamberlain LH. Identification of a Novel Sequence Motif Recognized by the Ankyrin Repeat Domain of zDHHC17/13 S-Acyltransferases. J Biol Chem. 2015;290:21939–21950. doi: 10.1074/jbc.M115.657668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Lobo S, Greentree WK, Linder ME, Deschenes RJ. Identification of a Ras Palmitoyltransferase in Saccharomyces cerevisiae. Journal of Biological Chemistry. 2002;277:41268–41273. doi: 10.1074/jbc.M206573200. [DOI] [PubMed] [Google Scholar]
- Martin BR. Nonradioactive analysis of dynamic protein palmitoylation. Curr Protoc Protein Sci. 2013;73 doi: 10.1002/0471140864.ps1415s73. Unit14.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI. Self-association of the H3 region of syntaxin 1A. Implications for intermediates in SNARE complex assembly. J Biol Chem. 2001a;276:13273–13282. doi: 10.1074/jbc.M009636200. [DOI] [PubMed] [Google Scholar]
- Misura K, Gonzalez LC, May AP, Scheller RH, Weis WI. Crystal structure and biophysical properties of a complex between the N-terminal SNARE region of SNAP25 and syntaxin 1a. J Biol Chem. 2001b;276:41301–41309. doi: 10.1074/jbc.M106853200. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Mitchell G, Ling Y, Budde C, Deschenes RJ. Mutational Analysis of Saccharomyces cerevisiae Erf2 Reveals a Two-step Reaction Mechanism for Protein Palmitoylation by DHHC Enzymes. Journal of Biological Chemistry. 2010;285:38104–38114. doi: 10.1074/jbc.M110.169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Hamel LD, Ishizuka K, Mitchell G, Schaefer LM, Deschenes RJ. The Erf4 subunit of the yeast Ras palmitoyl acyltransferase is required for stability of the Acyl-Erf2 intermediate and palmitoyl transfer to a Ras2 substrate. J Biol Chem. 2012;287:34337–34348. doi: 10.1074/jbc.M112.379297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Hamel LD, Reddy KD, Farh L, Rettew LM, Sanchez PR, Deschenes RJ. Mutations in the X-linked intellectual disability gene, zDHHC9, alter autopalmitoylation activity by distinct mechanisms. J Biol Chem. 2014;289:18582–18592. doi: 10.1074/jbc.M114.567420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Nakano K, Umehara T, Kimura M, Hayashizaki Y, Tanaka A, Horikoshi M, Padmanabhan B, Yokoyama S. Structure of the Oncoprotein Gankyrin in Complex with S6 ATPase of the 26S Proteasome. Structure. 2007;15:179–189. doi: 10.1016/j.str.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Nie J, Xu C, Jin J, Aka JA, Tempel W, Nguyen V, You L, Weist R, Min J, Pawson T, et al. Ankyrin repeats of ANKRA2 recognize a PxLPxL motif on the 3M syndrome protein CCDC8. Structure. 2015;23:700–712. doi: 10.1016/j.str.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochimica Et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Riley KE, Cui G, Merz KM. An ab initio investigation of the interactions involving the aromatic group of the set of fluorinated N-(4-sulfamylbenzoyl)benzylamine inhibitors and human carbonic anhydrase II. J Phys Chem B. 2007;111:5700–5707. doi: 10.1021/jp067313m. [DOI] [PubMed] [Google Scholar]
- Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature. 1998;395:237–243. doi: 10.1038/26155. [DOI] [PubMed] [Google Scholar]
- Sanders SS, Hou J, Sutton LM, Garside VC, Mui KKN, Singaraja RR, Hayden MR, Hoodless PA. Huntingtin interacting proteins 14 and 14-like are required for chorioallantoic fusion during early placental development. Dev Biol. 2015;397:257–266. doi: 10.1016/j.ydbio.2014.11.018. [DOI] [PubMed] [Google Scholar]
- Sanders SS, Mui KKN, Sutton LM, Hayden MR. Identification of binding sites in Huntingtin for the Huntingtin Interacting Proteins HIP14 and HIP14L. PLoS ONE. 2014;9:e90669. doi: 10.1371/journal.pone.0090669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysj. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–U105. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate EW, Kalesh KA, Lanyon-Hogg T, Storck EM, Thinon E. Global profiling of protein lipidation using chemical proteomic technologies. Current Opinion in Chemical Biology. 2015;24:48–57. doi: 10.1016/j.cbpa.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Weber P, Batoulis H, Rink KM, Dahlhoff S, Pinkwart K, Söllner TH, Lang T. Electrostatic anchoring precedes stable membrane attachment of SNAP25/SNAP23 to the plasma membrane. Elife. 2017;6 doi: 10.7554/eLife.19394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. Journal of Molecular Biology. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, Schuck P. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci. 2013a doi: 10.1002/0471140864.ps2012s71. Chapter 20, Unit20.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ghirlando R, Piszczek G, Curth U, Brautigam CA, Schuck P. Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Analytical Biochemistry. 2013b;437:104–108. doi: 10.1016/j.ab.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.