

Summary
We leveraged IDH wild-type glioblastomas, derivative neurospheres, and single cell gene expression profiles to define three tumor-intrinsic transcriptional subtypes designated as proneural, mesenchymal, and classical. Transcriptomic subtype multiplicity correlated with increased intratumoral heterogeneity and presence of tumor microenvironment. In silico cell sorting identified macrophages/microglia, CD4+ T lymphocytes, and neutrophils in the glioma microenvironment. NF1 deficiency resulted in increased tumor-associated macrophages/microglia infiltration. Longitudinal transcriptome analysis showed that expression subtype is retained in 55% of cases. Gene signature-based tumor microenvironment inference revealed a decrease in invading monocytes and a subtype-dependent increase in macrophages/microglia cells upon disease recurrence. Hypermutation at diagnosis or at recurrence associated with CD8+ T cell enrichment. Frequency of M2 macrophages detection associated with short-term relapse after radiation therapy.
Graphical abstract
Wang et al. define three IDH wild-type glioblastoma-intrinsic gene expression subtypes, which are partly shaped by the tumor immune environment. NF1 deficiency results in increased macrophage/microglia infiltration. Comparison of matched primary and recurrent tumors reveals frequent expression subtype changes.
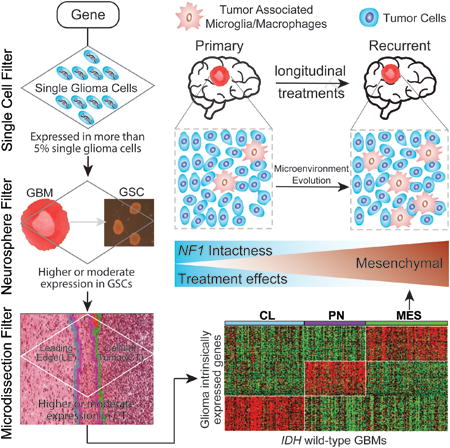
Introduction
The intrinsic capacity of glioblastoma (GBM) tumor cells to infiltrate normal brain impedes surgical eradication and predictably results in high rates of early recurrence. To better understand determinants of GBM tumor evolution and treatment resistance, The Cancer Genome Atlas Consortium (TCGA) performed high dimensional profiling and molecular classification of nearly 600 GBM tumors (Brennan et al., 2013; Cancer Genome Atlas Research, 2008; Ceccarelli et al., 2016; Noushmehr et al., 2010; Verhaak et al., 2010). TCGA identified common mutations in genes such as TP53, EGFR, IDH1, and PTEN as well as the frequent and concurrent presence of abnormalities in the p53, RB, and receptor tyrosine kinase pathways. Unsupervised transcriptome analysis additionally revealed four clusters, referred to as classical (CL), mesenchymal (MES), neural (NE), and proneural (PN), that were tightly associated with genomic abnormalities (Verhaak et al., 2010). The PN and MES expression subtypes have been most consistently described in the literature with PN relating to a more favorable outcome and MES relating to poor survival (Huse et al., 2011; Phillips et al., 2006; Zheng et al., 2012), but these findings were affected by the relatively favorable outcome of IDH mutant GBMs which are consistently classified as PN (Noushmehr et al., 2010; Verhaak et al., 2010). PN to MES switching upon disease recurrence has been implicated in treatment resistance in GBM relapse (Bao et al., 2006; Bhat et al., 2013; Ozawa et al., 2014; Phillips et al., 2006), but the frequency and relevance of this phenomenon in glioma progression remains ambiguous.
GBM tumor cells, along with the tumor microenvironment, together create a complex milieu that ultimately promotes tumor cell transcriptomic adaptability and disease progression (Olar and Aldape, 2014). The presence of tumor-associated stroma results in a MES tumor gene signature and poor prognosis in colon cancers (Isella et al., 2015). Furthermore, the association between MES gene expression signature and reduced tumor purity has been identified as a common theme across cancers (Martinez et al., 2015; Yoshihara et al., 2013). Tumor-associated macrophages (TAMs), including either of peripheral origin or representing brain-intrinsic microglia in glioma (Gabrusiewicz et al., 2016; Hambardzumyan et al., 2015), have been proposed as regulators of PN-to-MES transition through NF-κB activation (Bhat et al., 2013) and may provide growth factor-mediated proliferative signals which could be therapeutically targeted (Patel et al., 2014; Pyonteck et al., 2013; Yan et al., 2015).
Here, we explored the properties of the microenvironment in different GBM gene expression subtypes before and after therapeutic intervention.
Results
Transcriptomic analysis of glioma single cells, neurospheres, and tumor biopsies identifies GBM-specific intertumoral heterogeneity
We set out to elucidate the tumor-intrinsic and tumor microenvironment-independent transcriptional heterogeneity of GBMs by identifying genes uniquely expressed by glioma cells and not by tumor-associated host cells. We performed RNA sequencing of 133 single cells isolated from three GBMs (Lee et al., 2017) and compiled transcriptomes of an additional 672 single cells isolated from five GBMs (Patel et al., 2014). A set of 596 out of the 805 single cells passed quality control procedures and were determined to be single glioma cells (SGCs, Figure S1A). We observed that 14,656 of 22,870 unique genes were expressed in at least 5% of the 596 SGCs and were considered candidate bona fide glioma genes (BFGs) (Figure S1B).
To filter genes that were expressed by both GBM cells and the tumor microenvironment, we collected a cohort of 37 GBMs from which we derived glioma sphere-forming cell cultures (GSCs). Following RNA sequencing of this set, we performed pairwise gene expression comparison and identified 3,099 genes significantly overexpressed in GBM compared to their derivative GSCs (FDR adjusted t-test p value < 0.01). Removing these candidate microenvironment marker genes reduced the BFG list to 13,165 genes (Figure S1C).
Finally, we analyzed the RNA sequencing data of 30 cellular tumors and 19 matching leading edges of eight GBM surgery specimens from the Ivy Glioblastoma Atlas Project (Ivy GAP, http://glioblastoma.alleninstitute.org/). Cellular tumors are considered near 100% tumor cells versus no more than 10% tumor cells in the leading edge. We identified 5,978 genes significantly greater expressed in leading edge compared to matching cellular tumor (FDR adjusted t-test p values < 0.01), resulting in discarding 1,636 genes from the BFG list (Figure S1D). Of the 11,529 genes on the resulting BFG list, 7,425 genes are represented on the Affymetrix U133A microarray used to profile the TCGA GBM cohort. (Figure 1A, Figure S1E, Table S1).
Figure 1. Molecular classification of IDH-WT GBMs.
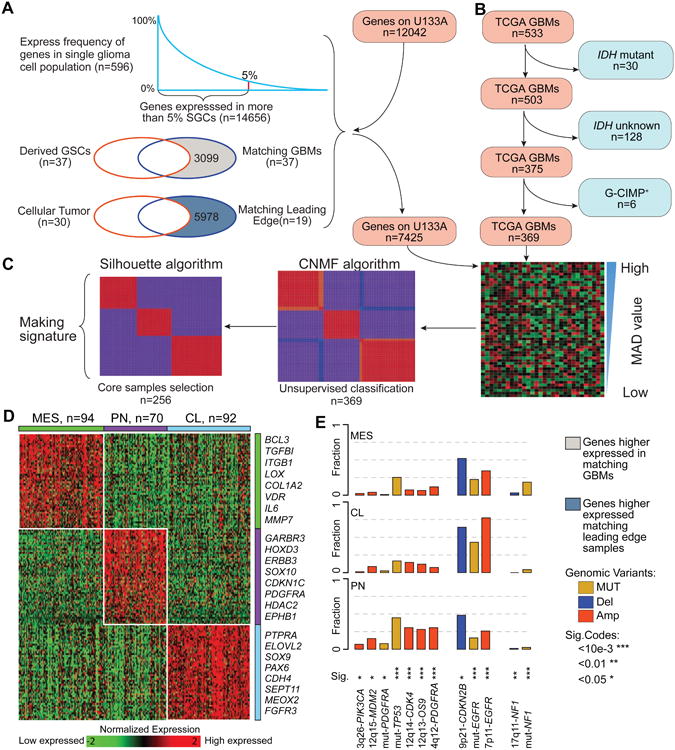
(A) Filtering tumor associated microenvironment genes. (B) Defining an IDH-WT GBM cohort in TCGA. (C) Overview of NMF clustering procedures. (D) Heatmap of 50-gene signatures by gene expression subtype. Representative genes are shown for each subtype. (E) Frequency of subtype related somatic genomic alterations. Chi-square test was used to calculate the distribution difference among three subtypes per genomic variant. See also Figure S1, Tables S1 and S2, and Method S1.
GBMs with IDH mutations (IDHmut) represent 5% of the cases and have distinct biological properties and confer favorable clinical outcomes compared to IDH wild-type (IDH-WT) GBMs (Brennan et al., 2013; Cancer Genome Atlas Research et al., 2015; Ceccarelli et al., 2016; Noushmehr et al., 2010). Using the filtered BFG/U133A set, we performed consensus non-negative matrix factorization clustering to identify three distinct subtypes comprising 369 IDH-WT GBMs (Figure 1B, Figure 1C, Table S1). When comparing the clustering result with the previously defined PN, NE, CL, and MES classification (Brennan et al., 2013; Verhaak et al., 2010), the three subtypes were strongly enriched for MES, PN, and CL GBMs, respectively (Figure S1F). Consequently, we designated the groups as MES, PN, and CL. None of the three subtypes were enriched for the NE subtype, suggesting the neural phenotype is non-tumor specific. The NE subtype has previously been related to the tumor margin where increased normal neural tissue is likely to be detected (Gill et al., 2014; Sturm et al., 2012), and such contamination might explain why the neural subtype was the only subtype to lack characteristic gene abnormalities (Brennan et al., 2013; Li et al., 2013). To be able to classify external GBM samples, we implemented a single sample gene set enrichment analysis (ssGSEA) based equivalent distribution resampling classification strategy using 50-gene signatures for each subtype (Figure 1D, Table S1) to assign each sample three empirical classification p values by which we determined the significantly activated subtype(s). The overlap between 50-gene signatures and the previously reported transcriptional subtype signatures (Verhaak et al., 2010) ranged from 42% to 54% (Figure S1G). We prepared an R-library to enable others to evaluate our approach (Method S1). To assess the robustness of our GBM subtype classification method, we compared cluster assignments of 144 TCGA GBM samples profiled using both RNA sequencing and Affymetrix U133A microarrays, and the assessment revealed 93% concordance (Figure S1H, Table S2). The 93% concordance was an improvement over the 77% subtype concordance determined using previously reported methods (Verhaak et al., 2010). In fact, classifying TCGA GBMs using the updated 50-gene signatures resulted in classification concordances ranging from 93% to 100% when comparing across different batches, different compositions of IDH-WT/IDHmut cases, and different RNAseq expression measurements (Figure S1I-S1K). Notably, we found high classification stability in small sample sizes, such as 85% concordance in cohorts of ten randomly selected samples (Figure S1L). We additionally evaluated the distribution of somatic variants across the three molecular subtypes (Figure 1E) and confirmed the strong associations between subtypes and genomic abnormalities in previously reported driver genes (Brennan et al., 2013; Verhaak et al., 2010).
Multi-activation of subtype signatures associated with intratumoral heterogeneity
We observed that 29/369 (8%) TCGA samples showed significant enrichment of multiple ssGSEA scores (empirical classification p value < 0.05), suggesting that these cases activate more than one transcriptional subtype (Figure 2A). To quantify such transcriptional heterogeneity, a score ranging from 0 to 1 was defined to quantitatively evaluate the simplicity of subtype activation based on order statistics of ssGSEA score. Samples with high simplicity scores activated a single subtype and those with lowest simplicity scores activated multiple subtypes. All multi-subtype TCGA samples scored simplicities of less than 0.05 (Figure 2A). To determine whether transcriptional heterogeneity associated with genomic intratumoral heterogeneity, we correlated simplicity scores, total mutation rates, and subclonal mutation rates. Included in the analysis were 224 TCGA GBMs with available whole exome sequencing data (Kim et al., 2015), and ABSOLUTE (Carter et al., 2012) determined high tumor purity (> 0.7) to equalize the mutation detection sensitivity (Aran et al., 2015). Although not significant (Wilcoxon rank test p value = 0.30), the total mutation rate was less in the bottom 50% of samples with lowest simplicity scores versus the top 50% with highest simplicity scores. Meanwhile, the subclonal mutation rate and fraction was significantly higher (Wilcoxon rank test p value = 0.03 and 0.02, respectively) in samples with lowest simplicity scores (Figure 2B, Table S3), suggesting that increased intratumoral heterogeneity associates with increased transcriptional heterogeneity.
Figure 2. Multi activation of transcriptional subtypes associated with intratumoral heterogeneity.

(A) The expression profiles of 369 IDH-WT GBMs were analyzed using Affymetrix U133A. The -log(empirical p value) of raw ssGSEA enrichment scores at each signature are shown as heatmaps, with dark blue representing no activation and bright red as highly activated. Yellow star indicates the secondary activated subtype (empirical p value < 0.05). For each panel, the first row shows simplicity score, and the second row indicates transcriptional subtype. (B) Comparison of mutation rate, subclonal mutation rate and subclonal mutation fraction between IDH-WT GBMs with high and low simplicity scores. p values were calculated using Wilcoxon rank test and shown at the top of each panel. Boxplots represent 25th and 75th percentiles, with midlines indicating the median values and points within the boxes indicating the mean values. Whiskers extend to the lowest/highest values of the data sample exculding outliers. The notch displays the 95% confidence interval around the median. (C) Kaplan-Meier survival curve by subtype. (D) Transcriptome classification of five bulk tumor samples and 501 single GBM cells derived from them. The top two row of each panel show the dominant and secondary subtype of the GBM tumor bulk. The heatmap of each panel shows the empirical -log(p value) of the ssGSEA scores of the derived single GBM cells on each of the three subtype signatures. The bottom row shows the subtype distribution of derived single GBM cells within the same GBM tumor of origin. See also Figure S2 and Tables S3 and S4.
We compared outcomes amongst the three transcriptional groups and observed no significant differences (Figure S2A). However, upon restricting the analysis to those samples with high simplicity scores (> 0.99, n = 74, top 20% cases), we discovered median survival of 11.5, 14.7, and 17.0 months in MES, CL, and PN cases, respectively, which revealed a significant survival difference between MES and non-MES cases (log rank test, p value = 0.03) (Figure 2C, Table S4). Consistent with this trend, greater simplicity scores correlated with relatively favorable outcome within the PN subtype while outcome remained unchanged within the CL subtype and the MES subtype (Figure S2B, C, D).
Single GBM cell RNA sequencing recently suggested that GBMs are comprised of a mixture of tumor cells with variable GBM subtype footprints (Patel et al., 2014). Within this dataset, we compiled the RNA sequenced transcriptomes of 501 single glioma cells in addition to the bulk tumor derived from five primary GBMs (Table S4) to investigate the intratumoral transcriptional heterogeneity at the single glioma cell level. In four of five cases (MGH26, MGH28, MGH29 and MGH30), the bulk tumor samples were classified in the same primary subtype as the majority of their single cells (Figure 2D). Our analysis suggests that the heterogeneity observed at the single cell level is captured in the expression profile of the bulk tumor.
Transcriptional subtypes differentially activate the immune microenvironment
Despite restricting the cluster analysis to genes exclusively expressed by GBM cells, we found that tumor purity predictions based on ABSOLUTE were significantly reduced in GBM classified as MES (Figure 3A). This was corroborated by gene expression based predictions of tumor purity using the ESTIMATE method (Student T-test p value < 2.2e-16; Figure 3B) (Yoshihara et al., 2013). The ESTIMATE method has been optimized to quantify tumor-associated fibroblasts and immune cells (Yoshihara et al., 2013), and the convergence of decreased ABSOLUTE and decreased ESTIMATE tumor purity confirms previous indications of increased presence of macrophages/microglia and neuroglial cells in MES GBM (Bao et al., 2006; Engler et al., 2012; Gabrusiewicz et al., 2016; Ye et al., 2012). The mean simplicity score of samples classified as MES was 0.48 which was significantly less than mean simplicity scores of samples classified as PN (Wilcoxon rank test p value < 0.003) and CL (Wilcoxon rank test p value < 1.13e-5), confirming increased transcriptional heterogeneity.
Figure 3. Transcriptional subtypes differentially activate the immune microenvironment.
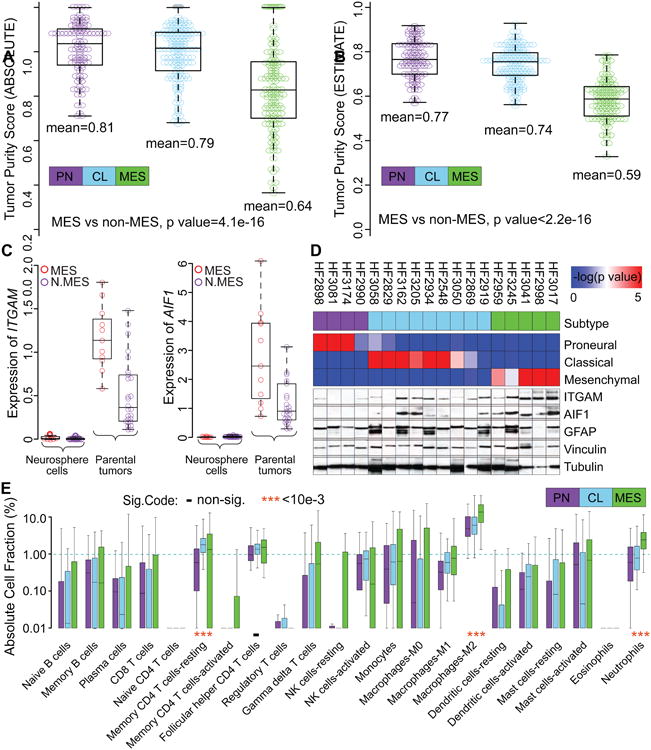
(A, B) Tumor purity of 364 and 369 TCGA IDH-WT GBM samples was determined by ABSOLUTE (A) and ESTIMATE (B), respectively. The difference in tumor purity between subtypes was evaluated using a two-sample Student t-test. (C) Comparison of ITGAM and AIF1 gene expression levels between GBM and derived neurosphere models. N.MES indicates non-mesenchymal cases. (D) The upper panel shows ssGSEA enrichment scores and associated expression subtype classifications. Bottom panels display protein expression of the microglial markers ITGAM and AIF1, astrocyte marker glial fibrillary acidic protein (GFAP) and the loading control tubulin and vinculin. (E) Comparison of immune cell fractions among subtypes. Immune cell fractions were estimated using CIBERSORT and corrected using ABSOLUTE purity scores per sample. The distribution of immune cell fractions of 86 PN, 136 CL and 104 MES IDH-WT GBMs with simplicity score>0.05 were shown by purple, skyblue and green boxplots, respectively. Median value difference of cell fraction among subtypes was evaluated using Mood's test. Boxplots represent 25th and 75th percentiles, with midlines indicating the median values and points within the boxes indicating the mean values. Whiskers extend to the lowest/highest values of the data sample excluding outliers (A-C, E). See also Figure S3 and Table S5.
Tumor-associated macrophages are a major source of tumor-associated non-neoplastic cells. In the brain, macrophages can be categorized as microglia, the resident macrophages in the central nervous system, and circulation-derived monocytes. Comparison of transcriptional levels of the macrophages/microglial markers integrin subunit alpha M (ITGAM, also known as CD11B) and allograft inflammatory factor 1 (AIF1, also known as IBA1) in 37 GBM-neurosphere pairs revealed that the two genes are not expressed in cultured cells but were highly transcribed in GBM, in particular in those classified as mesenchymal (Figure 3C). We found increased ITGAM and AIF1 protein expression in six GBM transcriptionally characterized as mesenchymal, compared to 12 non-MES GBM (Figure 3D). An unbiased quantification of macrophage/microglial (AIF1+) percentage using the Caliper Vectra image system and InForm software (Figure S3A, S3B) in twelve of these cases showed that the frequency of AIF1+ cells was significantly greater in MES (n = 4) vs. non-MES (n = 8) (median 38% vs. 16%, Wilcoxon rank test p value < 2.2e-16). (Figure S3A, S3B).
To comprehensively determine the cellular components of the tumor microenvironment across different transcriptional subtypes, we used the CIBERSORT in silico cytometry method (Newman et al., 2015). Upon filtering samples with classification simplicity scores less than 0.05, we evaluated 22 different immune cell types in 86 PN, 137 CL, and 106 MES samples (Table S5). We found that the tumor promoting M2 macrophage gene signature (Hambardzumyan et al., 2015) showed a greater association with the MES subtype (13%) relative to the PN (5%) and CL (6%) subtypes (Figure 3E), consistent with previous analysis of the TCGA database (Doucette et al., 2013; Gabrusiewicz et al., 2016). In addition to the M2 macrophage gene signature, there was also a significantly greater fraction of MES samples that expressed the proinflammatory M1 macrophage (Wilcoxon rank test p value = 0.004) and neutrophil (Wilcoxon rank test p value = 1.31e-12) gene signatures. Meanwhile, the activated natural killer cell gene signature (Wilcoxon rank test p value = 5.5e-4) was significantly reduced in the MES subtype and the resting memory CD4+ T cell gene signature (Wilcoxon rank test p value = 2.56e-7) was significantly reduced in the PN subtype. The association of the tumor-intrinsic MES GBM subtype with increased levels of M2 macrophages may imply that these GBMs are candidates for therapies directed against tumor-associated macrophages (Pyonteck et al., 2013). Likewise, the activated dendritic cell gene signature (Wilcoxon rank test p value = 3.0e-4) (Figure 3E) was significantly greater in the CL subtype, suggesting this subtype may benefit from dendritic cell vaccines (Palucka and Banchereau, 2012). This result contrasts a previous study suggesting that MES GBM patients treated with dendritic cell vaccination were likely to benefit (Prins et al., 2011).
Molecular classification based on tumor-specific DNA methylation profiles identified six distinct subtypes of primary glioma, including three IDHmut classes (M1 – M3) and three IDH-WT groups (M4 – M6) (Ceccarelli et al., 2016). We compared the frequency of immune cells across the three IDH-WT DNA methylation subtypes consisting of 87, 157, and 33 cases in LGm4, LGm5, and LGm6, respectively (Figure S3C). Among the 22 immune cell types we examined, we observed increases in neutrophils (Mood's test p value < 0.01) and M2 macrophages in LGm5. LGm5 was found to be enriched for the MES expression subtype in the original publication.
NF1 deficiency of tumor cells increases infiltration of tumor associated macrophages/microglia
MES GBMs frequently deactivate NF1 through genomic copy loss or somatic mutations (Verhaak et al., 2010). Formation of dermal neurofibromas in the context of NF1 loss of heterozygosity has been reported to be context and microenvironment dependent, suggesting that NF1 may play a role in organizing the microenvironment (Le et al., 2009). NF1 deleted/mutated GBMs showed reduced tumor purity compared to GBMs with wild-type NF1 (Wilcoxon rank test p value = 7e-4), and specifically within the MES subtype (Wilcoxon rank test p value = 0.017) (Figure S4A). Consequently, the M2 macrophage gene signature is significantly greater in NF1 deficient cases (Figure S4B, Wilcoxon rank test p value = 5e-3 and 0.084). To further establish the association of NF1 deficiency with infiltration of tumor associated macrophages/microglia, we performed immunofluorescence (IF) staining of AIF1 and NF1 on six TCGA IDH-WT GBMs. We observed an inverse correlation between NF1 and AIF1 staining (Wilcoxon rank test p values < 0.05, Figure 4A, B). We quantified this effect by measuring the distance between NF1 deficient tumor cells and macrophages/microglia through an immunohistochemistry (IHC) based automated quantitative pathology imaging system. We found that AIF1+ cells were in significantly closer proximity to NF1- cells than NF1+ cells across 30 human GBMs (Wilcoxon rank test p values < 1e-3, Figure 4C, D). Consequently, the number of AIF1+ cells significantly decreased with increasing NF1 expression (Wilcoxon rank test p values < 0.01, Figure 4E). These results suggest that NF1 deactivation may promote macrophages/microglia recruitment in tumors. We evaluated the effect of NF1 deactivation on macrophages/microglia recruitment in vitro. We knocked down NF1 using two different hairpins in TS603 human NF1 wild-type glioma cells (Figure 4F, G)(Rohle et al., 2013), and measured the recruitment of both human cultured microglial cells and freshly isolated GBM associated macrophages using a transwell assay (Figure 4H, I, J, K). NF1 knock down resulted in a 47% and 56% increase in recruitment of freshly isolated GBM associated macrophage cells for shRNA #5 and #8, respectively (Figure 4K, paired Student t-test p values < 0.05). Consistent with this, recruitment of human microglia cells increased by 44% and 21% following NF1 knockdown by shRNA #5 and #8, respectively (Figure 4J, paired Student t-test p values < 0.05). These data substantiate the hypothesis that NF1 deactivation in glioma cells results in macrophages/microglia attraction.
Figure 4. Decreased NF1 expression enhances the recruitment of macrophages/microglia in GBM.
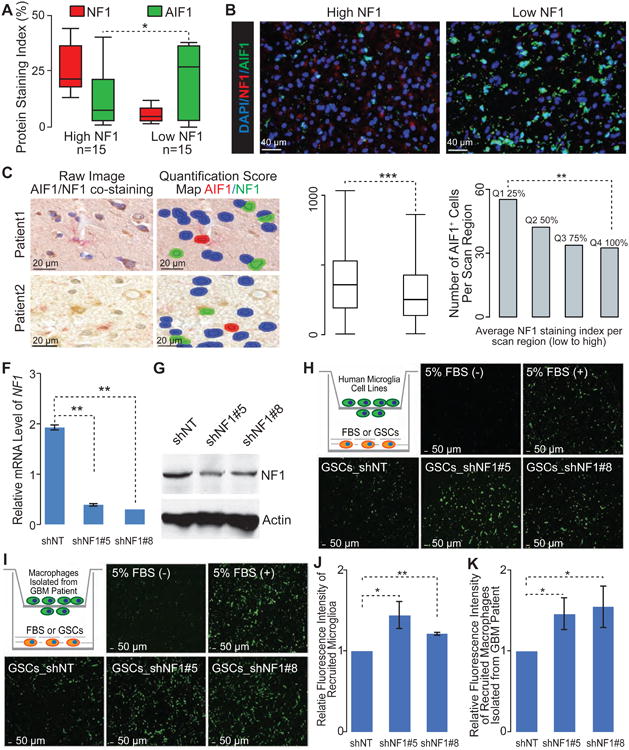
(A) Quantification of NF1 and AIF1 staining by Immunofluorescence (IF) in 6 GBMs from TCGA (IDH-WT). At least 3 regions from each tumor were analyzed (n=30 regions). (B) Representative IF images show NF1 and AIF1 staining TCGA IDH-WT GBMs. (C) Representative images with IHC double-staining and cell segmentation obtained from Caliper InForm analysis software show the close proximity of AIF1+ cells (red) and NF1- cells (blue) compared with NF1+ cells (green) in tumor sections from two GBM patients. (D) Boxplot of distances from AIF1+ cells to the nearest NF1- and NF1+ cells, respectively (4022 AIF1+ cells from 30 GBMs). (E) The appearance of AIF1+ cells within tumor sections with the various level of NF1 expression from 30 GBMs. (F) qRT-PCR for NF1 mRNA levels in patient-derived GSCs (TS603) by the indicated short hairpins (shNT, non-targeting short hairpin as control). Error bars represent SD of mean, n=3. (G) Immunoblot analysis of NF1 protein level in TS603 with short hairpins knocking down. (H, I) Representative IF images show the recruited human microglia (H) or GBM patient derived macrophages (I) by TS603 with NF1 knocking down in transwell assay. Cartoon depicting the experimental approach. (J, K) Quantification of recruited human microglia (J) or GBM patient derived macrophages (K) by TS603 with NF1 knocking down in transwell assay. Error bars represent SD of means from three independent experiments (J) or three biological replications (K). *, **, *** indicated the Student t-test / Wilcoxon rank test p value <0.05, 0.01, and 0.001, respectively, by paired student t test. Boxplots represent 25th and 75th percentiles, with midlines indicating the median values and points within the boxes indicating the mean values. Whiskers extend to the lowest/highest values of the data sample excluding outliers (A, D). See also Figure S4.
Taken together, our in silico analyses and biological validations demonstrate that genomic alteration-induced NF1 deficiency of tumor cells drives macrophages/microglia infiltration into the tumor-associated microenvironment.
Phenotypic plasticity upon GBM recurrence
GBM has long been hypothesized to progress along a PN to MES axis (Phillips et al., 2006). To determine the relevance of this transition process in IDH-WT glioma evolution, we performed a longitudinal analysis of the subtype classification and tumor-associated microenvironment in sample pairs obtained at diagnosis and first disease recurrence from 124 glioma patients. The cohort included 96 initial GBM and first recurrence pairs, eight pairs of primary low grade glioma and matching secondary GBM, and 20 pairs of primary and recurrent low grade glioma. Gene expression profiles of 78 tumor pairs were generated using transcriptome sequencing, and profiles of the remaining pairs were generated using Affymetrix (n = 31) and Illumina (n = 15) microarrays. To facilitate exploration of this dataset, we have made it available through a webportal (http://recur.bioinfo.cnio.es/) associated with GlioVis (Bowman et al., 2017). We used a gene expression signature (Baysan et al., 2012) to determine that 33 of 124 cases were IDHmut or hypermethylated (known as the GCIMP phenotype) at presentation and recurrence (Table S6). We used the renewed gene signatures and classification method to determine molecular subtype of the 91 pairs of IDH-WT cases and found that expression class remained consistent after disease recurrence for 50 of 91 IDH-WT cases (55%) (Figure 5A). The MES subtype was most stable (65%) while the CL (51%) and PN (41%) subtypes were less frequently retained. Ten, thirteen, and eighteen post-treatment tumors switched subtypes to become CL, MES and PN at disease recurrence, respectively, indicating that PN and MES increased in frequency after recurrence while the CL subtype was least frequently found (Figure 5A). The CL expression subtype was previously found to be most sensitive to intensive therapy, suggesting that therapy provides a competitive advantage for non-CL cells (Verhaak et al., 2010), which may be tied into the reduced EGFR expression frequency at recurrence (van den Bent et al., 2015), that may explain the reduced post-treatment incidence of the CL subtype. Despite a numerical increase in frequency of the MES subtype from primary disease (n = 37) to recurrent tumor (n = 42), the difference was not significant (Proportion test p value = 0.27).
Figure 5. Microenvironment transition between 91 primary and paired recurrent IDH-WT GBM.
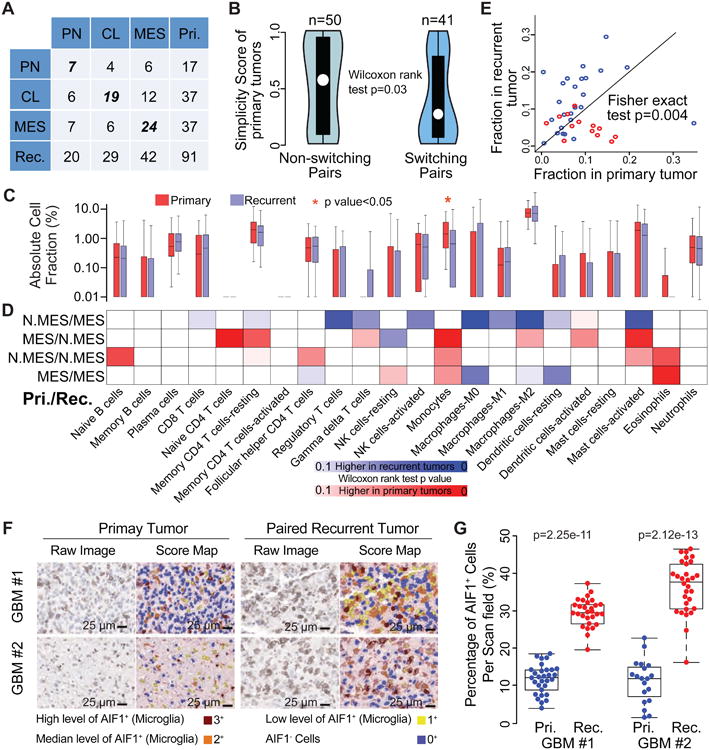
(A) Rows and columns of the cross table represents subtype distribution frequency of primary and paired recurrent tumors, respectively. (B) Violin plots show the distribution of simplicity scores of pairs without (left) and with (right) subtype transition. (C) Red and blue boxplots represent the immune cell fraction distribution of each immune cell type. Immune cell fraction was calculated using CIBERSORT and adjusted using ESTIMATE purity scores. Difference between cell fraction of primary and paired recurrent tumors was calculated using Wilcoxon rank test. (D) The blue-to-red heatmap represents immune cell fraction changes upon tumor recurrence per subtype transitions which were list on the left of the heatmap. Fisher exact test was used to evaluate the distribution difference between patients with higher/lower immune cell fractions at tumor recurrence per subtype transition. N.MES indicates non-mesenchymal case. (E) Each dot represents a pair of primary and recurrent GBM with axes indicating M2 macrophage cell fraction. (F) Representative images of AIF1 IHC staining and corresponding score map obtained by InForm image analysis in two matched pairs of primary and recurrent GBM. Scale bar, 25 μm. (G) Unbiased quantification of AIF1+ percentage in primary and recurrent GBMs, statistical testing was performed using Wilcoxon rank test. Boxplots represent 25th and 75th percentiles, with midlines indicating the median values and points within the boxes indicating the mean values. Whiskers extend to the lowest/highest values of the data sample exculding outliers (B, C, G). See also Figure S5 and Table S6.
We observed a significant difference in transcriptional simplicity between primary GBM retaining their expression subtype and those that switched to a different phenotype (Figure 5B). GBMs with a primary tumor simplicity score greater than average (0.46), indicating lower transcriptional heterogeneity, were classified as the same subtype in 30 of 45 (67%) cases, compared to 20 of 46 (43%) cases with primary tumor simplicity scores below average (Fisher exact test p value = 0.02) that switched subtype.
For comparison, we additionally performed the M1 – M6 DNA methylation based classification on 27 pairs of primary/recurrent gliomas (Table S6). DNA methylation subtype switched upon tumor progression in four of 27 cases. Of the four cases that switched DNA methylation class, one has been described as an example of extreme divergent evolution (Kim et al., 2015), with overlap in mutations between primary and recurrent tumor sample of less than 10%. The other three cases with switched methylation classes showed substantial differences in DNA copy number profiles between primary and recurrent tumors (Figure S5A), similarly suggesting significant genomic divergence. These results suggest that DNA methylome based classification has higher consistency between different time points compared to transcriptome subtypes (15% of 27 pairs per methylome subtype vs. 55% of 91 pairs per transcriptome subtype, Fisher's exact test p value = 1.7e-4).
Microenvironment transitions upon GBM recurrence
Debulking surgery, radiotherapy, and chemotherapy provide therapeutic benefit but induce tumor evolution by providing a competitive advantage to therapy resistant glioma cells. The tumor microenvironment plays an important role in providing nutrients and may influence the evolutionary process. We explored this possibility by comparing the tumor-associated microenvironment in primary and recurrent GBMs using CIBERSORT (Table S6) (Newman et al., 2015). A comparison between 91 primary and recurrent IDH-WT tumors revealed a decrease in monocyte gene signature expression at recurrence, suggesting relative depletion of circulation derived monocytes (Figure 5C). Next, we dissected microenvironment fluctuations between diagnosis and recurrent tumors across different subtype combinations. Primary non-MES (CL or PN) tumors showed relatively high tumor purity and, consequently, recurrent MES tumors classified as non-MES demonstrated a relatively global decrease of immune cells while non-MES cases transitioning to MES at recurrence represented increased immune cell fractions (Figure 5D). In contrast to the trend of monocyte depletion, the imputed M2 macrophage frequency was significantly higher at recurrence in cases transitioning to MES (Figure 5E). This observation converges with the higher predicted fraction of M2 macrophages in primary MES GBM relative to primary non-MES GBM. We validated the increase in macrophages using immunostaining of AIF1 expression in two primary-recurrent GBM pairs which were classified as CL to MES (Figure 5F). AIF1 immunoexpression was restricted to macrophages/microglia, cells exhibiting either ameboid or ramified morphology, with no expression in glioma tumor cells (Figure 5F, Figure S3). Quantitative analysis of microglia frequency using Inform software for automated pathology imaging processing confirmed a significantly higher presence (p value = 2.25e-11 and 2.12e-13 for patient #1 and #2, respectively) of AIF1+ cells at MES recurrence (Figure 5G, Figure S3). These findings further solidify the association between MES GBM and macrophage/microglia and extend this mutual relationship to disease recurrence. Compared to primary MES tumors, MES tumors at recurrence showed an increase in transcriptional activity associated with non-polarized M0 macrophages, which has been previously described (Gabrusiewicz et al., 2016), but also dendritic cells. In contrast, primary PN GBM were found to contain significantly higher fractions of five immune cell categories compared to recurrent PN GBM, indicating a relative absence of immune infiltration in PN GBM upon recurrence.
We evaluated the effect of transcriptional subtype on patient survival. The analysis was restricted to 54 cases for whom annotation on overall survival (OS) time and time to disease progression (PFS) were available and with high simplicity scores, indicating low transcriptional heterogeneity. Patients whose primary tumor was classified as MES trended towards adverse overall survival (logrank test p = 0.09 with HR = 1.68) (Figure 6A). This pattern was retained in patients whose recurrent glioma was classified as MES, again trending towards significance (logrank test p = 0.11 with HR = 1.60) (Figure 6B). Consequently, cases for whom both primary and recurrent tumor were classified as MES subtype showed the least favorable outcome, suggesting an additive effect of transcriptional subtype at different time points (Figure 6C, Figure S5B, S5C, S5D).
Figure 6. Survival analysis of paired IDH-WT GBM.

(A) OS and PFS analyses between samples with different primary subtype. (B) Difference of survival time after secondary surgery between patients with non-MES and MES in primary tumors (left) and in recurrent tumors (right). (C) OS and PFS analyses between samples with difference recurrent subtype. See also Figure S5.
Treatment-induced immunological microenvironment changes upon GBM recurrence
Temozolomide (TMZ) treatment of gliomas can induce hypermutation (Hunter et al., 2006; Kim et al., 2015). Missense mutations may generate neoantigens that can be recognized by CD8+ T cells (Schumacher and Schreiber, 2015). Exome mutation data were accessible for 45 pairs in our cohort. Six recurrent GBM (HMR) were identified as being hypermutated (>=400 SNVs). The mutation spectrum of the six HMRs were characteristic of a typical TMZ induced mutational signature (Figure S6) (Alexandrov et al., 2013). For the five pairs of HMR with transcriptomes available, the predicted frequency of CD8+ T cells was significantly increased at recurrence in comparison to their primary tumors (Wilcoxon rank test p value = 8e-3) (Figure 7A). This observation was corroborated by comparing seven hypermutated primary GBMs to 238 non-hypermutated GBMs (Wilcoxon rank test p value=0.031) (Figure 7B). The observation suggests that patients with hypermutated tumors may have a more immunological reactive microenvironment that may be responsive to immune checkpoint inhibitors (Sharma and Allison, 2015).
Figure 7. Immune cell frequency comparison.

(A) Blue and red diamond indicate individual primary and recurrent tumors. Dash line connects paired primary and recurrent tumors. (B) Blue and red circle indicate non-hypermutated and hypermutated primary samples. (C) Sky blue/dark blue and orange/red boxplots indicate short- and long- term relapsed tumors, respectively. y-axis stands for immune cell fraction. Wilcoxon rank tests were used to examine the significance of the differences between groups, and p values were shown at top of each panel. Boxplots represent 25th and 75th percentiles, with midlines indicating the median values and points within the boxes indicating the mean values. Whiskers extend to the lowest/highest values of the data sample exculding outliers (A-C). See also Figure S6 and Table S7.
Preclinical studies suggested radiation may increase the recruitment of T cells in the tumor microenvironment (Deng et al., 2014; Zeng et al., 2013). As such, we compared the microenvironment of primary GBM treated with radiation therapy and separated short term relapses (PFS < 6 months, n = 27) from late relapses (PFS > 12 months, n = 21) (Table S7). We observed no significant differences between primary tumors with short-term and long-term relapse. When comparing relapse GBMs, we found that short-term relapse GBMs showed a significantly higher predicted presence of M2 macrophages and CD4+ T cells (CD4+ T memory resting and CD4+ follicular helper cells) after radiation, compared to long-term relapse tumors (Figure 7C). M2 macrophages have been speculated to play a role in resistance to radiotherapy (Meng et al., 2010; Ruffell and Coussens, 2015) and macrophage targeting immunotherapy (Pyonteck et al., 2013; Ries et al., 2014) may play a radiosensitizing role. The increase of CD4+ T cells at recurrence of short term relapse tumors points towards inhibiting CTLA-4 as adjuvant therapy to radiation.
Discussion
Transcriptome profiling of tumor samples is a commonly used modality for interrogating pathway functionality and phenotype based patient classification. The transcriptional footprint left by the tumor microenvironment, which may constitute 10-80% of cells in a tumor biopsy (Yoshihara et al., 2013), can obscure the true activity of the signaling network (Isella et al., 2015; Kim and Verhaak, 2015). Here, we employed in silico methods to integrate mRNA expression profiles from glioma samples and glioma cell culture models to provide insights into glioma-intrinsic pathway activities and classification, and to deconvolute the glioma associated stroma into its immunological cellular components.
GBM expression subtype classification has emerged as an important concept to better understand the biology of this devastating disease (Dunn et al., 2012; Huse et al., 2011; Sturm et al., 2014). Robust classification of new GBM tumors is therefore critical to ensure consistency in reporting between different studies. Our updated methods, released through a R-library, were found to be highly robust and provide a standardized strategy for classification of gliomas. The transcriptional glioma subtypes defined through clustering based on tumor-intrinsic genes strongly overlapped with the proneural, classical, and mesenchymal subtypes but identified the neural subtype as normal neural lineage contamination. We observed that the mesenchymal GBM subtype continued to associate with the presence of tumor-associated glial and microglia cells. Mesenchymal glioma cell differentiation status has been found to correlate with enrichment of macrophages/microglia (Bhat et al., 2013; Kreutzberg, 1996). Our results confirm that a macrophage/microglia rich microenvironment can shape a mesenchymal glioma cell phenotype. However, we also found that genetic deactivation of NF1 associated with attracting macrophages/microglia, suggested that a two-way interaction exists between tumor cells and microenvironment. Further studies are needed to identify the mechanism of NF1 regulated chemotaxis, which may result in the development of agents that are able to repress the recruitment of macrophages/microglia as a therapeutic modality.
Longitudinal analysis of tumor samples is complicated by the lack of tissue collections including such pairs. Through aggregation of several datasets, we compiled a cohort of 124 glioma pairs, including 91 pairs of IDH-WT tumors. Comparison of pairs of initial gliomas and first disease recurrence did not identify the trend of proneural GBM transitioning to a mesenchymal phenotype that has often been suspected (Phillips et al., 2006). Mesenchymal subtype at diagnosis and at disease recurrence correlated with relatively poor outcome. The immune microenvironment of recurrent IDH-WT GBM showed a reduction in blood derived monocytes which may reflect lower penetration through the blood brain barrier as a result of radiotherapy (van Vulpen et al., 2002). While the frequency of M2 macrophage/microglia was increased in recurrent mesenchymal GBM compared to primary non-mesenchymal GBM, the overall fraction of M2 macrophage/microglia remained stable. This possibly suggests that the majority of these cells are derived from resident CNS macrophages rather than through active recruitment from the circulation. Our analysis resulted in two insights with potentially important therapeutic implications. We showed an association between the presence of M2 macrophages and poor response to ionizing radiotherapy. While further optimization of macrophage inhibiting agents is required before they become clinically useful (Quail et al., 2016), our results may provide an imperative for combination of radiotherapy and M2 macrophage inhibiting agents (Pyonteck et al., 2013; Xu et al., 2014). Second, we identified a correlation between hypermutation and increased frequency of CD8+ lymphocytes, both at primary diagnosis and at disease recurrence, albeit with different mutational signatures. This subset of patients may be responsive to checkpoint inhibition blockade, to which limited successes have been reported (Bouffet et al., 2016; Johanns et al., 2016).
In summary, our study defines a strategy to determine transcriptional subtype and associates expression subtypes to the tumor-associated immuno-environment. Our findings may aid in the implementation of immunotherapy approaches (Blank et al., 2016) in a disease type with very limited treatment options. Collectively, our results improve our understanding of determinants of GBM subtype classification, elucidate the critical impact of the tumor microenvironment, and provide handles on the interpretation of transcriptional profiling of glioma.
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact: Roel G.W. Verhaak (roel.verhaak@jax.org)
Experimental Model and Subject Details
Cell lines and cell culture
Patient-derived glioma stem cells (GSCs, TS603) were described previously (Hu et al., 2016). The GSCs were cultured in NSC proliferation media (Millipore Corporation, Billerica, MA) with 20 ng/ml EGF and 20 ng/ml bFGF. Human microglia cells were purchased from ScienCell and were cultured in microglia completed media as recommended by vendor. GBM patient-derived macrophages were isolated by CD14 microbeads using immunomagnetic method as described previously(Gabrusiewicz et al., 2016).
The pLKO.1 shRNAs for targeting NF1 were purchased from Sigma. Lentiviruses were produced in 293T cells (ATCC) with packaging system (pCMVR8.74, pMD2.G, Addgene) as per Vendor's instruction.
Human GBM biospecimens for RNA sequencing
The GBM tumor tissues were collected and named in the order that they were acquired. The primary tumors matched sphere cell lines were isolated from fresh tissues. Each tissue was enzymatically and mechanically dissociated into single cells and grown in DMEM/F12 media supplemented with B27 (Invitrogen), EGF (20 ng/ml), and bFGF (20 ng/ml), resulting in neurosphere growth. All cell lines were tested to exclude the presence of Mycoplasma infection. Human tissue collection was performed with written informed consent from patients, using a protocol approved by the institutional review board (IRB) of The University of Texas M.D. Anderson Cancer Center (LAB04-0001, PA16-0408),.
Tissues from nine pairs of initial and matched recurrent GBMs were obtained from Henry Ford Hospital in accordance with institutional policies and all patients provided written consent, with approval from the Institutional Review Boards (Henry Ford Hospital IRB protocol #402).
The frozen tissue from 44 patients with initial and recurrent GBM that received resection at Samsung Medical Center and Seoul National University Hospital were provided by Dr. Nam's lab. Surgery specimens were obtained in accordance to the Institutional Review Board (IRB) of the Samsung Medical Center (No. 2010-04-004) and Seoul National University Hospital (No. C-1404-056-572), with written informed consent from all patients.
Method Details
Generation of the primary and recurrent glioma RNA-Seq datasets
U133A array profiles for 533 primary GBM, and RNA-Seq data for 166 primary and 13 recurrent GBM were obtained from the TCGA portal https://tcga-data.nci.nih.gov/tcga/. Mutation calls and DNA copy number profiles were obtained for all samples, where available. All non-TCGA data used in this study is publicly available as raw data. Processed primary/recurrence expression data can be analyzed through GlioVis portal http://recur.bioinfo.cnio.es/.
Regarding the pairs of primary and recurrent gliomas from from Henry Ford Hospital (n = 12), all RNA samples tested were obtained from frozen specimens. All of the recurrent GBMs had been previously treated with chemotherapy and radiation. Three cases had a history of lower grade astrocytoma prior to the first GBM (HF-2869/HF-3081/HF-3162). Tumors were selected solely on the basis of availability. RNA-Seq libraries were generated using RNA Truseq reagents (Illumina, San Diego, CA, USA) and paired-end sequenced using standard Illumina protocols. Read length was 76 base pairs for cases sequenced by TCGA and from Henry Ford (processed at MD Anderson).
RNA-Seq data on frozen tissue from 44 patients with initial and recurrent GBM from Samsung Medical Center and Seoul National University Hospital were provided by Dr. Nam's lab. Affymetrix CEL files of 39 pairs of initial and recurrent glioma were retrieved from the Gene Expression Omnibus (GEO accession GSE4271, GSE42670, GSE62153)(Joo et al., 2013; Kwon et al., 2015; Phillips et al., 2006). The expression profiles of the 23 pairs from GSE4271 were determined using Affymetrix HG-U133 GeneChips, the 1 pairs from GSE42670 were analyzed using the Affmetrix HuGene-1-0-st platform, the 15 pairs from GSE62153 were analyzed using Illumina Human HT-12 V4.0 expression BeadChip. The RNA sequencing data of 14 and 5 pairs of primary and recurrent low grade glioma were from TCGA LGG cohort and available through the European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/), EGAS00001001255, (Mazor et al., 2015), respectively. The RNA sequencing data of 9 pairs of primary and recurrent glioblastoma have been submitted to EGA under accession number EGAS00001001033. Genome wide DNA copy number profiling and exome sequencing on thirteen TCGA tumor pairs and nine of ten Henry Ford tumor pairs were performed and data was analyzed using standard protocols and pipelines as previously described (Kim et al., 2015)
Evaluating datasets integrating stability of the classification system
To evaluate the cross platform classification stability, we collected RNA sequencing data of 162 primary GBMs (Brennan et al., 2013) for which an Affymetrix HT-U133A gene expression profile was also available from the TCGA dataset. We observed a low Pearson Correlation Coefficient (< 0.15) between RNA sequencing based reads per kilobase of transcript per million reads (RPKM) and Affymetrix HT-U133A profiles in eighteen cases and these were removed from further analysis. This results in 144 pairs of transcriptomes to assess the concordance between classification results of the newly developed 50-gene signatures based classification system between RNAseq and microarray based profiling platform. As shown in Figure S1H, a total of 134 (93%) cases received the same subtype across different platforms.
There are several commonly used metrics for quantifying the expression on RNA-seq platform, such as reads per kilobase per million mapped reads (RPKM), k-mers per kilobase per million mapped k-mers (KPKM) and transcripts per million (TPM)(Patro et al., 2014). To evaluate the subtyping concordance when using different expression quantification metrics, we collected a set of 46 samples with transcriptome profiled and quantified outside of our lab with three metrics RPKM, KPKM and TPM. As shown in Figure S1I, our classification system achieved a 100% consistence while using different quantification metrics.
For some of the GBM datasets, not every IDH status was determined when using the classification system. We evaluated the interference of embedded non-IDH-WT samples in a GBM cohort. A set of 369 cases in the TCGA GBM cohort are IDH-WT and GCIMP-, 42 and 122 cases are IDHmut/GCIMP+ and status unknown, respectively. We performed the classification system on the 369 IDH-WT cases and entire 533 GBMs separately. Then we compared the classification results of the 369 IDH-WT cases in two different runs. As shown in Figure S1J, 97% IDH-WT GBMs received the same subtyping results.
Integrating datasets from various sources may be impacted by batch effects. We have 111 and 179 RNA sequenced profiles of GBMs collected independently from TCGA dataset and Samsung Medical Center. We combined the 290 samples from the two datasets together and performed the classification. While comparing the subtyping results with the subtypes identified in separate dataset, as shown in Figure S1K, 13 cases showed different subtypes, resulting in a 93% concordance.
As the classification system using resampling method to build a background distribution for determining the empirical p values for classification, the sample size of inputted dataset for performing the classification has an impact on the subtyping results. To evaluate this effect, we randomly selected samples from the 533 TCGA GBMs profiled using microarray or the 111 in house (SMC samples) RNA sequenced GBMs and performed the classification system independently, and we repeated the random selection procedures 50 times. Then we compared the subtyping results within the randomly generated sample sets with the classification results received using the entire 533 TCGA or 111 SMC GBMs. As shown in Figure S1L (left), the newly developed classification achieved a 90% concordance between sample sets with size >= 20 and the entire 533 TCGA or 111 SMC GBMs. Upon removing non-simplex samples from the original sample sets, the classification performance per sample size improves (Figure S1L, right).
Transcriptome data processing
The latest version of custom CDF files (Version19, http://brainarray.mbni.med.umich.edu) (Dai et al., 2005; Sandberg and Larsson, 2007) were used to map probes from the Affymetrix HG-U133A and HuGene-1_0-st GeneChip platforms to the Ensemble transcript database, combined in one probe set per gene, and normalized using the AROMA package with default parameters, resulting in RMA normalized and log transformed gene expression values (Bengtsson et al., 2009). All RNA sequencing data was processed by the PRADA pipeline (Torres-Garcia et al., 2014). Briefly, reads were aligned using BWA against the genome and transcriptome. After initial mapping, the aligned reads were filtered out if their best placements are only mapped to unique genomic coordinates. Quality scores are recalibrated using the Genome Analysis Toolkit (GATK), and duplicate reads are flagged using Picard. Mapped features were quantified and normalized per kilo base of transcript per million reads (RPKM) and were converted to a log2 scale to represent a gene expression level. RPKM values measuring the same gene that mapped to the Ensemble transcript with longest size were selected to obtain one expression value per gene and sample. RPKM values were converted to a log2 scale to represent gene expression level. The statistical environment R was used to perform all the statistical analysis and graph plots.
Deriving GBM intrinsic subtype signatures
We performed consensus non-negative matrix factorization (CNMF) clustering method to identify distinct subtypes among the 369 IDH-WT primary GBMs based on the 7,425 BFGs we established using single glioma cells, GBM-neurosphere pairs and comparison of core vs leading edge tumors. Genes were ranked according to their maximum absolute deviation (MAD) values from high to low in the 369 by 7425 expression matrix. When the 369 IDH-WT GBMs were clustered into three subtypes, we received the highest average clustering cophenetic scores (Table S1), resulting the identification of three distinct subtypes among the 369 IDH-WT GBMs. A set of 256 GBMs was recognized as core samples based on a positive silhouette width resulting 94, 70 and 92 cases were clustered as subtype1, subtype2 and subtype3, respectively, which were later names as MES, PN and CL subtype. Signature genes per cluster were selected on the basis of differences in gene expression level and were considered significant if they reached the cut-off value with t-test p value < 1e-3 for higher expressed in this subtype, while also showing a significant lower expression with t-test p value < 1e-3 in the other two subtypes. 64, 50 and 330 genes were identified as candidate signature genes for MES, PN and CL, respectively. In the original gene signatures, genes could be either down-regulated or up-regulated, while only up-regulated genes (n = 50 per gene signature) were selected for revised gene signatures by keeping the same size of signature genes for each subtype as well.
Molecular classifications based on ssGSEA enrichment scores
Single sample gene set enrichment analysis was performed as follows. For a given GBM sample, gene expression values were rank-normalized and rank-ordered. The Empirical Cumulative Distribution Functions (ECDF) of the signature genes and the remaining genes were calculated. A statistic was calculated by integration of the difference between the ECDFs, which is similar to the one used in GSEA but is based on absolute expression rather than differential expression (Barbie et al., 2009). Since the ssGSEA test is based on the ranking of genes by expression level, the uncentered and log-transformed U133A and RPKM expression levels were used as input for ssGSEA. Since the scores of the three signatures were not directly comparable, we performed a resampling procedure to generate null distributions for each of the four subtypes. First we generated a large number of virtual samples in which each gene obtains its expression level by randomly selecting an expression value of the same gene in the remainder of the samples. Then, the three ssGSEA scores for each signature were calculated. Following this procedure, we generated a large number (>= 1,00,000) of random ssGSEA scores for each subtype, to build the null distribution and to give empirical p values for the raw ssGSEA scores of each sample. By testing on multiple datasets with different sample sizes, we found the resampling generated distribution could be replaced with Student-T distribution (sample size > 30) or Normal distribution (sample size > 50) for getting very similar results. R-library with the code and expression matrices used is provided as Method S1.
Evaluate the simplicity of subtype activation
For a single sample, we decreased rank the empirical p values for each subtype to generate order statistics as RN–1, RN–2 … R1, R0. In particular, R0 equals to the minimum empirical p value and points to the dominant subtype, i.e., the most significantly activated subtype. The accumulative distance to the dominant subtype (ADDS) was defined as:
Similarly, the accumulative distance between non-dominant subtypes (ADNS) as:
Obviously, the ADDS and ADNS are positive and negative correlated with single activation, respectively. Hence, we defined the simplicity score by combing ADDS and ADNS together and corrected with a constant as follows:
Establishment of datasets of primary tumors and matched sphere cell lines
To minimize any batch effect the downstream molecular analyses were performed on identical cell culture batches. Total RNA from formalin fixed, paraffin embedded tumor tissues and matching neurospheres was prepared using the Masterpure complete DNA and RNA isolation kit (Epicenter) after proteinase K digestion per to the instructions from the manufacturer. Paired-end Illumina HiSeq sequencing assays were performed resulting in a medium number of 50 million 75bp paired end reads per sample. We employed the PRADA pipeline to process the RNA sequencing data (Torres-Garcia et al., 2014). In short, Burroughs-Wheeler alignment, Samtools, and Genome Analysis Toolkit were used to map short reads to the human genome (hg19) and transcriptome (Ensembl 64) and RPKM gene expression values were generated for each of the 135,994 transcripts of 21,165 protein coding genes in Ensembl database.
RNA isolation and qRT-PCR
GSCs (TS603) were infected by lentivirus carrying short hairpins to target NF1 gene. The pLKO.1 shRNAs were purchased from Sigma. The cells were harvested after 48 post-infection and RNA was isolated with RNeasy® Mini Kit (Qiagen), and then used for first-strand cDNA synthesis using random primers and SuperScriptIII Reverse Transcriptase (Invitrogen). qRT–PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems). The sequences of NF1 primers are: forward-CAGAGAGCCTTGAGGAAAACC, reverse- CTGGCTAACCACCTGGTATAAAC. The relative expression of genes was normalized using ribosomal protein L39 (forward-CAGCTTCCCTCCTCTTCCTT, reverse- GCCAGGAATCGCTTAATCC) as a housekeeping gene.
Immunoblotting (IB), immunohistochemistry (IHC) and immunofluorescence (IF)
For immunoblotting, cells were harvested, washed with phosphate buffered saline, lysed in RIPA buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 1.0% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]; Sigma); and frozen tumor sections were lysed using RPPA lysis buffer (1% Triton X-100 50mM HEPES pH 7.4, 150mM NaCl, 1.5mM MgCl2, 1mM EGTA, 100mM NaF, 10mM Na pyrophosphate, 1mM Na3VO4, 10% glycerol). Both types of lysate buffer contain with protease inhibitor cocktail tablet complete mini (Roche Diagnostics), phosphatase inhibitor cocktail 2 (Sigma) and 1 μM DTT, and centrifuged at 10,000 × g at 4°C for 15 min. Protein concentration was measured using the BCA kit (Thermo Scientific - Pierce #23225). Protein lysates were subjected to SDS-polyacrylamide gel electrophoresis on 4-12% gradient polyacrylamide gel (NuPage, Thermo Fischer Scientific), transferred onto nitrocellulose membranes which were incubated with indicated primary antibodies, washed, and probed with HRP-conjugated secondary antibodies. The primary antibodies include ITGAM (CD11B) (Sigma Aldrich, #HPA002274), AIF1 (IBA1) (Wako, #016-20001), GFAP (Cell Signaling, #3670), NF1 (clone McNFn27b, GeneTex, #GTX15776), Actin (Sigma Aldrich, A5044), Vinculin (EMD Millipore, # 05-386) and Tubulin (Cell Signaling, #2128).
For IHC staining, brain tumor sections were incubated with the primary antibodies for 1 hour at room temperature (RT) or overnight at 4°C after deparaffinization, rehydration, antigen retrieval, quenching of endogenous peroxidase and blocking. The sections were incubated with horseradish peroxidase (HRP)-conjugated polymer (DAKO) for 40 minutes and then Diaminobenzidine using Ultravision DAB Plus Substrate Detection System (Thermo Fischer Scientific) for 1-10 minutes at RT, followed by hematoxylin staining. For IHC double staining, the tumor sections were incubated with MACH 2 Double Stain 2 (Biocare Medical) for 20-30min; and then the sections were stained by Biocare's Warp Red (Biocare Medical) and subsequently by DAB detection system (Thermo Fischer Scientific) for 1-10 minutes at RT, followed by hematoxylin staining. The primary antibodies for IHC staining include AIF1 (Abcam, ab178864) and NF1 (clone McNFn27b, GeneTex #GTX15776).
For IF staining, OCT frozen brain sections were thawed at RT for 30 minutes, rinsed and rehydrated with phosphate buffered saline 3 times. After blocking with PBS buffer containing 10% FBS, 1% BSA and 0.3% Triton, the sections were incubated with the primary antibodies (AIF1, ab178864; NF1, GTX15776) overnight at 4°C. The samples were then incubated with species-appropriate donkey secondary antibodies coupled to AlexaFluor dyes (488, 594, Invitrogen) for 1 hour at RT. VECTASHIELD with DAPI (Vector Laboratories) was used to mount cover slips. The slides were analyzed using Leica DMi8 microsystems.
Transwell assay
Transwell assays were performed in BD FluoroBlok 96-multiwell insert systems (8.0 μm pore sizes) as per manufacturer's protocol (BD biosciences). Human microglial cells or patient-derived microphages were seeded in transwell inserts at 1×104 cells/well. After 4 hours starvation in DMEM/F-12 basal media (Thermo Fischer Scientific) at 37°C, 5% CO2 incubator, the inserts were transferred into the basal chambers containing 5% FBS or GSC (TS603) with NF1 knocking down by shRNA in NSC growth media. After 24 hours incubation, the inserts were transferred into a second 96-well plate containing 4 μg/mL Calcein AM (BD biosciences) in DPBS. Incubate for 1 hour at 37°C, 5% CO2, fluorescence of invaded cells was read at wavelengths of 494/517 nm (Ex/Em) on fluorescent plate reader.
Single-cell isolation and RNA sequencing data processing
We generated single cell RNA sequencing data as follows. From a single patient, we obtained two initial tumor samples from the right and left frontal lobes and a recurrent tumor from the left frontal lobe that emerged after concurrent chemoradiotherapy (CCRT) and EGFR-targeted treatment. After these three tumor tissues were dissociated, the C1™ Single-Cell Auto Prep System (Fluidigm, San Francisco, CA, USA) with the SMARTer kit (Clontech, Mountain View, CA, USA) was used to capture single cells and to isolate cDNA from the cells. With the Nextera XT DNA Sample Prep Kit (Illumina) and HiSeq 2500, we sequenced the transcriptomes of 29, 60, and 44 single cells from left, right and relapse tumors, respectively. From the sequenced reads (100bp paired-end), 5′-end 30 nucleotides were trimmed and mapped on human genome assembly (hg19). For all 133 single cells, RPKM values of 23660 genes were obtained by DEGseq (R package) (Wang et al., 2010). To identify whether the in-house sequenced single cells were tumor cells, we first performed a gene signature based filtering as follows. We compiled the transcriptomes of four tumor astrocytes, six fetal astrocytes, twelve mature astrocytes, one neuron, five oligodendrocytes, three macrophages/microglia, and two endothelial cells from published datasets(Zhang et al., 2016). Gene signatures of these human brain cell types were established by using CIBERSORT based on the compiled transcriptomes, and glioma cell signature as well. Then we assigned seven fraction scores for each of the 133 single cells using CIBERSORT based on the signature matrix of seven cell types. A single cell was assigned to the cell type with highest fraction score, hence to determine a single cell to be tumor or non-tumor cell (Newman et al., 2015). As a result, 98 of 133 cells were classified as tumor cells whereas the rest were identified as oligodendrocyte or microglia. Expression profiles of these 98 glioma cells were used for further analysis. A set of 95 of the 98 single cells with more than 5,000 genes detected were retained in the consequent analyses. We also compiled 672 single cells from a published dataset (Patel et al., 2014). 502 of the 672 single cells expressed more than 5,000 genes.
For more conservative identification of non-tumor-like case(s) in the 597 single cells, we performed an expression based CNV profiling analysis which is similar to the method used in previous publications (Patel et al., 2014). We identified 3,262 significantly differentially expressed genes (FDR<0.01) between TCGA tumors vs. normal cases. We then plotted the average expression per 60 gene window along genomic coordinates to infer the CNV. We clustered the gene expression estimated CNV profile of the 502 or 95 single cells together with eight non-tumor cells (human brain tissue isolated microglia, GSE80338)(Szulzewsky et al., 2016). For the 502 single cells, the unsupervised hierarchical clustering roughly identified eight major subgroups (group size>2), and all the non-tumor cells were clustered together with only one single cell (Figure S1A, left). For the 95 single cells, the eight non-tumor cells and single cells we clustered into two separate clusters without mixture (Figure S1A, right). Notably, the CNV profile of the single cell subgroups shows a typical CNV pattern of gliomas such as amplification of chromosome 7/12 and deletion of chromosome 9/10. We removed the single cell which was grouped together with non-tumor cells, and hence resulted a 596 single glioma cells population.
We analyzed the distribution of the gene express frequency in the 596 single glioma cells, and identified an abnormal low express frequency peak at 5%, while the frequency of genes which expressed in more than 5% single cells are approximately followed a uniform distribution. We reasoned that the genes expressed in less than 5% are highly suspected to be non-tumor cell intrinsically expressed and hence yield the abnormal distribution peak. By discarding the 8,214 genes expressed in less than 5% single cells, the retained 14,656 genes were considered as the candidate bona fide glioma genes.
Quantification and Statistical Analysis
For quantification of protein staining and measurement of distance between AIF1+ cells to NF1+ and NF- cells in GBM tumor sections, the IHC stained images were captured using Caliper Vectra Image System and analyzed data were generated using InForm software as described previously(Hu et al., 2016). In short, thirty scan fields were selected across the entire tumor section. In one tumor slide, the primary tumor of patient #2, nineteen scan fields were selected due to the small size of the tumor section. Percentages of the median and high levels (2+, 3+) of AIF1 were used for the comparison. AIF1+ cells were first located and the nearest NF1+ and NF1- cells within 40 pixels (∼28 μm) of each respective AIF1+ cell was defined and calculated using R package. Statistical analysis was performed by using Wilcoxon rank test.
Comparisons of neurosphere formation and gene expression by qRT-PCR were performed using the unpaired student's t-test. For all experiments with error bars, standard deviation was calculated to indicate the variation within each experiment and data, and values represent mean ± SD. All other statistical analyses were performed using R package (Version 3.2.5), and the detailed information about statistical methods were specified in figures/tables.
Data and Software Availability
Data Resources
The accession numbers for the whole transcriptome sequencing data generated in this study are EGAS00001001033 and EGAS00001002429.
Supplementary Material
Table S1. Glioma intrinsic genes and subtype signatures. Related to Figure 1.
Table S2. Classification concordance cross platforms. Related to Figure 1.
Table S3. Transcriptional subtype simplicity vs. subclonality. Related to Figure 2.
Table S4. Classification of TCGA IDH-WT GBMs and single cells. Related to Figure 2.
Table S5. Immune cell fraction of TCGA IDH-WT GBMs. Related to Figure 3.
Table S6. Clinical data, methylation subgroup switching, and immune cell fraction of primary/recurrent pairs. Related to Figure 5.
Table S7. Treatment annotations. Related to Figure 7.
Method S1. GBM.classification.zip. Related to Figure 1.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Anti-AIF1 | Abcam | Cat#:ab178846 |
| Mouse monoclonal anti-GFAP (GA5) | Cell Signaling Technology | Cat# 3670 RRID:AB_10831828 |
| Mouse monoclonal Anti-beta-Tubulin | Cell Signaling Technology | Cat# 2128, RRID:AB_823664) |
| Mouse monoclonal Anti-Vinculin | EMD Millipore | Cat# 05-386, RRID:AB_309711 |
| Mouse monoclonal anti-NF1 (McNFn27b) | GeneTex | Cat#:GTX15776 |
| Rabbit polyclonal anti-ITGAM | Sigma-Aldrich | Cat# HPA002274 RRID:AB_1078435 |
| Mouse monoclonal Anti-Bovine alpha-Actinin | Sigma-Aldrich | Cat# A5044 RRID:AB_476737 |
| Rabbit polyclonal Anti-Iba-1 | Wako | Cat# 016-20001 RRID:AB_839506 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EGF | Sigma-Aldrich | Cat#: E9644 |
| bFGF | Sigma-Aldrich | Cat# F0291 |
| Calcein AM | BD Biosciences | Cat#564061 |
| Critical Commercial Assays | ||
| RNA Truseq library prep kit | Illumina | Cat #.15042173 |
| MasterPure™ Complete DNA and RNA Purification Kit | Epicenter | Cat# MC85200 |
| BD FluoroBlok™ System | BD Biosciences | Cat# BD351161 |
| MACH 2 Double Stain 2 | Biocare Medical | Cat# MRCT525G |
| SMARTer® PCR cDNA Synthesis Kit | Clontech | Cat# 634926 |
| Nextera XT DNA Sample Prep Kit | Illumina | Cat#FC-131-1096 |
| Deposited Data | ||
| RNA sequencing data | European Genome-phenome Archive (EGA) | EGAS00001001033 |
| RNA sequencing data | European Genome-phenome Archive (EGA) | EGAS00001002429 |
| Software and Algorithms | ||
| InForm software | ||
| Pannoramic Viewer | ||
| Integrative Genomics Viewer (IGV, Version2.3) | ||
| R package (Version 3.2.5) | ||
Significance.
Glioblastoma expression subtypes have been related to genomic abnormalities, treatment response, and differences in tumor microenvironment. We defined tumor-intrinsic gene expression subtypes, which establishes a role for the tumor immune environment in shaping the tumor cell transcriptome. Notably, NF1 inactivation resulted in chemoattraction of macrophages/microglia. Comparison of matching primary and recurrent gliomas elucidated treatment-induced phenotypic tumor evolution, including expression subtype switching in nearly half of our cohort, as well as associations between microenvironmental components and treatment response. Characterization of the evolving glioblastoma transcriptome and tumor microenvironment aids in designing more effective immunotherapy trials. Our study provides a comprehensive transcriptional and cellular landscape of IDH wild-type glioblastoma during treatment modulated tumor evolution. All expression datasets are accessible through http://recur.bioinfo.cnio.es/.
Highlights.
- GBM-intrinsic transcriptional subtypes: proneural, classical, mesenchymal
- NF1 deficiency drives recruitment of tumor associated macrophages/microglia
- Resistance to radiotherapy may associate with M2 macrophage presence
- CD8+ T cells are enriched in temozolomide-induced hypermutated GBMs at recurrence
Acknowledgments
The authors thank Katherine Stemke-Hale for assistance in manuscript editing. The results published here are in part based upon data generated by The Cancer Genome Atlas project established by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) of the National Institutes of Health. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov. This work is supported by grants from the National Institutes of Health grants P50 CA127001 (EPS, FLM, RGWV), R01 CA190121 (EPS, RGWV), P50 CA097257 (MSB, JJP), RO1 CA120813 (ABH) and Cancer Center Support Grants P30CA16672 and P30CA034196; Cancer Prevention & Research Institute of Texas (CPRIT) grant number R140606, the University Cancer Foundation via the Institutional Research Grant program at the University of Texas MD Anderson Cancer Center (RGWV); the National Brain Tumor Association Defeat GBM project (EPS, RGWV), the National Brain Tumor Association Oligo Research Fund (RGWV); the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C3418, DHN); the Seve Ballesteros Foundation supported (MS); CPRIT RP140612 and American Cancer Society Research Scholar Grant RSG1514501CDD (FLM).
Footnotes
Author Contributions: Q.W., X.H., and R.G.W.V. designed the project; Q.W., X.H. and R.G.W.V analyzed data; E.P.S provided annotations and interpretations of clinical data; B.H. designed and performed the experimental validations; H.K., S.Z, E.M-L., E.E., and F.B. provided assistance for TCGA data analysis; M.S. provided implement of visualization of longitudinal datasets; L.S., A.C.D., S.L., P.L., Y.L., Z.D.L., G.F., J.J.P., and M.S.B. provided access to GBM biospecimens; K.R.G., G.W., E.C., Y-H.L., N.S., C-E.G.S., and A.O. provided assistance in cell culture, molecular biochemical and pathological experiments; H.J.C. and D-H.N. performed single cell sequencing and data analysis; E.P.S. provided access to molecular data of neurospheres and parental tumor bulks; A.B.H., J.H., F.M., T.M., J.J.P., M.S.B., R.A.D., D-H.N, and E.P.S. provided intellectual contributions in this study; Q.W., B.H., and R.G.W.V. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysan M, Bozdag S, Cam MC, Kotliarova S, Ahn S, Walling J, Killian JK, Stevenson H, Meltzer P, Fine HA. G-cimp status prediction of glioblastoma samples using mRNA expression data. PloS one. 2012;7:e47839. doi: 10.1371/journal.pone.0047839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H, Ray A, Spellman P, Speed TP. A single-sample method for normalizing and combining full-resolution copy numbers from multiple platforms, labs and analysis methods. Bioinformatics. 2009;25:861–867. doi: 10.1093/bioinformatics/btp074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The “cancer immunogram”. Science. 2016;352:658–660. doi: 10.1126/science.aaf2834. [DOI] [PubMed] [Google Scholar]
- Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol. 2016;34:2206–2211. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017;19:139–141. doi: 10.1093/neuonc/now247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucette T, Rao G, Rao A, Shen L, Aldape K, Wei J, Dziurzynski K, Gilbert M, Heimberger AB. Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res. 2013;1:112–122. doi: 10.1158/2326-6066.CIR-13-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, James CD, Molinaro A, Phillips JJ. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7:e43339. doi: 10.1371/journal.pone.0043339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1 doi: 10.1172/jci.insight.85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A, Yun J, Samanamud J, Sims JS, Banu M, et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc Natl Acad Sci U S A. 2014;111:12550–12555. doi: 10.1073/pnas.1405839111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2015;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wang Q, Wang YA, Hua S, Sauve CG, Ong D, Lan ZD, Chang Q, Ho YW, Monasterio MM, et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell. 2016;167:1281–1295 e1218. doi: 10.1016/j.cell.2016.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006;66:3987–3991. doi: 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59:1190–1199. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, Uppaluri R, Ferguson C, Schmidt RE, Dahiya S, et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016;6:1230–1236. doi: 10.1158/2159-8290.CD-16-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, Yang H, Choi YL, Park WY, Kong DS, et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013;3:260–273. doi: 10.1016/j.celrep.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Kim H, Verhaak RG. Transcriptional mimicry by tumor-associated stroma. Nat Genet. 2015;47:307–309. doi: 10.1038/ng.3255. [DOI] [PubMed] [Google Scholar]
- Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, Grimsby J, Sougnez C, Muller F, Hu J, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25:316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kwon SM, Kang SH, Park CK, Jung S, Park ES, Lee JS, Kim SH, Woo HG. Recurrent Glioblastomas Reveal Molecular Subtypes Associated with Mechanistic Implications of Drug-Resistance. PLoS One. 2015;10:e0140528. doi: 10.1371/journal.pone.0140528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–463. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Wang J, Sa JK, Ladewig E, Lee HO, Lee IH, Kang HJ, Rosenbloom DS, Camara PG, Liu Z, et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet. 2017;49:594–599. doi: 10.1038/ng.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, Yang JY, Broom BM, Verhaak RG, Kane DW, et al. TCPA: a resource for cancer functional proteomics data. Nat Methods. 2013;10:1046–1047. doi: 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E, Yoshihara K, Kim H, Mills GM, Trevino V, Verhaak RG. Comparison of gene expression patterns across 12 tumor types identifies a cancer supercluster characterized by TP53 mutations and cell cycle defects. Oncogene. 2015;34:2732–2740. doi: 10.1038/onc.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJ, Smirnov IV, Reis GF, Phillips JJ, Barnes MJ, et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell. 2015;28:307–317. doi: 10.1016/j.ccell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, Weichselbaum RR. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olar A, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. The Journal of pathology. 2014;232:165–177. doi: 10.1002/path.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng Y, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nature reviews Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF, Liau LM. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17:1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1. R inhibition in gliomas Science. 2016;352:aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Larsson O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics. 2007;8:48. doi: 10.1186/1471-2105-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Szulzewsky F, Arora S, de Witte L, Ulas T, Markovic D, Schultze JL, Holland EC, Synowitz M, Wolf SA, Kettenmann H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia. 2016;64:1416–1436. doi: 10.1002/glia.23014. [DOI] [PubMed] [Google Scholar]
- Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014;30:2224–2226. doi: 10.1093/bioinformatics/btu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bent MJ, Gao Y, Kerkhof M, Kros JM, Gorlia T, van Zwieten K, Prince J, van Duinen S, Sillevis Smitt PA, Taphoorn M, French PJ. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol. 2015;17:935–941. doi: 10.1093/neuonc/nov013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vulpen M, Kal HB, Taphoorn MJ, El-Sharouni SY. Changes in blood-brain barrier permeability induced by radiotherapy: implications for timing of chemotherapy? (Review) Oncol Rep. 2002;9:683–688. [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- Xu S, Wei J, Wang F, Kong LY, Ling XY, Nduom E, Gabrusiewicz K, Doucette T, Yang Y, Yaghi NK, et al. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Kong LY, Hu J, Gabrusiewicz K, Dibra D, Xia X, Heimberger AB, Li S. FGL2 as a Multimodality Regulator of Tumor-Mediated Immune Suppression and Therapeutic Target in Gliomas. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, Xiao HL, Wang B, Yi L, Wang QL, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol. 2012;189:444–453. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Chheda MG, Verhaak RG. Studying a complex tumor: potential and pitfalls. Cancer J. 2012;18:107–114. doi: 10.1097/PPO.0b013e3182431c57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Glioma intrinsic genes and subtype signatures. Related to Figure 1.
Table S2. Classification concordance cross platforms. Related to Figure 1.
Table S3. Transcriptional subtype simplicity vs. subclonality. Related to Figure 2.
Table S4. Classification of TCGA IDH-WT GBMs and single cells. Related to Figure 2.
Table S5. Immune cell fraction of TCGA IDH-WT GBMs. Related to Figure 3.
Table S6. Clinical data, methylation subgroup switching, and immune cell fraction of primary/recurrent pairs. Related to Figure 5.
Table S7. Treatment annotations. Related to Figure 7.
Method S1. GBM.classification.zip. Related to Figure 1.