

Abstract
Metabolic syndrome (MetS) is a group of cardio-metabolic risk factors that includes obesity, insulin resistance, hypertension, and dyslipidemia; these are also a combination of independent coronary artery disease (CAD) risk factors. Alarmingly, the prevalence of MetS risk factors are increasing and a leading cause for mortality. In the vasculature, complications from MetS and type 2 diabetes (T2D) can be divided into microvascular (retinopathy and nephropathy) and macrovascular (cardiovascular diseases and erectile dysfunction). In addition to vascular and endothelial dysfunction, vascular remodeling and stiffness are also hallmarks of cardiovascular disease (CVD), and well-characterized vascular changes that are observed in the early stages of hypertension, T2D, and obesity (1-3). In the heart, the link between obstructive atherosclerosis of coronary macrovessels and myocardial ischemia (MI) is well established. However, recent studies show that abnormalities in the coronary microcirculation are associated with functional and structural changes in coronary microvessels (classically defined as being <150-200 μm internal diameter), which may cause or contribute to MI even in the absence of obstractive CAD. This suggests a prognostic value of an abnormal coronary microcirculation as an early sub-clinical culprit in the pathogenesis and progression of heart disease in T2D and MetS. The aim of this review is to summarize recent studies investigating the coronary microvascular remodeling in an early pre-atherosclerotic phase of MetS and T2D, and to explore potential mechanisms associated with the timing of coronary microvascular remodeling relative to that of the macrovasculature.
Graphical abstract
Introduction
Metabolic Syndrome (MetS) is a cluster of metabolic disorders that includes at least three of the following: hyperglycemia, insulin resistance, abdominal obesity, hypertriclyceridemia/hyercholesterolemia, and hypertension. Much like type 2 diabetes (T2D), it is a risk factor for coronary heart disease and stroke. According to the American Heart Association (AHA), an estimated 34% of adult Americans have MetS, and its prevalence is increasing globally (4). T2D is a chronic disease characterized by hyperglycemia and insulin resistance, and it also is a major public health problem of considerable magnitude. T2D affects children, adolescents and adults, and according to the Centers for Disease Control and Prevention (CDC), 29.1 million people or 9.3% of the U.S. population had diabetes in 2014 (5). The total economic cost of diagnosed diabetes in 2012 was $245 billion, which is a 41% increase from our previous estimate of $174 billion (in 2007 dollars) (6). Both T2D and MetS confer a 2-to 4-fold increased risk of cardiovascular morbidity and mortality. CAD is the main cause of death in patients with T2D, accounting for about 70% of cases (7). However, improvements in therapy and management have contributed to a decrease in cardiovascular mortality, resulting in diabetics who live longer, thus experiencing more diabetes-related cardiovascular complications. Understanding the mechanisms associated with cardiovascular complications of T2D and MetS is crucial in order to develop therapeutics aiming to improve the health outcomes in people with T2D and MetS.
MetS is characterized by state of a chronic low-grade inflammation, which plays an important role in the progression of the disease. In fact, in diseases such as obesity and T2D, circulating levels of inflammatory markers such as C-reactive protein (CRP), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) are elevated (8, 9). These circulating inflammatory markers can induce endothelial dysfunction by mean of increasing reactive oxygen species (ROS) generation and decreasing nitric oxide (NO) bioavailability.
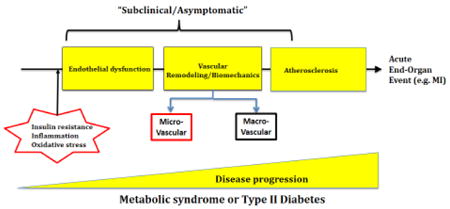
With the loss of a healthy endothelial layer, there is dysregulation in the ability of the endothelium to regulate vascular tone. Blood vessels then undergo vascular remodeling, which may be a compensatory mechanism to restore wall tension and normalize wall stress. Although signaling pathways involved in both macro- and microvascular remodeling are similar, the patterns and the progression of remodeling are different. In hypertension, small resistance arteries undergo eutrophic or hypertrophic remodeling. In contrast, large arteries undergo hypertrophic remodeling associated with increased media to lumen ratio and increased medial cross-sectional area (CSA) (10). Furthermore, macrovessels are prone at a later stage to atherosclerosis-induced remodeling as a consequence of endothelial and vascular wall injury and chronic inflammation. In T2D and MetS, animal studies from our and other laboratories have showed that while coronary microvessels undergo inward hypertrophic remodeling, mesenteric arterioles undergo outward hypertrophic remodeling, suggesting a different patterns of hypertrophic remodeling that is vascular bed specific. In early stages of disease progression, we found an absence of overt passive structural changes in macrovessels (aorta) (3, 11-14). We can conclude from these studies that microvessels may undergo vascular remodeling at an early stage of the disease progression and may present an early subclinical culprit in pathogenesis of coronary microvascular disease (CMD) in T2D and MetS.
Current dogma suggests that myocardial ischemia (MI) results from abnormalities of the epicardial coronary arteries, namely occlusive coronary artery disease as a result of atherosclerosis. However, recent studies have shown that coronary microvascular dysfunction and remodeling can contribute to MI in patients who were found to have normal or near normal coronary arteries during an angiography. This demonstrates that structural and functional CMD may have a prognostic value in clinical setting (15, 16). Of note, it is important to distinguish between CMD and CAD; while CAD is caused by atherosclerotic plaque build-up inside the coronary arteries at later stages of the disease, CMD is characterized by structural and functional changes in coronary resistance microvessels (CRM) that appear to start earlier during disease progression. This can occur in the presence or absence of atherosclerosis. The purpose of this review is to discuss the timing of macro- vs. micro-vascular disease, and the potential mechanisms and key players that may contribute to coronary microvascular disease in MetS and diabetes. In some cases where direct evidence from T2D or MetS is lacking, paralells are drawn from lessons learned in hypertension.
Physiology and Pathophysiology of the Vasculature
Due to pulsatile blood pressure and flow, the vascular wall is constantly subjected to hemodynamic forces such as cyclic stretch and shear stress. Pressure induces a rapid but sustained vasoconstriction called myogenic tone, whereas flow-mediated dilation results from shear stress. In fact, shear stress is a one of the regulators of the expression and production of vasodilatory agents such as nitric oxide (NO), prostacyclin and endothelium-derived hyperpolarizing factor (EDHF), in addition to vasoconstrictive agents such as endothelin-1 (ET-1) and angiotensin II (AngII) (17-19). Under physiological conditions where the vasculature is partially constricted due to sympathetic adrenergic influences, these vasoactive agents control vascular tone to regular blood flow; however, in disease conditions where chronic change in pressure or flow is observed, these vasoactive substances were shown to contribute to vascular remodeling by regulating VSMCs and ECM function.
Vascular remodeling is an adaptation response to physiological and pathophysiological stimuli, with the ultimate goal of restoring wall tension and normalizing wall stress. It is characterized by structural and functional alterations of the vascular wall in response to injury, disease or aging. The vessel wall is composed of endothelial cells (EC), vascular smooth muscle cells (VSMC) and fibroblasts, housed within an organized network of extracellular matrix (ECM). The ability of the vascular wall to reorganize its cellular and extracellular components is paramount for function. Vascular remodeling can be classified based on the change in lumen diameter (inward or outward) and wall area (eutrophic, hypotrophic or hypertrophic) (20, 21). Cells in the vascular wall respond to a plethora of stimuli such as mechanical forces, hormonal and vasoactive molecules, as well as inflammatory mediators.
ECs in the tunica intima, due to their location at the interface between the blood and vessel wall, act as sensors for mechanical stimuli (flow and pressure), as well as circulating humoral and inflammatory factors. In response to pressure change and circulating vasoactive substances in disease states, ECs release various mediators that regulate migration, proliferation, and apoptosis of VSMCs as well as the composition of the ECM (22).
Under normal conditions, VSMCs function as contractile cells that produce and organize the surrounding ECM support. The vascular wall maintains a contractile VSMC phenotype accompanied with slow ECM turnover. This net balance of normal VSMC function is altered in metabolic diseases, leading to phenotypic switch to either a “migro-proliferative” or “matrigenic” phenotype, with the first resulting in VSMC migration and/or proliferation producing thickening of the intima, while the latter resulting in VSMC differentiation to a secretory phenotype altering ECM composition (23-25). The ECM, regulated primarily by VSMCs, supports the structural integrity of the blood vessel wall, with fibrillar collagens providing most of the tensile support in conjunction with elastin providing elastic recoil properties. The relative mechanical contributions of elastin and collagen are thought to be responsible for the characteristic stress-strain curve of the vascular wall. However, literature suggests that the changes in both collagen and elastin were insufficient to explain the observed changes in vascular stiffness, suggesting contribution of intrinsic VSMC stiffness to overall vascular stiffness (26). The mechanisms involved in vascular remodeling in disease states are closely interrelated, culminating in an alteration of both the cellular and the non-cellular elements of the vascular wall.
Endothelial function in T2D/MetS
MetS is characterized by an early onset of endothelial dysfunction, which actually precedes vascular remodeling (see Graphical abstract) (12, 27). At an early stage of the disease, increased circulating inflammatory markers and oxidative stress lead to endothelial dysfunction. With the loss of the endothelium role in regulating vascular tone, the blood vessels undergo vascular remodeling to restore normal vascular wall tension.
Endothelium-induced coronary vasodilation was shown to be markedly attenuated in obese Ossabaw swine model of MetS (28). Furthermore, in a mouse model of T2D (db/db mice), endothelial dysfunction in CRMs is present as early as 12 weeks (29-31). Data from our laboratory demonstrate that CRM microvascular remodeling starts only at 16 weeks of age. In another model of MetS (Zucker rats), decreased endothelium-mediated relaxation to acetylcholine was observed as early as 11 weeks in the femoral artery (macrovessel), with no change in vascular remodeling (32). This suggests that endothelial dysfunction proceeds vascular macro and microvascular remodeling.
Factors such as hyperglycemia, elevated circulating free fatty acids, inflammatory markers (TNF-α, IL-6), and adipokines (leptin and resistin) can promote endothelial dysfunction in T2D and MetS, with increased oxidative stress playing a central role (27). In diseases such as hypertension and diabetes, endothelial dysfunction is characterized functionally by a decrease in endothelium-mediated vasorelaxation of the arteries and arterioles, partially due to decreased NO bioavailability (31, 33, 34). Decreased NO bioavailability can result from a either reduced eNOS activity or from direct reductions in NO via ROS scavenging (31). NO scavenging by superoxide (O2-) release can then cause further increases in ROS levels through the formation of peroxynitrate (ONOO-), also leading to eNOS uncoupling (35, 36). eNOS uncoupling then results in O2- generation by the enzyme rather than NO (35, 36). In T2D, hyperglycemia can induce ROS generation by direct glycation and inactivation of superoxide dismutase (SOD), as well as activation of the advanced glycation end products (AGE)-mediated activation of 1,2-diacylglycerol (DAG)/ protein kinase C (PKC) pathway, with subsequent activation of NAPDH oxidase (27, 37). PKC is critical in normal vascular function, yet can also contribute to endothelial dysfunction by increasing thromboxane A2 levels and reducing prostacyclin (PGI2) in a cyclooxygenase-2 (COX-2)-dependent manner (38). In addition, AGEs reduce eNOS mRNA half-life through an increased rate of mRNA degradation (37). This process also contributes to the reduction in NO bioavailability. The pro-inflammatory milieu present amplifies endothelial dysfunction through increased ROS generation and activation of VSMC pro-inflammatory pathways. These signaling pathways were shown to contribute to both endothelial/vascular dysfunction and remodeling, however, their contribution may differ depending on vascular bed and size of the vessel.
Macro vs. microvascular remodeling
Macrovessels (>150-200 μm internal diameter) and microvessels (<150-200 μm internal diameter) differ both in structure and function. Macrovessels, due to their large diameter, serve as low-resistance conduits supplying blood to peripheral organs. Due to their elastic properties, conduit arteries such as the aorta also regulate the pressure and flow pulsation resulting from intermittent ventricular ejection, which allow to transform oscillatory hemodynamics into a steady flow and pressure. In contrast, microvessels account for the majority of the resistance to blood flow, playing an important role in maintaining blood pressure and adequate tissue perfusion and nutrient delivery.
As mentioned above, vascular remodeling can be influenced by VSMC cell proliferation and/or hypertrophy, and alterations in the ECM. However, VSMC cytoskeleton and cell–cell/cell-ECM linkages are also integral to the remodeling process. These events can all be differentially regulated between micro- and macrovessels and demonstrate a time-dependent progression (37).
In MetS, the central pathological mechanism in macrovascular remodeling, including in the coronary circulation, is largly dictated by the process of atherosclerosis, which leads to narrowing of arterial walls throughout the body at later stages of disease. Nevertheless, pre-atherosclerotic structural changes in macrovessels can be associated with changes in wall thickness in hypertension (39) and/or ECM dysregulation associated with increased stiffness in T2D and MetS (40). The increased macrovascular stiffness leads to greater pulsatile stress and strain resulting in atherogenesis later during disease progression (41).
In contrast, microvascular remodeling is a hallmark of metabolic diseases, the onset of which occurs prior to the appearance of occlusive atherosclerosis. In hypertension, depending on the severity of hypertension, small resistance arteries undergo either inward eutrophic (moderate) or inward hypertrophic remodeling (severe), reliant on the changes in media CSA (42, 43). In diabetes and MetS, recent studies from our laboratory have shown that microvascular remodeling in diabetes depends on the type of diabetes (T1DM or T2DM), is dynamic, and is vascular bed specific (3, 12, 13). For instance, we reported that in a db/db mouse model of T2D that mesenteric arteries undergo outward hypertrophic remodeling, while CRMs exhibited inward hypertrophic remodeling that was associated with reduced stiffness (12, 13). Interestingly, while changes in coronary microvascular structure were observed, the coronary macrovascular (left descending coronary arteries) structure was preserved in swine and rat models of T2D and MetS (44, 45). This suggests that CRM remodeling preceeds coronary macrovascular remodeling in an early stage of T2D and MetS, advocating the prognostic value of CRM remodeling as an early sub-clinical culprit in the pathogenesis of heart disease in T2D and MetS.
Coronary microvascular remodeling- What are the potential key players in diabetes and metabolic syndrome?
Coronary microvascular remodeling in T2D and MetS
Most studies investigating vascular remodeling have used the aorta and mesenteric resistance vessels as universal surrogates to elucidate canonical mechanisms of macrovascular and microvascular disease, respectively (46, 47). While these are important studies, few studies have investigated remodeling in a vascular-bed-specific manner to account for physiological demand, in particular, microvascular remodeling in the coronary vasculature. This bed is unique in that it fills primarily during diastole and flow is dictated significantly by a tightly regulated combination of local metabolites, autonomic function, and structural considerations (structure-function relationships). Furthermore, deviations from this tight regulation lead to coronary artery disease, the number one contributor to heart failure. Poiseuille's Law states that flow is proportional to the radius to the 4th power; this law defines our understanding of the crucial importance of change of vascular diameter where small changes in vessel radius have a important effect on flow. Using mouse and swine models of MetS and T2D, we showed that CRMs undergo inward hypertrophic remodeling associated with decreased lumen diameter and increased wall thicknes and wall to lumen ratio (3, 11, 12, 44), associated with a decrease in CRM vascular stiffness. Interestingly, similar patterns of decreased stiffness and hypertrophy was recently observed in a rat model of aging (48). The structural changes in CRMs that we observed were accompanied with a decrease in coronary blood flow (CBF) and coronary flow reserve (CFR), suggesting that microvascular remodeling accounts for at least some of the deleterious ischemic events in T2D and MetS. In fact, in the absence of direct and precise quantification of microvascular function, CFR was proposed as a prognostic tool to predict coronary microvascular dysfunction in patients with angiographically normal arteries (15, 16).
Of note, we also reported that CRMs from T1D mice (streptozotocin model) did not exhibit any alterations in structural remodeling or mechanics (3). This is different in the CRMs between T1D and T2D, and may be due to to the distinct etiology of the two diseases. In fact, T2D is preceeded by a pre-diabetic phase characterized by early metabolic imbalance, increased insulin resistance and a slight increase in fasting glucose levels. These events increase and activate systemic innate immune cells and the respective inflammatory pathways in the vasculature, particularly the coronary arteries. In contrast, T1D-associated microvascular and cardiac complications occur a while after the onset of diabetes with a strong correlation between the CVD and the duration of T1D (49).
Furthermore, in hypertension, coronary macrovessels exhibited lower distensibility and a greater elastic modulus when compared to that of their normotensive controls (50, 51). While most studies showed a hypertrophic remodeling with increased wall thickness, the lumen diameter and W/L ratio and CSA were either increased or unchanged (50-53). The discrepancy between these studies may result from use of different coronary vessels (for example; LAD vs. septal coronary artery) or different methods of assessing vascular structure- histology (50, 51) vs. pressure myograph (52, 53). Thus, for future studies, it is imperative to differentiate and understand the differences between the coronary macrovascular (e.g. LAD) and coronary microvessels, when discussing coronary remodeling.
While the signaling mechanisms (s) associated with CRMs remodeling in T2D and MetS are still under investigation, they are usually closely interrelated, culminating in alterations of both the cellular and the non-cellular elements of the vascular wall. The remainder of this review will discuss potential key players in microvascular remodeling, as well as the possible contribution of these players in coronary microvascular remodeling.
Nitric oxide
As discussed above, nitric oxide synthase (NOS) -mainly eNOS- is the enzyme that catalyzes the conversion of L-arginine to L-citrulline and NO. NO can be generated in response to shear stress or through the activation of membrane receptors by vasoactive molecules such as acetylcholine and adenosine. In a rat model of MetS (Zucker rats), CMRs exhibited endothelial dysfunction associated with a decrease in coronary eNOS expression and NO production when compared to that of the lean control rats (54). In addition to regulation of the vascular tone, NO is an important regulator of vascular remodeling through the inhibition of VSMC proliferation (55) and regulation of extracellular matrix protein expression (56-58). Furthermore, NO regulates VSMC stiffness by inducing a reduction in VSMC Elastic modulus (E-modulus) and adhesion to collagen I, thus maintaining vascular wall's elasticity and preventing stiffness (59).
Several studies have focused on the role of NO in regulating vascular remodeling. Pharmacological inhibition of NOS in rats resulted in hypertension associated with coronary microvascular remodeling characterized by increased wall-to-lumen ratio and perivascular fibrosis (60). Additionally, the effect of NOS inhibition on microvascular remodeling seems to be predominantly from the decrease in NO bioavailability, as the normalization of blood pressure with hydralazine did not reverse vascular remodeling (60). Furthermore, genetic deletion of eNOS in mice was shown to cause an increase in wall thickness accompanied by a hyper-plastic response of the carotid arterial wall (55). Additionally, asymmetric dimethylarginine (ADMA) – an endogenous inhibitor of NO- was increased in hypertensive animals and associated with aortic and coronary microvascular remodeling (53, 61). The decrease in ADMA was associated with attenuation of vascular remodeling of aorta and coronary artery, through an increase in NO bioavailability and superoxide dismutase (SOD) activity (52, 53, 61).
Another cellular regulator of NO bioavailability is arginase. Arginase is expressed in the blood vessels where it competes with NOS for its substrate L-arginine, resulting in decreased NO production and bioavailability. Arginase expression and activity is elevated in disease states such as hypertension and diabetes (34, 39, 62, 63). Increased arginase expression was shown to contribute to endothelial dysfunction by reducing l-arginine availability to eNOS for NO production and thus to CRM vasodilation (64). In deoxycorticosterone acetate-salt hypertension (DOCA) model, genetic deletion of arginase reversed the increased coronary perivascular fibrosis observed in wild type mice (63). In an AngII-model of hypertension, an increase in wall to lumen ratio was associated with increased arginase expression, decreased NO bioavailability, increased proliferation in aorta, as well as coronary fibrosis with increased collagen expression (39). Pharmacological inhibition of arginase also shown to inhibit VSMC proliferation (39). This suggests a potential role of arginase in vascular remodeling.
Reactive oxygen species
In the vasculature, the major source of ROS are NADPH oxidases (NOX), xanthine oxidase, eNOS uncoupling, and mitochondrial electron transport chain, with O2- being the predominant effector. In normal conditions, ROS are generated at low levels and not only contribute to the regulation of vascular function but seem to be compulsory. In disease conditions, increased ROS generation is associated with endothelial dysfunction, as well as vascular remodeling (65, 66). The increased ROS levels may result from either increased ROS-generating pathways such as NADPH oxidase, or decreased ROS-scavenging pathways such as SOD (65, 66). ROS are central to numerous signaling pathways that regulate vascular remodeling. Indeed, ROS was shown to be involved in remodeling mediated by growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β). Finally, the role of mitochondrial ROS in mediateding microvascular remodeling in mesenteric arteriols was also observed in reduced renal mass hypertension model (67).
In addition, ROS modulates vascular tone and is associated with increased VSMC proliferation and migration, as well as phenotypic switching and/or ECM composition alteration (66, 68). Preliminary data from our laboratory demonstrated that inhibition of mitochondrial ROS prevented CRMs remodeling in T2D mouse model, suggesting a crutial role of ROS in microvascular remodeling.
Renin-Angiotensin system (RAS)
Angitensin II (AngII) is the classically-predominate bioactive component of the renin-angiotensin aldosterone system (RAAS), which aids in the maintenance of systemic BP through various mechanisms in the cardiovascular and renal systems (69, 70). Although AngII is crucial for renal function, this peptide is also implicated in the cardiovascular system, more specifically in vascular smooth muscle contraction. AngII also strongly regulates vascular remodeling via VSMC hypertrophy and hyperplasia (70). In addition, AngII increases VSMC stiffness via increased VSMC E-modulus and adhesion to ECM proteins (59, 71). AngII-mediated vascular remodeling has been extensively studied and is mediated by a plethora of intracellular pathways downstream of the AngII type 1 receptor (AT1R); this triggers proproliferative, profibrotic, and pro-inflammatory signals, all contributing to progression of the disease. The AT1R/NOX/ROS signaling pathways all seem to be a primary contributor to AngII-mediated vascular remodeling, as these effects can be attenuated by apocynin, a NOX inhibitor (66).
Mitogen activated protein kinase (MAPK), which is distinctive for its role in growth factor and cytokine activation, is activated by AT1R activation (70). The inhibition of the MAPK pathway improved vascular function and remodeling by reducing vascular growth, and through increased expression of the membrane metalloproteinases (MMP) (72). Furthermore, a recent study showed that both angiotensin-converting enzyme (ACE) inhibition and AT1R blockade prevented arterial wall hypertrophy in mesenteric arteries (73).
TGF-β, a potent regulator of VSMC phenotype and important modulator of ECM composition also contributes to AngII/AT1R-mediated remodeling in diabetic rats (74). Indeed, studies from our laboratory demonstrate that AngII/AT1R activation may contribute to the vascular remodeling observed in T2D (11). Additionally, this AT1R-mediated activation of the VSMC by pro-inflammatory signaling may contribute to remodeling present in metabolic diseases (75, 76).
Of note, AT2 receptor (AT2R) activation has opposing effects on the vasculature, as compared to AT1R activation. In line with this, AngII-mediated AT2R activation triggers anti-proliferative, anti-fibrotic, anti-inflammatory, and antioxidant effects; These findings make the AT2R a potential target for reversing vascular remodeling observed in diseases where canonical AT1R activation may play a role (77, 78). Thus, activation of this signaling pathway seems to counteract the more canonical AT1R-mediated events (70). In addition, another biologically active component of the RAS is Angiotensin (1-7) [Ang-(1-7)]. Ang-(1-7) is produced by Angiotensin converting enzyme 2 (ACE2) which convert AngI to Ang-(1-9), that is subsequently cleaved to generate Ang-(1-7), or by converting AngII directly to Ang-(1-7) (79, 80). Through the activation of its receptor MAS, Ang-(1-7) was shown to counteract most of the deleterious actions of the Ang II/AT1 receptor axis (76, 79, 80). Furthermore, Ang-(1-7)/MAS axis was to prevent vascular remodeling by inhibiting proliferative signaling pathway such as pkc, PDGF and TGF-β signaling pathways (79, 81).
Endothelin-1
In addition to AngII, ET-1 is another potent modulator of vascular tone and also contributes to vascular remodeling in diseases such as hypertension and diabetes (82). Similar to AngII, ET-1 signals through two distinguishable and divergent receptor subtypes, ETA and ETB receptors. In the vasculature, the ETA receptor is mainly located on VSMC. However, the ETB receptor is primarily located on endothelial cells, but may also be present on VSMC. Under physiological conditions, ETA receptor signaling mediates vasoconstriction, which is partly counteracted by endothelial ETB receptor-mediated release of NO (83). In pathophysiological conditions, there are increases in plasma ET-1 levels as well as expression and signaling of ET-1 receptors in the VSMC (83). Plasma ET-1 levels were shown to be elevated in humans with MetS (84, 85). In a prediabetic MetS dog model, Knudson et al. showed that ETA-mediated coronary arteriole vasocontriction was not different from the control group despite a significant decrease in ETA receptor expression and no change in circulating ET-1 levels (85), suggesting an early phase of coronary endothelial dysfunction due to the sensitization of ET-1 signaling (85).
ET-1-mediated intracellular signaling is very similar to that of AngII, resulting in activation of multiple signaling cascades that include PKC, MAPKs, ROS, and TGF-β. These pathways lead to subsequent increases in vascular proliferation, migration and fibrosis (70). Furthermore, ET-1 can also contribute to vascular remodeling by regulation of MMP activity (86) and changes in ECM composition, such as increased collagen levels (86, 87).
Advanced glycation end products (AGE)/receptor for advanced glycation end products (RAGE) pathway
AGEs are proteins or lipids that become glycated after exposure to sugars. AGEs contribute to a variety of microvascular and macrovascular complications via molecular cross-linking in the basement membrane of the ECM. It was shown to covalently cross-link ECM proteins, making these proteins resistant to hydrolytic turnover. This process results in the excessive accumulation of collagen and augmentation of stiffening in tissues such as the heart and aorta (88-92). In addition to directly binding ECM proteins, AGEs regulate ECM turnover and arterial stiffness via RAGE activation (93, 94). Under normal conditions, all polypeptides can be non-enzymatically glycated; however, the net effect of this phenomenon is substantially augmented in disease states such as diabetes (88). Increased vascular AGE causes endothelial dysfunction associated with increases RAGE expression and vascular cell adhesion molecule-1 (VCAM-1), as well as increased stiffness associated with increased pulse wave velocity (PWV), aortic wall thickness, and elastin disruption (93). AGE production and RAGE expression were shown to be increased in db/db mouse model of T2D and MetS, which were associated with enthothelial dysfunction in CRMs. Inhibition of AGE/RAGE signaling using soluble RAGE (sRAGE) improved endothelial function in diabetic mice, suggesting an important role of AGE/RAGE signaling in mediating CRMs endothelial dysfunction (95). In an experimental model of hypertension, both vascular AGE and RAGE levels were increased, which was associated with endothelial dysfunction, vascular hypertrophy in the aorta possibly due to increased VSMC proliferation and collagen deposition (96). This vascular effect of AGE/RAGE was mediated by increased tissue RAAS system activation, increased ROS and activation of pro-inflammatory nuclear factor NF-κB. AGE signaling inhibition improved vascular structure and function, and this effect was also blood pressure-independent (96). Furthermore, RAGE activation was shown to stimulate increased RAGE expression in a positive feedback manner that was ROS/NF-κB dependent. This reveals the feed-forward cycle of endothelial and vascular dysfunction, vascular remodeling and inflammation; all these factors further contribute to the progression and detriment of vascular pathology.
Using a mouse model of T2D (db/db mice), data from our laboratory showed a correlation between RAGE expression and vascular remodeling (unpublished data). While we did not observe any effect on diabetes on RAGE expression and remodeling in the aorta, diabetic CRMs underwent inward hypertrophic remodeling associated with increased RAGE expression. This hypertrophic remodeling was mitigated by genetic deletion of RAGE in db/db mice, suggesting that the observed microvascular remodeling is RAGE-dependent. AGE/RAGE signaling can induce VSMC phenotypic changes, proliferation and migration associated with increased MMP activation (97).
Inflammation
Low grade vascular inflammation plays an important role in progression of disease states. Immune cells such as T-cells and macrophages play a central role in mediating endothelial dysfunction and vascular remodeling (46, 47, 98, 99). These cells produce pro-inflammatory cytokines (such as TNF-α) and interleukins (IL) that contribute to vascular remodeling in a variety of ways (98). Lee et al. showed that cardiac TNF-α protein expression and serum IL-6 were significantly increased in a db/db mice, and were associated with endothelial dysfunction in the coronary microvasculature (100). In another study, plasma concentration and the expression of TNF-α and its receptor (TNFR1) were elevated in coronary arterioles of db/db mice resulting in coronary endothelial dysfunction (101). Furthermore, treatment with a TNF-α neutralizing antibody or TNF-α gene knock-out improved endothelial function in diabetic mice's coronary arterioles (101, 102).
In addition to increased circulating pro-inflammatory markers, increased expression of cell adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1) and VCAM-1 are strongly associated with vascular remodeling (46, 47). In diabetes and hypertension, activation of pro-inflammatory pathways, especially via NF-κB, is central to several pathways associated with vascular remodeling. The AngII, ET-1, AGE/RAGE and ROS pathways all culminate in increased NF-κB levels. In fact, NF-κB activation was shown to directly cause phenotype switching and increase of VSMC proliferation (97, 103-105). NF-κB also contributes to the disease progression by increasing expression of pro-inflammatory cytokines (TNF-α, IL-6, IL-8, IL-1) as well as cell adhesion molecules (ICAM-1 and VCAM-1) (106).
Conclusions
Recent studies have shed light on the functional and structural changes observed at an early stage of metabolic diseases. In T2D and MetS, our data and others show that microvascular endothelial dysfunction precedes the remodeling, which may be a compensatory mechanism to restore normal microvascular wall tension. Macrovascular remodeling occurs at a later stage, and can result from changes in VSMCs migration/proliferation as well as ECM tournover and/or in response to an atherosclerotic plaque.
In the heart, future studies aiming to unveil potential mechanistic and therapeutic approaches are crutial to our understanding of CMD, with the ultimate goal of preventing or halting the progression of cardiovascular complications. In clinical settings, several measurements relying on the quantification of CBF and CFR are used to describe the function of coronary microvasculature (15). However, the development of direct and quantitative methods to detect or predict CMD are critical, especially for patients with no obstructive CAD, and can be used to guide intervention aimed to reverse microvascular dysfunction and reduce the burden of risk factors.
Acknowledgments
Funding: This work was supported by the National Institutes of Health (R00HL116769 and S10OD023438 to AJT) and Nationwide Children's Hospital (to AJT).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prasad AM, Ketsawatsomkron P, Nuno DW, Koval OM, Dibbern ME, Venema AN, Sigmund CD, Lamping KG, Grumbach IM. Role of CaMKII in Ang-II-dependent small artery remodeling. Vascul Pharmacol. 2016 doi: 10.1016/j.vph.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bender SB, Castorena-Gonzalez JA, Garro M, Reyes-Aldasoro CC, Sowers JR, DeMarco VG, Martinez-Lemus LA. Regional variation in arterial stiffening and dysfunction in Western diet-induced obesity. Am J Physiol Heart Circ Physiol. 2015;309(4):H574–582. doi: 10.1152/ajpheart.00155.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trask AJ, Delbin MA, Katz PS, Zanesco A, Lucchesi PA. Differential coronary resistance microvessel remodeling between type 1 and type 2 diabetic mice: impact of exercise training. Vascul Pharmacol. 2012;57(5-6):187–193. doi: 10.1016/j.vph.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Heart Association. Available from: https://www.heart.org/idc/groups/heart-public/@wcm/@hcm/documents/downloadable/ucm_300322.pdf.
- 5.Centers for Disease Control and Prevention. Available from: https://www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf.
- 6.American Diabetes A. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013;36(4):1033–1046. doi: 10.2337/dc12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sacks FM. Reducing cardiovascular risk in metabolic syndrome and type 2 diabetes mellitus beyond low-density lipoprotein cholesterol lowering: a role for fenofibrate. Introduction. Am J Cardiol. 2008;102(12A):1L–3L. doi: 10.1016/j.amjcard.2008.09.066. [DOI] [PubMed] [Google Scholar]
- 8.Calabro P, Golia E, Maddaloni V, Malvezzi M, Casillo B, Marotta C, Calabro R, Golino P. Adipose tissue-mediated inflammation: the missing link between obesity and cardiovascular disease? Intern Emerg Med. 2009;4(1):25–34. doi: 10.1007/s11739-008-0207-2. [DOI] [PubMed] [Google Scholar]
- 9.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 10.Duprez DA. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens. 2006;24(6):983–991. doi: 10.1097/01.hjh.0000226182.60321.69. [DOI] [PubMed] [Google Scholar]
- 11.Husarek KE, Katz PS, Trask AJ, Galantowicz ML, Cismowski MJ, Lucchesi PA. The angiotensin receptor blocker losartan reduces coronary arteriole remodeling in type 2 diabetic mice. Vascul Pharmacol. 2016;76:28–36. doi: 10.1016/j.vph.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz PS, Trask AJ, Souza-Smith FM, Hutchinson KR, Galantowicz ML, Lord KC, Stewart JA, Jr, Cismowski MJ, Varner KJ, Lucchesi PA. Coronary arterioles in type 2 diabetic (db/db) mice undergo a distinct pattern of remodeling associated with decreased vessel stiffness. Basic Res Cardiol. 2011;106(6):1123–1134. doi: 10.1007/s00395-011-0201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Souza-Smith FM, Katz PS, Trask AJ, Stewart JA, Jr, Lord KC, Varner KJ, Vassallo DV, Lucchesi PA. Mesenteric resistance arteries in type 2 diabetic db/db mice undergo outward remodeling. PLoS One. 2011;6(8):e23337. doi: 10.1371/journal.pone.0023337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anghelescu M, Tonniges JR, Calomeni E, Shamhart PE, Agarwal G, Gooch KJ, Trask AJ. Vascular Mechanics in Decellularized Aortas and Coronary Resistance Microvessels in Type 2 Diabetic db/db Mice. Ann Biomed Eng. 2015;43(11):2760–2770. doi: 10.1007/s10439-015-1333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356(8):830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 16.Spoladore R, Fisicaro A, Faccini A, Camici PG. Coronary microvascular dysfunction in primary cardiomyopathies. Heart. 2014;100(10):806–813. doi: 10.1136/heartjnl-2013-304291. [DOI] [PubMed] [Google Scholar]
- 17.Matrougui K, Levy BI, Henrion D. Tissue angiotensin II and endothelin-1 modulate differently the response to flow in mesenteric resistance arteries of normotensive and spontaneously hypertensive rats. Br J Pharmacol. 2000;130(3):521–526. doi: 10.1038/sj.bjp.0703371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uematsu M, Ohara Y, Navas JP, Nishida K, Murphy TJ, Alexander RW, Nerem RM, Harrison DG. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am J Physiol. 1995;269(6 Pt 1):C1371–1378. doi: 10.1152/ajpcell.1995.269.6.C1371. [DOI] [PubMed] [Google Scholar]
- 19.Malek A, Izumo S. Physiological fluid shear stress causes downregulation of endothelin-1 mRNA in bovine aortic endothelium. Am J Physiol. 1992;263(2 Pt 1):C389–396. doi: 10.1152/ajpcell.1992.263.2.C389. [DOI] [PubMed] [Google Scholar]
- 20.Mulvany MJ. Vascular remodelling of resistance vessels: can we define this? Cardiovasc Res. 1999;41(1):9–13. doi: 10.1016/s0008-6363(98)00289-2. [DOI] [PubMed] [Google Scholar]
- 21.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension. 1989;13(6 Pt 2):968–972. doi: 10.1161/01.hyp.13.6.968. [DOI] [PubMed] [Google Scholar]
- 22.Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330(20):1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 23.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 24.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 25.Stegemann JP, Hong H, Nerem RM. Mechanical, biochemical, and extracellular matrix effects on vascular smooth muscle cell phenotype. J Appl Physiol. 2005;98(6):2321–2327. doi: 10.1152/japplphysiol.01114.2004. [DOI] [PubMed] [Google Scholar]
- 26.Sehgel NL, Vatner SF, Meininger GA. “Smooth Muscle Cell Stiffness Syndrome”-Revisiting the Structural Basis of Arterial Stiffness. Front Physiol. 2015;6:335. doi: 10.3389/fphys.2015.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picchi A, Capobianco S, Qiu T, Focardi M, Zou X, Cao JM, Zhang C. Coronary microvascular dysfunction in diabetes mellitus: A review. World J Cardiol. 2010;2(11):377–390. doi: 10.4330/wjc.v2.i11.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bratz IN, Dick GM, Tune JD, Edwards JM, Neeb ZP, Dincer UD, Sturek M. Impaired capsaicin-induced relaxation of coronary arteries in a porcine model of the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2008;294(6):H2489–2496. doi: 10.1152/ajpheart.01191.2007. [DOI] [PubMed] [Google Scholar]
- 29.Bagi Z, Koller A, Kaley G. Superoxide-NO interaction decreases flow- and agonist-induced dilations of coronary arterioles in Type 2 diabetes mellitus. Am J Physiol-Heart C. 2003;285(4):H1404–H1410. doi: 10.1152/ajpheart.00235.2003. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Belmadani S, Picchi A, Xu XB, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang CH. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007;115(2):245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- 31.Kassan M, Choi SK, Galan M, Bishop A, Umezawa K, Trebak M, Belmadani S, Matrougui K. Enhanced NF-kappaB activity impairs vascular function through PARP-1-, SP-1-, and COX-2-dependent mechanisms in type 2 diabetes. Diabetes. 2013;62(6):2078–2087. doi: 10.2337/db12-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu X, Guo XM, Karathanasis SK, Zimmerman KM, Onyia JE, Peterson RG, Kassab GS. Rosiglitazone reverses endothelial dysfunction but not remodeling of femoral artery in Zucker diabetic fatty rats. Cardiovasc Diabetol. 2010;9 doi: 10.1186/1475-2840-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wynne BM, Labazi H, Tostes RC, Webb RC. Aorta from angiotensin II hypertensive mice exhibit preserved nitroxyl anion mediated relaxation responses. Pharmacol Res. 2012;65(1):41–47. doi: 10.1016/j.phrs.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toque HA, Nunes KP, Yao L, Xu Z, Kondrikov D, Su Y, Webb RC, Caldwell RB, Caldwell RW. Akita spontaneously type 1 diabetic mice exhibit elevated vascular arginase and impaired vascular endothelial and nitrergic function. PLoS One. 2013;8(8):e72277. doi: 10.1371/journal.pone.0072277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, Belin de Chantemele E, Feher A, Romero MJ, Bagi Z. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes. 2014;63(4):1381–1393. doi: 10.2337/db13-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB. High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci. 2003;44(7):3135–3143. doi: 10.1167/iovs.02-1022. [DOI] [PubMed] [Google Scholar]
- 37.Delbin MA, Trask AJ. The diabetic vasculature: Physiological mechanisms of dysfunction and influence of aerobic exercise training in animal models. Life Sci. 2014;102(1):1–9. doi: 10.1016/j.lfs.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 38.Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Gatti CD, Joch H, Volpe M, Luscher TF. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells - Role of protein kinase C and reactive oxygen species. Circulation. 2003;107(7):1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- 39.Bhatta A, Yao L, Toque HA, Shatanawi A, Xu Z, Caldwell RB, Caldwell RW. Angiotensin II-induced arterial thickening, fibrosis and stiffening involves elevated arginase function. PLoS One. 2015;10(3):e0121727. doi: 10.1371/journal.pone.0121727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayden MR, Sowers JR, Tyagi SC. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: the matrix preloaded. Cardiovasc Diabetol. 2005;4 doi: 10.1186/1475-2840-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Payne RA, Webb DJ. Arterial blood pressure and stiffness in hypertension: is arterial structure important? Hypertension. 2006;48(3):366–367. doi: 10.1161/01.HYP.0000237668.31786.1f. [DOI] [PubMed] [Google Scholar]
- 42.Schiffrin EL. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension. 2012;59(2):367–374. doi: 10.1161/HYPERTENSIONAHA.111.187021. [DOI] [PubMed] [Google Scholar]
- 43.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38(3 Pt 2):581–587. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 44.Trask AJ, Katz PS, Kelly AP, Galantowicz ML, Cismowski MJ, West TA, Neeb ZP, Berwick ZC, Goodwill AG, Alloosh M, Tune JD, Sturek M, Lucchesi PA. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol (1985) 2012;113(7):1128–1140. doi: 10.1152/japplphysiol.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villalba N, Martinez P, Briones AM, Sanchez A, Salaices M, Garcia-Sacristan A, Hernandez M, Benedito S, Prieto D. Differential structural and functional changes in penile and coronary arteries from obese Zucker rats. Am J Physiol Heart Circ Physiol. 2009;297(2):H696–707. doi: 10.1152/ajpheart.01308.2008. [DOI] [PubMed] [Google Scholar]
- 46.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25(10):2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 47.Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, Schiffrin EL. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol. 2007;292(4):H1789–1795. doi: 10.1152/ajpheart.01118.2006. [DOI] [PubMed] [Google Scholar]
- 48.Hanna MA, Taylor CR, Chen B, La HS, Maraj JJ, Kilar CR, Behnke BJ, Delp MD, Muller-Delp JM. Structural remodeling of coronary resistance arteries: effects of age and exercise training. J Appl Physiol (1985) 2014;117(6):616–623. doi: 10.1152/japplphysiol.01296.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lipes MA, Galderisi A. Cardiac autoimmunity as a novel biomarker, mediator, and therapeutic target of heart disease in type 1 diabetes. Curr Diab Rep. 2015;15(5):30. doi: 10.1007/s11892-015-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pourageaud F, Crabos M, Freslon JL. The elastic modulus of conductance coronary arteries from spontaneously hypertensive rats is increased. J Hypertens. 1997;15(10):1113–1121. doi: 10.1097/00004872-199715100-00009. [DOI] [PubMed] [Google Scholar]
- 51.Roque FR, Briones AM, Garcia-Redondo AB, Galan M, Martinez-Revelles S, Avendano MS, Cachofeiro V, Fernandes T, Vassallo DV, Oliveira EM, Salaices M. Aerobic exercise reduces oxidative stress and improves vascular changes of small mesenteric and coronary arteries in hypertension. Br J Pharmacol. 2013;168(3):686–703. doi: 10.1111/j.1476-5381.2012.02224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnalich-Montiel A, Gonzalez MC, Delgado-Baeza E, Delgado-Martos MJ, Condezo-Hoyos L, Martos-Rodriguez A, Rodriguez-Rodriguez P, Quintana-Villamandos B. Short-term esmolol improves coronary artery remodeling in spontaneously hypertensive rats through increased nitric oxide bioavailability and superoxide dismutase activity. Biomed Res Int. 2014;2014:531087. doi: 10.1155/2014/531087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quintana-Villamandos B, Arnalich-Montiel A, Arribas S, Luneburg N, Boger RH, Delgado-Martos MJ, Fernandez-Criado C, Delgado-Baeza E, Gonzalez MC. Early regression of coronary artery remodeling with esmolol and DDAH/ADMA pathway in hypertensive rats. Hypertens Res. 2016 doi: 10.1038/hr.2016.57. [DOI] [PubMed] [Google Scholar]
- 54.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99(1):69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 55.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest. 1998;101(4):731–736. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hale SA, Weger L, Mandala M, Osol G. Reduced NO signaling during pregnancy attenuates outward uterine artery remodeling by altering MMP expression and collagen and elastin deposition. Am J Physiol Heart Circ Physiol. 2011;301(4):H1266–1275. doi: 10.1152/ajpheart.00519.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myers PR, Tanner MA. Vascular endothelial cell regulation of extracellular matrix collagen: role of nitric oxide. Arterioscler Thromb Vasc Biol. 1998;18(5):717–722. doi: 10.1161/01.atv.18.5.717. [DOI] [PubMed] [Google Scholar]
- 58.Tronc F, Mallat Z, Lehoux S, Wassef M, Esposito B, Tedgui A. Role of matrix metalloproteinases in blood flow-induced arterial enlargement: interaction with NO. Arterioscler Thromb Vasc Biol. 2000;20(12):E120–126. doi: 10.1161/01.atv.20.12.e120. [DOI] [PubMed] [Google Scholar]
- 59.Hong Z, Reeves KJ, Sun Z, Li Z, Brown NJ, Meininger GA. Vascular smooth muscle cell stiffness and adhesion to collagen I modified by vasoactive agonists. PLoS One. 2015;10(3):e0119533. doi: 10.1371/journal.pone.0119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Numaguchi K, Egashira K, Takemoto M, Kadokami T, Shimokawa H, Sueishi K, Takeshita A. Chronic inhibition of nitric oxide synthesis causes coronary microvascular remodeling in rats. Hypertension. 1995;26(6 Pt 1):957–962. doi: 10.1161/01.hyp.26.6.957. [DOI] [PubMed] [Google Scholar]
- 61.Quintana-Villamandos B, Gonzalez MC, Delgado-Martos MJ, Condezo-Hoyos L, Boger RH, Luneburg N, Pazo-Sayos L, Gutierrez-Arzapalo PY, Delgado-Baeza E. Short-term esmolol attenuates remodeling of the thoracic aorta in hypertensive rats by decreasing concentrations of ADMA down-regulated by oxidative stress. Eur J Pharmacol. 2016;791:502–509. doi: 10.1016/j.ejphar.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 62.Elms SC, Toque HA, Rojas M, Xu Z, Caldwell RW, Caldwell RB. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia. 2013;56(3):654–662. doi: 10.1007/s00125-012-2789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toque HA, Nunes KP, Rojas M, Bhatta A, Yao L, Xu Z, Romero MJ, Webb RC, Caldwell RB, Caldwell RW. Arginase 1 mediates increased blood pressure and contributes to vascular endothelial dysfunction in deoxycorticosterone acetate-salt hypertension. Front Immunol. 2013;4:219. doi: 10.3389/fimmu.2013.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang W, Hein TW, Zhang C, Zawieja DC, Liao JC, Kuo L. Oxidized low-density lipoprotein inhibits nitric oxide-mediated coronary arteriolar dilation by up-regulating endothelial arginase I. Microcirculation. 2011;18(1):36–45. doi: 10.1111/j.1549-8719.2010.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cho YE, Basu A, Dai A, Heldak M, Makino A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am J Physiol Cell Physiol. 2013;305(10):C1033–1040. doi: 10.1152/ajpcell.00234.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22(3):535–542. doi: 10.1097/00004872-200403000-00016. [DOI] [PubMed] [Google Scholar]
- 67.Wang C, Luo Z, Kohan D, Wellstein A, Jose PA, Welch WJ, Wilcox CS, Wang D. Thromboxane prostanoid receptors enhance contractions, endothelin-1, and oxidative stress in microvessels from mice with chronic kidney disease. Hypertension. 2015;65(5):1055–1063. doi: 10.1161/HYPERTENSIONAHA.115.05244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Staiculescu MC, Foote C, Meininger GA, Martinez-Lemus LA. The role of reactive oxygen species in microvascular remodeling. Int J Mol Sci. 2014;15(12):23792–23835. doi: 10.3390/ijms151223792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nunes KP, Labazi H, Webb RC. New insights into hypertension-associated erectile dysfunction. Curr Opin Nephrol Hypertens. 2012;21(2):163–170. doi: 10.1097/MNH.0b013e32835021bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wynne BM, Chiao CW, Webb RC. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J Am Soc Hypertens. 2009;3(2):84–95. doi: 10.1016/j.jash.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hong Z, Sun Z, Li Z, Mesquitta WT, Trzeciakowski JP, Meininger GA. Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc Res. 2012;96(1):73–80. doi: 10.1093/cvr/cvs239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Potthoff SA, Stamer S, Grave K, Konigshausen E, Sivritas SH, Thieme M, Mori Y, Woznowski M, Rump LC, Stegbauer J. Chronic p38 mitogen-activated protein kinase inhibition improves vascular function and remodeling in angiotensin II-dependent hypertension. J Renin Angiotensin Aldosterone Syst. 2016;17(3) doi: 10.1177/1470320316653284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cousin M, Custaud MA, Baron-Menguy C, Toutain B, Dumont O, Guihot AL, Vessieres E, Subra JF, Henrion D, Loufrani L. Role of angiotensin II in the remodeling induced by a chronic increase in flow in rat mesenteric resistance arteries. Hypertension. 2010;55(1):109–115. doi: 10.1161/HYPERTENSIONAHA.108.127456. [DOI] [PubMed] [Google Scholar]
- 74.Sun H, Zhao Y, Bi X, Li S, Su G, Miao Y, Ma X, Zhang Y, Zhang W, Zhong M. Valsartan blocks thrombospondin/transforming growth factor/Smads to inhibit aortic remodeling in diabetic rats. Diagn Pathol. 2015;10:18. doi: 10.1186/s13000-015-0246-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakashima T, Umemoto S, Yoshimura K, Matsuda S, Itoh S, Murata T, Fukai T, Matsuzaki M. TLR4 is a critical regulator of angiotensin II-induced vascular remodeling: the roles of extracellular SOD and NADPH oxidase. Hypertens Res. 2015;38(10):649–655. doi: 10.1038/hr.2015.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bihl JC, Zhang C, Zhao Y, Xiao X, Ma X, Chen Y, Chen S, Zhao B, Chen Y. Angiotensin-(1-7) counteracts the effects of Ang II on vascular smooth muscle cells, vascular remodeling and hemorrhagic stroke: Role of the NFsmall ka, CyrillicB inflammatory pathway. Vascul Pharmacol. 2015;73:115–123. doi: 10.1016/j.vph.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Foulquier S, Steckelings UM, Unger T. Impact of the AT(2) receptor agonist C21 on blood pressure and beyond. Curr Hypertens Rep. 2012;14(5):403–409. doi: 10.1007/s11906-012-0291-6. [DOI] [PubMed] [Google Scholar]
- 78.Paulis L, Becker ST, Lucht K, Schwengel K, Slavic S, Kaschina E, Thone-Reineke C, Dahlof B, Baulmann J, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation in Nomega-nitro-L-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension. 2012;59(2):485–492. doi: 10.1161/HYPERTENSIONAHA.111.185496. [DOI] [PubMed] [Google Scholar]
- 79.Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216(2):R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 80.Trask AJ, Ferrario CM. Angiotensin-(1-7): pharmacology and new perspectives in cardiovascular treatments. Cardiovasc Drug Rev. 2007;25(2):162–174. doi: 10.1111/j.1527-3466.2007.00012.x. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Z, Chen L, Zhong J, Gao P, Oudit GY. ACE2/Ang-(1-7) signaling and vascular remodeling. Sci China Life Sci. 2014;57(8):802–808. doi: 10.1007/s11427-014-4693-3. [DOI] [PubMed] [Google Scholar]
- 82.Sachidanandam K, Hutchinson JR, Elgebaly MM, Mezzetti EM, Dorrance AM, Motamed K, Ergul A. Glycemic control prevents microvascular remodeling and increased tone in type 2 diabetes: link to endothelin-1. Am J Physiol Regul Integr Comp Physiol. 2009;296(4):R952–959. doi: 10.1152/ajpregu.90537.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. 2007;76(1):8–18. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 84.Ferri C, Bellini C, Desideri G, Baldoncini R, Properzi G, Santucci A, De Mattia G. Circulating endothelin-1 levels in obese patients with the metabolic syndrome. Exp Clin Endocrinol Diabetes. 1997;105(Suppl 2):38–40. doi: 10.1055/s-0029-1211794. [DOI] [PubMed] [Google Scholar]
- 85.Knudson JD, Rogers PA, Dincer UD, Bratz IN, Araiza AG, Dick GM, Tune JD. Coronary vasomotor reactivity to endothelin-1 in the prediabetic metabolic syndrome. Microcirculation. 2006;13(3):209–218. doi: 10.1080/10739680600556894. [DOI] [PubMed] [Google Scholar]
- 86.Sachidanandam K, Portik-Dobos V, Kelly-Cobbs AI, Ergul A. Dual endothelin receptor antagonism prevents remodeling of resistance arteries in diabetes. Can J Physiol Pharmacol. 2010;88(6):616–621. doi: 10.1139/Y10-034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pu Q, Neves MF, Virdis A, Touyz RM, Schiffrin EL. Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodeling. Hypertension. 2003;42(1):49–55. doi: 10.1161/01.HYP.0000078357.92682.EC. [DOI] [PubMed] [Google Scholar]
- 88.Schmidt AM, Yan SD, Wautier JL, Stern D. Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999;84(5):489–497. doi: 10.1161/01.res.84.5.489. [DOI] [PubMed] [Google Scholar]
- 89.Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232(4758):1629–1632. doi: 10.1126/science.3487117. [DOI] [PubMed] [Google Scholar]
- 90.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(5):932–943. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 91.Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, deGroof RC, Lakatta EG. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104(13):1464–1470. doi: 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- 92.Aronson D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens. 2003;21(1):3–12. doi: 10.1097/00004872-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 93.Grossin N, Auger F, Niquet-Leridon C, Durieux N, Montaigne D, Schmidt AM, Susen S, Jacolot P, Beuscart JB, Tessier FJ, Boulanger E. Dietary CML-enriched protein induces functional arterial aging in a RAGE-dependent manner in mice. Mol Nutr Food Res. 2015;59(5):927–938. doi: 10.1002/mnfr.201400643. [DOI] [PubMed] [Google Scholar]
- 94.Serban AI, Stanca L, Geicu OI, Munteanu MC, Dinischiotu A. RAGE and TGF-beta1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS One. 2016;11(3):e0152376. doi: 10.1371/journal.pone.0152376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gao X, Zhang H, Schmidt AM, Zhang C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2008;295(2):H491–498. doi: 10.1152/ajpheart.00464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Y, Yu M, Zhang L, Cao Q, Song Y, Liu Y, Gong J. Soluble receptor for advanced glycation end products mitigates vascular dysfunction in spontaneously hypertensive rats. Mol Cell Biochem. 2016;419(1-2):165–176. doi: 10.1007/s11010-016-2763-5. [DOI] [PubMed] [Google Scholar]
- 97.Chaabane C, Heizmann CW, Bochaton-Piallat ML. Extracellular S100A4 induces smooth muscle cell phenotypic transition mediated by RAGE. Biochim Biophys Acta. 2015;1853(9):2144–2157. doi: 10.1016/j.bbamcr.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 98.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107(2):263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee S, Park Y, Zhang C. Exercise Training Prevents Coronary Endothelial Dysfunction in Type 2 Diabetic Mice. Am J Biomed Sci. 2011;3(4):241–252. doi: 10.5099/aj110400241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang C, Park Y, Picchi A, Potter BJ. Maturation-induces endothelial dysfunction via vascular inflammation in diabetic mice. Basic Res Cardiol. 2008;103(5):407–416. doi: 10.1007/s00395-008-0725-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Park Y, Capobianco S, Gao X, Falck JR, Dellsperger KC, Zhang C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;295(5):H1982–1988. doi: 10.1152/ajpheart.01261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heiss EH, Liu R, Waltenberger B, Khan S, Schachner D, Kollmann P, Zimmermann K, Cabaravdic M, Uhrin P, Stuppner H, Breuss JM, Atanasov AG, Dirsch VM. Plumericin inhibits proliferation of vascular smooth muscle cells by blocking STAT3 signaling via S-glutathionylation. Sci Rep. 2016;6:20771. doi: 10.1038/srep20771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ogawa A, Firth AL, Yao W, Rubin LJ, Yuan JX. Prednisolone inhibits PDGF-induced nuclear translocation of NF-kappaB in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2008;295(4):L648–657. doi: 10.1152/ajplung.90245.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Singh P, Zheng XL. Dual regulation of myocardin expression by tumor necrosis factor-alpha in vascular smooth muscle cells. PLoS One. 2014;9(11):e112120. doi: 10.1371/journal.pone.0112120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.De Martin R, Hoeth M, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000;20(11):E83–88. doi: 10.1161/01.atv.20.11.e83. [DOI] [PubMed] [Google Scholar]