

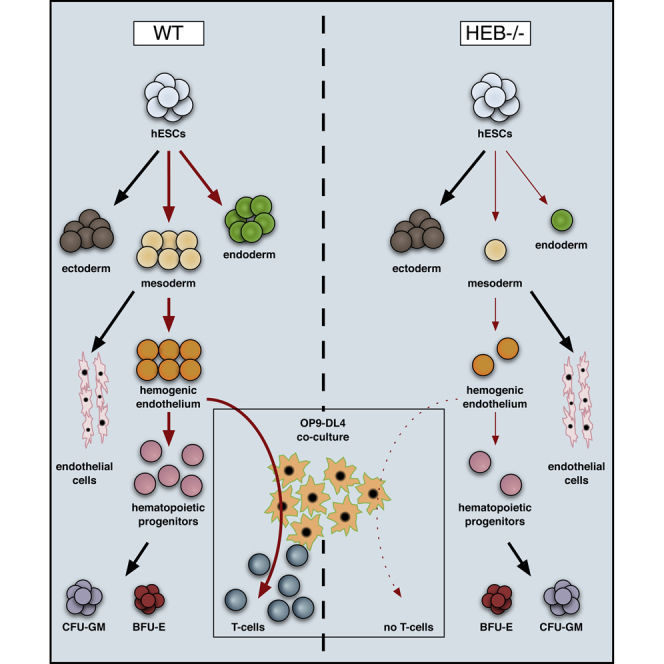
Summary
Hematopoietic stem cells arise from mesoderm-derived hemogenic endothelium (HE) during embryogenesis in a process termed endothelial-hematopoietic transition (EHT). To better understand the gene networks that control this process, we investigated the role of the transcription factor HEB (TCF12) by disrupting the TCF12 gene locus in human embryonic stem cells (hESCs) and inducing them to differentiate toward hematopoietic outcomes. HEB-deficient hESCs retained key features of pluripotency, including expression of SOX2 and SSEA-4 and teratoma formation, while NANOG expression was reduced. Differentiation of HEB−/− hESCs toward hematopoietic fates revealed a severe defect in mesodermal development accompanied by decreased expression of regulators of mesoendodermal fate choices. We also identified independent defects in HE formation at the molecular and cellular levels, as well as a failure of T cell development. All defects were largely rescued by re-expression of HEB. Taken together, our results identify HEB as a critical regulator of human mesodermal and hematopoietic specification.
Keywords: human embryonic stem cell, genome editing, transcriptional regulation, HEB, mesoderm, hemogenic endothelium, hematopoiesis, Runx1, Notch1, T cell development
Graphical Abstract
Highlights
-
•
Genome editing targeting TCF12 in hESCs to study human embryonic development
-
•
HEB is required for NANOG and TGFβ signaling but not for hESC pluripotency
-
•
Requirement for HEB in mesoderm development and pre-hematopoietic events
-
•
HEB is required for expression of Notch1 and Runx1 in endothelial cells
Li et al. establish the first E protein knockout in hESCs and find that, although pluripotency is intact, HEB factors are required for the expression of NANOG and TGFβ family members. Their results also reveal essential roles for HEB at multiple stages of pre-hematopoietic development, placing HEB at a top tier of the gene regulatory network for human hematopoiesis.
Introduction
During embryogenesis, multiple waves of mesodermal progenitors generate hematopoietic cells with different characteristics. Two early waves of hematopoietic progenitors give rise to primitive progenitors, followed by distinct myeloid and erythroid populations known as EMPs (Ditadi et al., 2017). At a later time point, hematopoietic stem cells (HSCs), which are distinguished by their self-renewing capacity, arise from mesodermal precursors with endothelial characteristics. This process, termed the endothelial-to-hematopoietic transition (EHT), involves a transient developmental intermediate known as hemogenic endothelium (HE) (Gritz and Hirschi, 2016). Early mesodermal specification from embryonic stem cells (ESCs) and generation of HE are both dependent on signaling through transforming growth factor β (TGFβ) family members (Hadjimichael et al., 2016, Hong et al., 2011, Monteiro et al., 2016). As mesodermal precursors differentiate toward the hematopoietic lineage, the transcription factors Runt-related transcription factor 1 (Runx1) and Notch1 are upregulated, and these factors are essential for HE generation in an evolutionarily conserved manner (Burns et al., 2005, Butko et al., 2016, Ditadi et al., 2015). Without Runx1, HE does not form (Yzaguirre et al., 2017). Furthermore, Runx1 and its target Spi1 (Huang et al., 2008) are among four factors that can reprogram adult endothelial cells into HSCs with long-term engrafting and lymphoid potential (Lis et al., 2017). The expression of a specific isoform of Runx1 also marks HE as distinct from arterial vascular endothelium in human ESC (hESC)-derived progenitors (Ditadi et al., 2015).
Notch1 is also a key regulator of HE. Notch1 directly upregulates Runx1 and controls the HSC-associated factor GATA3 (Burns et al., 2005, Butko et al., 2016, Ditadi et al., 2015, Frelin et al., 2013). Consequently, the generation of HE and the process of EHT are severely compromised in the absence of Notch signaling (Butko et al., 2016). The transcription factor HEB operates in the context of Notch1 and Runx1 during T cell development (Braunstein and Anderson, 2012), and has been shown to act cooperatively with the SMAD factors, downstream of TGFβ family signaling, in mouse ESCs (mESCs) (Yoon et al., 2015). Furthermore, HEB and Notch1 operate in a positive feedback loop during early T cell development (Braunstein and Anderson, 2012). In addition, HEB has been implicated in mesodermal development from mESCs (Yoon et al., 2015), potentially placing it upstream of HE formation.
HEB belongs to the E protein transcription factor family, which also includes E2A (TCF3) and E2-2 (TCF4) (Murre et al., 1989). E proteins regulate transcription of their target genes as obligate dimers, dimerizing with each other or with class II basic helix-loop-helix factors. HEB factors are the products of the TCF12 gene locus, which encodes both the canonical HEB protein (HEBCan) and a shorter variant (HEBAlt) by way of alternative transcriptional initiation and alternative splicing (Hu et al., 1992, Wang et al., 2006). HEB is important in various developmental processes, including T-lymphopoiesis, neurogenesis, and myogenesis (Barndt et al., 1999, Parker et al., 2006, Ravanpay and Olson, 2008). Among the E proteins E2A has been well studied, but far less is known about HEB.
To address potential roles for HEB factors in the generation of HE, we knocked out HEB protein expression in hESCs by targeting the TCF12 locus using the CRISPR/Cas9 gene-editing approach, and performing directed in vitro differentiation assays to assess their lineage potential (Kennedy et al., 2012). Our findings revealed that although undifferentiated HEB−/− hESCs retained pluripotency, the expression of NANOG and several TGFβ signaling factors were decreased. Furthermore, HEB deficiency had a profoundly negative impact on mesoderm formation, followed by independent downstream effects on HE formation and T cell development. These defects were largely corrected upon ectopic HEB expression, indicating that HEB plays critical roles in the gene networks that direct mesoderm formation, and additional roles in the generation of HE and T cell precursors during human development.
Results
CRISPR/Cas9-Mediated Targeting of HEB Transcription Factors in hESCs
To evaluate the role of HEB factors in the formation of HE, we used CRISPR/Cas9 gene editing to target exon 9 of the TCF12 gene locus, disrupting both HEBAlt and HEBCan (Figure S1A). hESCs were transfected with a plasmid encoding the TCF12 targeting guide RNA, the Cas9 enzyme, and GFP. Transfected GFP+ hESCs were single-cell sorted and cultured. After expanding individual clones, we identified two out of eight that contained unique insertion-deletions with biallelic mutations (KO-4 and KO-8) (Figure S1B). Western blot analysis confirmed an absence of detectable HEB protein in both KO-4 and KO-8 (Figure S1C). We selected KO-4 as our primary HEB−/− hESC for further analysis, and key experiments were repeated using KO-8, as shown in Supplemental Information.
Characterization of HEB−/− hESC Pluripotency
To assess whether HEB−/− hESCs maintained their pluripotent stem cell (PSC) characteristics, we evaluated colony morphology, growth rate, gene expression, and teratoma formation. Colony morphology and growth rate were indistinguishable between wild-type (WT) and HEB−/− hESCs (Figures 1A and 1B). Immunofluorescence staining of WT and HEB−/− hESCs showed similar levels of OCT4, SOX2, SSEA-4, TRA-1-60, and TRA-1-81 protein expression. NANOG was only expressed in a small proportion of sparsely distributed cells in the HEB−/− hESC colonies, suggesting heterogeneity in these cells, perhaps due to epigenetic changes (Figures 1C and S1D). Western blot analysis confirmed that HEB−/− hESCs had similar levels of OCT4 and SOX2 protein expression compared with WT, and decreased levels of NANOG (Figures 1D and S1E). To functionally test pluripotency, we injected HEB−/− and WT hESCs into immune-compromised (NOD-SCID) mice to allow for teratoma development (Figure 1E). Teratomas containing all three germ layers formed from both WT and HEB−/− hESCs (Figure 1F), indicating that, despite the decrease in NANOG, HEB−/− hESCs retained the key features of pluripotency.
Figure 1.

CRISPR/Cas9-Targeted TCF12 in Human ESCs Results in Ablation of HEB Proteins but Does Not Disrupt Pluripotency
(A and B) Morphology (A) and cell proliferation (B) of wild-type (WT) and HEB−/− (KO-4) hESC colonies.
(C) Immunofluorescent staining of pluripotency markers (green or red) in WT and HEB−/− hESCs. Nuclei (blue) were visualized with DAPI.
(D) Western blot analysis for the expression of HEB, OCT4, SOX2, NANOG, and the αTUBULIN loading control.
(E) Teratomas collected from four mice per hESC genotype.
(F) Representative H&E staining for ectoderm (neuroepithelium), mesoderm (cartilage and muscle), and endoderm (gut-like glandular epithelial tissue) are indicated. Arrows indicate distinguishing features of each tissue type.
Images in (A), (C), and (D) and graph in (B) are representative of three independent experiments. Error bars represent mean ± SD (n=3 independent experiments). Scale bars in (A), (C) and (F), 100 μm. See also Figure S1.
Global Transcriptome Analysis of WT versus HEB−/− hESCs
To examine genome-wide changes in mRNA expression in WT versus HEB−/− hESCs, we performed RNA sequencing (RNA-seq) using biological triplicates (Figure 2A). There were 274 significant changes in mRNA expression (p < 0.05) between WT and HEB−/−. As compared with WT, HEB−/− hESCs had lower expression of 126 transcripts and higher expression of 148 transcripts (Table S2). To determine whether other E protein family members were upregulated to compensate for HEB loss, as has been previously observed (Ravanpay and Olson, 2008), we examined TCF4 (E2-2) and TCF3 (E2A) mRNA expression (Figure 2B). We found that they were not significantly different between WT and HEB−/− hESCs. However, NANOG mRNA expression was greatly decreased in the HEB−/− hESCs (Figures 2C and S2A), whereas OCT4 and SOX2 transcripts were similar (Figure 2C). Gene ontology analysis of the RNA-seq data revealed that the loss of HEB perturbed the expression of genes involved in heart, neural crest, mesenchymal, and endothelial development (Figure S2B). These genes, which are expressed at the transcript level in undifferentiated ESCs (Brown et al., 2011), were for the most part decreased in the absence of HEB, suggesting an important role for HEB in the differentiation of these lineages. We also found a striking perturbation in the expression of genes involved in TGFβ signaling, including NODAL, the NODAL inhibitor LEFTY1, and the bone morphogenic protein (BMP) factor growth differentiation factor 3 (GDF3) (Figures 2D and S2A), which are known to play roles in mesodermal lineage outcomes, including HE (Monteiro et al., 2016).
Figure 2.

Global Transcriptome Profiling by RNA-Seq Reveals Differences between WT and HEB−/− hESCs
(A) Heatmap showing differential gene expression patterns of three replicates each for WT and HEB−/− hESCs (clone KO-4).
(B–D) Expression of E proteins (B), genes associated with pluripotency (C), and TGFβ family members (D) in undifferentiated WT and HEB−/− hESCs, as determined by RNA-seq. FPKM, fragments per kilobase of exon per million fragments mapped. Error bars represent mean ± SD (n = 3 biological replicates). ∗∗p < 0.01 by Student's t test. See also Figures S2 and S3; Table S2.
Direct Binding of HEB to Gene Loci Involved in Early Mesoendoderm Formation
To establish whether the differentially expressed genes in HEB−/− hESCs are direct targets of HEB, we analyzed chromatin immunoprecipitation sequencing (ChIP-seq) data in the ENCODE database using the Integrative Genomics Viewer browser (Robinson et al., 2011). Specifically, we visualized occupancy of HEB, NANOG, and p300 on gene loci encoding factors that were identified as HEB dependent by RNA-seq and are known to be involved in mesoendodermal development (Figure S3). Strikingly, there were clearly defined peaks for HEB binding that coincided with NANOG peaks as well as the transcriptional activator p300 on the regions upstream of the transcriptional start sites for NANOG, NODAL, and LEFTY1. In each case, the HEB peaks were located slightly upstream of the transcriptional start site, consistent with promoter binding. In contrast, the GDF3 locus did not exhibit major peaks for HEB, NANOG, or p300. Therefore, it appears that HEB coordinately binds with NANOG on the promoters of several genes involved in TGFβ signaling, whereas the regulation of GDF-3 expression by HEB is more likely indirect.
Defects in Mesoendodermal Specification and Development from HEB−/− hESCs
WT and HEB−/− hESCs were next differentiated into embryoid bodies (EBs) using a protocol that allows the generation of definitive hematopoietic precursors (Figure 3A) (Kennedy et al., 2012). Whereas HEBCan and HEBAlt were expressed at the mRNA level throughout the 8 days of WT EB culture (Figure 3B), only HEBCan was expressed in sorted day-8 (d8) WT CD34+ cells. We used qRT-PCR to measure the expression of key developmental markers of mesoderm, endoderm, and ectoderm in undifferentiated WT and HEB−/− hESCs and in d4 EBs (Figures 3C and S4A). In WT EBs, OCT4 was downregulated between d0 and d4, coupled with an upregulation of genes associated with endodermal (GATA4 and GATA6), mesodermal (T [brachyury] and MESP1), and ectodermal (NEUROD and CGB7) fates. By contrast, HEB−/− d4 EBs failed to upregulate GATA4, T, and MESP1 transcripts; GATA6 was also poorly upregulated. HEB deficiency did not decrease NEUROD or CGB7 expression. These results suggest that HEB plays important roles in mesoendodermal differentiation but is dispensable for ectodermal development.
Figure 3.

HEB−/− hESCs Display Defects in Mesoendodermal Induction and Early Hematopoietic Differentiation
(A) Experimental overview of embryoid body (EB) formation and differentiation. BMP4, bone morphogenetic protein 4; bFGF, basic fibroblast growth factor; VEGF, vascular endothelial growth factor; IL, interleukin; EPO, erythropoietin; SCF, stem cell factor; IGF1, insulin-like growth factor 1; FLT3L, FMS-like tyrosine kinase 3 ligand; TPO, thrombopoietin.
(B) Reverse-transcriptase PCR analysis of HEB transcript (HEBCan, canonical; HEBAlt, alternative) expression at various stages of EB differentiation, and in sorted day-8 (d8) CD34+ cells (last column). GAPDH was measured as a loading control.
(C) qRT-PCR analysis for the expression of pluripotency and differentiation markers in undifferentiated hESCs (day 0 [d0]) versus d4 EB-derived cells.
(D) Flow-cytometric analysis of CD34 and KDR, CD144, and CD31 expression on d8 EB-derived cells.
(E and F) Percentages (E) and numbers (F) of CD34+ cells in d8 EBs.
(G) qRT-PCR analysis of the expression of mesodermal and hematopoietic genes in CD34+ cells. For qRT-PCR graphs, mRNA levels are shown relative to GAPDH.
Error bars represent mean ± SD (n = 3 independent experiments). ∗∗p < 0.01; ∗∗∗p < 0.005 by Student's t test. Images in (B) and plots in (D) are representative of three independent experiments. See also Figure S4.
Severe Defect in the Generation of Early Mesodermal Precursors in HEB−/− EB Cultures
To assess the ability of HEB−/− hESCs to generate precursors with hematopoietic potential, d8 EB cultures were dissociated and stained for the expression of CD34, CD309 (KDR), CD31 (PECAM-1), and CD144 (VE-cadherin), all of which mark mesoderm-derived endothelial cells (Figure 3D). WT EBs contained robust populations of CD34+ cells that were also positive for KDR, CD144, and CD31, consistent with the presence of HE. In contrast, very few KDR+ CD34+ cells were present in HEB−/− EB cultures (Figures 3D–3F), indicating a significant block in mesodermal differentiation. CD34+ cells were also greatly depleted in KO-8 cultures (Figure S4B). To assess the lineage potential of the few KDR+ CD34+ cells that were generated in HEB−/− EB cultures, we performed gene expression analysis (Figures 3G and S4C). HEB−/− CD34+ cells expressed significantly less NOTCH1, RUNX1, and GATA3 mRNA than WT CD34+ cells, consistent with a potential loss of HE (Burns et al., 2005, Butko et al., 2016). Expression of the other NOTCH receptors was not significantly perturbed. These data suggest that HEB−/− cells that bypassed the early mesodermal block to become CD34+ cells were compromised in their hematopoietic potential. By contrast, Tie2 (TEK, CD202b) and Myocardin (MYOCD) expression were unchanged between HEB−/− and WT CD34+ cells (Figure 3G), suggesting preservation of non-hematopoietic mesodermal lineage potential.
Endothelial Potential Is Intact in the Absence of HEB
To decipher the relationship between the role of HEB in mesodermal specification and its role in the generation of CD34+ endothelium, we placed WT or HEB−/− (KO-4 and KO-8) hESCs in EB cultures for 4 days (Figure 4A). By d4 of EB culture, HEB−/− hESCs were already clearly compromised in their ability to give rise to KDR+ cells (Figure 4B). To bypass the initial mesodermal block, we isolated KDR+ cells from WT or HEB−/− d4 EBs by magnetic and flow-cytometric cell sorting (Figure 4C). Equal numbers of KDR+ cells were then placed into reaggregate cultures and analyzed at 2-day intervals (Figure 4D). CD34+ cells were present at the start (d4 + 0), and became the majority population after 2 days (Figure 4D). Importantly, there were no significant differences in the proportions of CD34+ cells between WT and HEB−/− KDR+ reaggregate cultures, at any time point, indicating that the ability to generate CD34+ cells was intact in KDR+ HEB−/− cells. To assess endothelial function, we sorted CD34+ cells from d8 WT or HEB−/− EBs and placed them on Matrigel-coated plates (Figure 4E). Following overnight culture, HEB−/− CD34+ cells formed vascular structures similar to those generated by WT CD34+ cells (Figure 4F). These results indicate that the early mesodermal defect in HEB−/− precursors was largely responsible for the decrease in CD34+ cells we observed in HEB−/− d8 EBs, and that bypassing that block allowed the development of CD34+ endothelial cells.
Figure 4.

HEB−/− Mesodermal Precursors Are Not Impaired in Their Ability to Give Rise to CD34+ Endothelial Cells
(A) Experimental overview for KDR+ cell isolation and reaggregation.
(B) Percentage of KDR+ cells in day-4 (d4) EBs derived from WT, KO-4, or KO-8 hESCs, before sorting.
(C) Pre-sort and post-sort purity of KDR+ cell enrichment after each sorting step (magnetic-activated cell sorting [MACS] followed by fluorescence-activated cell sorting), as determined by flow cytometry.
(D) Percentages of CD34+ cells in cultures at 2-day intervals after reaggregation of d4 EB-derived KDR+ cells. Representative flow-cytometry plots of CD34 and KDR expression are shown on the right.
(E) Experimental overview for endothelial tube formation in vitro.
(F) Images of endothelial vessel-like structures. Scale bar, 100 μm.
Error bars represent mean ± SD (n = 3 independent experiments). ∗∗p < 0.01, ∗∗∗p < 0.005 by Student's t test. Plot in (B) is compiled from six independent experiment, with one replicate per experiment, and plots in (C) and (D) and images in (F) are representative of three independent experiments.
Intact Myeloid Potential in HEB−/− CD34+ CD45+ Cells Derived from Day-18 EBs
Having established the endothelial potential of CD34+ HEB−/− precursors, we next assessed their hematopoietic potential. To do this, we developed a methylcellulose assay for measuring myeloerythropoietic potential in EB-derived cells (Figure 5A). A timeline analysis of WT EBs showed that very few hematopoietic colonies (colony-forming units—granulocyte-macrophage [CFU-GM] and blast forming units—erythrocytes [BFU-E]) developed from d8 and d13 EBs, whereas d18 EBs gave rise to a substantial number of colonies (Figures S5A and S5B). Therefore, the d18 EB time point was chosen to compare myeloerythroid potential in HEB−/− and WT precursors. Whole WT or HEB−/− d18 EBs were dissociated into single cells, which were plated in methylcellulose (Figure 5A). HEB−/− cells were clearly defective in their ability to generate CFU-GM and BFU-E when compared with WT cells (Figures 5B and S5C). We next examined d18 EBs by flow cytometry. WT cultures contained four populations: CD34+ CD45−, CD34+ CD45+, CD34− CD45+, and CD34− CD45− cells (Figures 5C and S5D). Sorting and culturing of these subsets in methylcellulose revealed that WT CD34+ CD45+ cells gave rise to both CFU-GM and BFU-E, whereas the other populations failed to yield any colonies (data not shown). Compared with WT, HEB−/− EBs exhibited a dramatic decrease in the frequency and numbers of the CD34+ CD45−, CD34+ CD45+ and CD34− CD45+ fractions (Figures 5D, 5E, and S5E). However, once sorted, HEB−/− CD34+ CD45+ cells could readily give rise to CFU-GM and BFU-E (Figures 5F and S5F). Therefore, myeloerythroid potential remained intact in the small proportion of HEB−/− mesodermal progenitors that had survived the early block.
Figure 5.

Inefficient Hematopoietic Outcomes from HEB−/− hESC-Derived Progenitors
(A) Experimental overview for the colony-forming unit (CFU) assay of erythromyeloid potential. Cell suspensions from day-18 (d18) EBs were obtained and placed into methylcellulose cultures either as an unfractionated population (top) or as four sorted subsets based on combinatorial expression of CD34 and CD45 (bottom).
(B) Numbers of erythroid (BFU-E) and myeloid (CFU-GM) arising from unfractionated WT and HEB−/− d18 EB-derived cells in methylcellulose cultures.
(C) Flow-cytometric analysis for CD34 and CD45 expression of unfractionated WT and HEB−/− d18 EB-derived cells.
(D and E) The percentages (D) and cell numbers (E) of CD34+ CD45−, CD34+ CD45+, and CD34− CD45+ subsets within WT and HEB−/− d18 EBs.
(F) Numbers of erythroid (BFU-E) and myeloid (CFU-GM) arising from CD34+ CD45+ cells sorted from WT and HEB−/− d18 EBs.
(G) Experimental overview for T cell differentiation. CD34+ cells were sorted from d8 EBs and co-cultured with OP9-DL4 cells.
(H and I) Percentages (H) and numbers (I) of CD45+ cells at successive days (d) of OP9-DL4 co-cultures.
Error bars represent mean ± SD (n = 3 independent experiments). ∗∗∗p < 0.005 by Student's t test. Plots in (C) are representative of three independent experiments. See also Figures S5 and S6.
Failure of T Cell Development from HEB−/− Precursors
To further test hematopoietic potential, we sorted CD34+ cells from d8 EBs and plated them at equal numbers on OP9-DL4 cells to induce T cell development (Figure 5G) (Mohtashami et al., 2010, Schmitt and Zuniga-Pflucker, 2002). In WT cultures, CD45+ cells arose by d6 and persisted until d30 (Figures 5H, 5I, and S6A). By d12, early (CD7+ CD5+) T cell precursors appeared (Figure S6B) and by d30, later-stage (CD4+ CD8+) T cell precursors were present in WT cultures (Figure S6C). By contrast, very few CD45+ cells were generated in HEB−/− cultures at any time point (Figures 5H, 5I, and S6A), and the few that were detectable did not exhibit a T-lineage phenotype (Figures S6B and S6C; and data not shown). Therefore, d8 EB-derived HEB−/− CD34+ cells not only lacked T cell potential but also failed to generate CD45+ cells in OP9-DL4 co-cultures.
Reconstitution of HEB−/− hESCs with Ectopically Expressed HEBCan
To ensure that the defects in mesodermal development and T cell development were not due to off-target effects caused by the CRISPR/Cas9 approach, we elected to reconstitute HEB−/− hESCs with HEBCan. We chose to focus specifically on HEBCan because WT CD34+ cells derived from d8 EB expressed HEBCan and not HEBAlt (Figure 3B). A lentiviral vector encoding HEBCan upstream of an IRES-GFP, under the control of a CAG promoter, was assembled, and HEB−/− hESCs (KO-4) were transduced. A GFP-only vector was used as a control for the effects of transduction in HEB−/− hESCs. Western blot and immunofluorescence analysis showed that transduction of HEBCan into HEB−/− cells restored HEBCan protein expression to near physiological levels in undifferentiated hESCs (Figure 6A), and that GFP expression was maintained until at least d8 of EB culture (Figure 6B).
Figure 6.

Ectopic Expression of HEBCan in HEB−/− hESCs Restores Lineage-Specific Gene Expression and Hematopoietic Specification
(A) Western blot analysis for HEB expression in WT, KO (HEB−/−), KO + GFP (HEB−/− hESCs transduced with GFP control vector) and KO + HEBCan (HEB−/− hESCs transduced with HEBCan-encoding vector) hESCs.
(B) Bright-field (top) and fluorescent (bottom) images of day-8 (d8) EBs derived from HEB−/− hESCs transduced with control or HEBCan-expressing lentiviral particles. Scale bar, 100 μm.
(C and D) qRT-PCR analysis for the expression of pluripotency-associated genes (C) and mesoendodermal genes (D) in WT, KO + GFP, and KO + HEBCan hESC-derived cells at d0 and d4 of EB culture. mRNA levels are shown relative to GAPDH.
(E) Flow-cytometric analysis of CD34 and KDR, CD144, and CD31 on WT, KO + GFP, and KO + HEBCan d8 EB-derived cells.
(F and G) Percentages (F) and numbers (G) of CD34+ cells in WT, KO + GFP, and KO + HEBCan d8 EBs.
(H) Flow-cytometric analysis for CD34 and CD45 on WT, KO + GFP, and KO + HEBCan d18 EB-derived cells.
(I and J) Percentages (I) and numbers (J) of CD34/CD45 subsets in WT, KO + GFP, and KO + HEBCan d18 EB-derived cells.
(K) Numbers of erythroid (BFU-E) and myeloid (CFU-GM) arising from unfractionated WT, KO + GFP, and KO + HEBCan d18 EBs.
Error bars represent mean ± SD (n = 3 independent experiments). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005 by Student's t test. Images in (A) and (B) and plots in (E) and (H) are representative of three independent experiments.
Rescue of Lineage-Specific Genes and Mesodermal Specification by HEBCan
To assess the consequences of adding HEBCan to HEB−/− cells, we first measured gene expression by qRT-PCR in undifferentiated HEB−/− hESCs transduced by GFP only (KO + GFP) or HEBCan (KO + HEBCan) (Figure 6C). Untransduced WT hESCs were analyzed in parallel as a positive control. Transcript levels of NANOG, NODAL, and LEFTY1 were significantly increased in HEBCan-transduced cells relative to control-transduced HEB−/− hESCs, while GDF3 transcripts were fully restored to WT levels. Next, we examined gene expression in d4 EBs and found that T, MESP1, GATA4, and GATA6 were all upregulated in response to HEBCan (Figure 6D), suggesting increased potential for mesoendodermal fates. To assess the appearance of mesodermal cells, we performed flow cytometry on d8 EB. KO + HEBCan cultures exhibited an increased frequency of CD34+ KDR+ CD144+ CD31+ cells as compared with KO + GFP cultures (Figures 6E–6G), and increased populations of CD34+ CD45−, CD34+ CD45+, and CD34− CD45+ cells in d18 EBs (Figures 6H–6J). Furthermore, the myeloid potential of unfractionated d18 EBs was also increased in the KO + HEBCan cultures relative to KO + GFP cultures (Figure 6K). Altogether, these results show that restoration of HEBCan expression in HEB−/− hESCs allowed for mesodermal differentiation, and restored the robust generation of precursors with myeloerythroid potential.
Ectopic HEBCan Enables HEB−/− Progenitors to Undergo T Cell Development
To evaluate whether HEBCan could alleviate the block in T cell development, we sorted CD34+ cells from d8 EBs derived from WT, KO + GFP, and KO + HEBCan hESCs. NOTCH1 mRNA expression was fully restored, and GATA3 and RUNX1 were significantly upregulated, in the KO + HEBCan cells relative to the KO + GFP cells (Figure 7A). Next, the CD34+ cells were placed in OP9-DL4 cell cultures. Strikingly, KO + HEBCan cultures readily gave rise to CD45+ cells in OP9-DL4 cultures, which were restored to at least WT levels, whereas KO + GFP cultures did not (Figures 7B and 7C). Moreover, KO + HEBCan CD34+ cells developed into T cell precursors, whereas KO + GFP cells were virtually absent, similarly to untransduced HEB−/− cells (Figures 7D and 7E). Together, these results suggest that ectopic expression of HEBCan in HEB−/− precursors provides a full rescue of CD45+ hematopoietic progenitors with T cell potential and permits them to develop into committed T cell precursors.
Figure 7.

HEBCan Rescues Hematopoiesis and T Cell Development in HEB−/− hESCs in OP9-DL4 Co-cultures
(A) qRT-PCR analysis for the expression of hematopoietic genes in CD34+ cells sorted from WT, KO + GFP, and KO + HEBCan day-8 (d8) EBs. mRNA levels are shown relative to GAPDH.
(B and C) Percentages (B) and numbers (C) of CD45+ cells in d12 and d18 OP9-DL4 co-cultures.
(D and E) Flow-cytometric analysis of T cell development from WT, KO + GFP, and KO + HEBCan d8 EB-derived CD34+ cells at d12 (D) and d18 (E) of OP9-DL4 co-culture. Cells are gated on the CD45+DAPI− population.
Error bars represent mean ± SD (n = 3 independent experiments). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005 by Student's t test. Plots in (B), (D), and (E) are representative of three independent experiments.
Discussion
Our generation of HEB−/− hESCs has provided a highly informative system for studying early human developmental processes as well as later hematopoietic outcomes. Here, we report that the absence of HEB results in a profound block in mesodermal development, associated with a failure to upregulate critical mesodermal regulatory genes in hESCs. Furthermore, HEB−/− CD34+ cells from d8 EBs were deficient in the expression of key HE regulators RUNX1 and NOTCH1, and failed to undergo hematopoiesis or T cell development in OP9-DL4 co-culture. Together, these discoveries unexpectedly place HEB at the top of a hierarchy of regulators responsible for directing mesodermal fate and pre-hematopoietic events during human development.
Our results also provide insights into roles for HEB factors in hESCs. We observed a significant decrease in NANOG expression in undifferentiated HEB−/− hESCs, whereas all other features of pluripotency appeared to be intact. Although NANOG is thought to be a critical member in the transcriptional network of pluripotency in WT ESCs (Boyer et al., 2005), it is not essential for their maintenance (Chambers et al., 2007) or for induction of PSCs (Schwarz et al., 2014, Takahashi et al., 2007). We also observed that the expression of the TGFβ superfamily members NODAL, LEFTY1, and GDF3 were markedly decreased in HEB−/− hESCs. These factors participate in a complex network downstream of ACTIVIN/NODAL signaling that serves to maintain pluripotency, inhibit neuroectoderm specification, and drive mesoendoderm differentiation (Brown et al., 2011, James et al., 2005, Vallier et al., 2005, Vallier et al., 2009, Xu et al., 2008). NANOG, which is a direct target of NODAL signaling, is required for upregulation of GDF3, which plays a role in pluripotency and in early mesoderm and endoderm development (Chen et al., 2006, Park et al., 2012). Therefore, the early defects we observed in HEB−/− hESC differentiation could be in part due to defects in NODAL signaling. However, the direct binding of HEB to NANOG and LEFTY1 gene loci indicates that HEB, like NODAL, is a key participant in the combinatorial code and series of feedforward loops that specify the mesodermal program.
Interestingly, our RNA-seq analysis of WT versus HEB−/− hESCs revealed that many genes involved in heart development were altered in HEB−/− hESCs, suggesting that HEB factors may be involved in cardiomyocyte specification. Our results also place HEB upstream of several additional regulators of mesodermal specification, including T (brachyury) (Herrmann et al., 1990, Schulte-Merker et al., 1994, Smith et al., 1991) and Mesp-1. Mesp-1 directs mesoderm into cardiac, hematopoietic, or skeletal myogenic progenitors in a context-dependent manner in mice (Chan et al., 2013). It is likely that those early defects undermined later developmental processes in HEB−/− differentiation in our assay systems. However, our results clearly show that HEB−/− KDR+ cells, once isolated from d4 HEB−/− EBs, were fully capable of producing CD34+ cells. These results are consistent with intrinsic differences between WT and HEB−/− CD34+ cells in their hematopoietic potential, in agreement with their defects in the mRNA expression of RUNX1, GATA3, and NOTCH1.
The onset of hematopoiesis during EB differentiation follows the induction of HE within the KDR+ CD34+ population (Ditadi et al., 2015, Kennedy et al., 2007, Kennedy et al., 2012, Wang et al., 2004, Woll et al., 2008, Zambidis et al., 2005). RUNX1, which is severely downregulated in HEB−/− CD34+ pre-hematopoietic cells, is essential for the formation of HE. Our results suggest that HE is transiently present in our EB culture system around d8, consistent with previous reports (Sugimura et al., 2017). Therefore, the HEB−/− CD45+ cells that arose in HEB−/− d18 EBs may have represented the few cells that bypassed the block in EHT earlier in EB development, or they may have been derived from different progenitors, perhaps those that give rise to EMPs (Chen et al., 2011). By contrast, HEB−/− CD34+ cells from d8 EBs failed to produce robust CD45+ populations in OP9-DL4 co-cultures at all. If the HEB-dependent event that led to the lack of CD45+ cells in OP9-DL4 co-culture was restricted to T cell development, the low levels of NOTCH1 in the HEB−/− CD34+ cells would be expected to allow myeloid and B cell development in these cultures, instead of a severe depletion of all CD45+ cells (Cho et al., 1999).
Remarkably, nearly all defects in mesodermal development and hematopoiesis observed in HEB−/− cells were restored by the sustained ectopic expression of HEBCan from the HEB−/− hESC stage onward. The magnitude of the rescue did not in all cases reach the same levels as untransduced WT cells, which could have resulted from uncontrolled levels of HEBCan driven by the lentiviral CAG promoter. However, it should be noted that the HEBCan-reconstituted cells were still missing HEBAlt. Whether HEBAlt and HEBCan work together to increase total HEB protein dosage, or whether they have distinct functions, has yet to be determined. Overall levels of HEB may be critical for its function during EHT, since Inhibitor of DNA binding 1 (ID1) and ID3 are strong negative regulators of this process (Hong et al., 2011). Regardless, the full restoration of CD45+ cells observed in OP9-DL4 cultures suggests that ectopic expression of HEBCan alone is sufficient for the rescue of EHT as well as downstream T-lymphoid progenitors.
Recently, a breakthrough has been achieved in deriving HSCs from cultured human PSCs (Sugimura et al., 2017). In this study, PSC-derived HE was transduced with combinations of transcription factors, and seven were identified to combinatorially confer long-term multilineage reconstitution potential, as assayed in immunodeficient mice. These included RUNX1 and its target SPI1 (PU.1). It will be of interest to examine the expression of additional regulators of HE such as GFI1 (Lis et al., 2017, Thambyrajah et al., 2016), EVI1 (Konantz et al., 2016), and ERG in our system to see whether these factors are also subject to control by HEB. Furthermore, understanding the signals that regulate these factors, including HEB, will be critical for allowing the generation of long-term reconstituting HSCs from hESCs without genetic manipulation.
In summary, our work shows that HEB factors are essential for inducing mesodermal gene networks in hESCs without disrupting pluripotency. Moreover, our results indicate that HEB is selectively needed for the generation of pre-hematopoietic cells, upstream of RUNX1 and NOTCH1. Together, these results identify HEB as a top-tier regulator of the gene networks that regulate mesoderm and hematopoietic specification during human embryogenesis.
Experimental Procedures
hESC Culture
Human H1 ESCs (Thomson et al., 1998) were maintained and expanded on plates coated with growth-factor-reduced Matrigel (Corning) in serum-free, defined TeSR-E8 medium (STEMCELL Technologies). Cells were passaged by non-enzymatic dissociation using 0.5 mM EDTA.
Generation of TCF12−/− (HEB−/−) hESCs
The pD1321-GFP expression vector containing cassettes for GFP, Cas9 endonuclease, and a CRISPR chimeric cDNA including the guide RNA moiety designed to target exon 9 of HEB (CCA GTA TGT TCG CTA GCA CTT TC), was custom synthesized (DNA2.0). hESCs were dissociated into single cells using Accutase (Sigma), and transfected with the CRISPR/Cas9 vector using the Nucleofector 2B device, program A013 (Amaxa). GFP+ cells were sorted 48 hr after transfection using a FACSAria II cell sorter (BD Biosciences) at the Sunnybrook Research Institute (SRI) Center for Flow Cytometry & Microscopy. Single cells were cultured on Matrigel-coated plates in TeSR-E8 medium, which was supplemented with Rho kinase (ROCK) inhibitor Y-27632 (Tocris) for the first 24 hr. Individual colonies were picked and expanded under the feeder-free/TeSR-E8 conditions. Aliquots of the cells were collected for purification of genomic DNA using the Genomic DNA kit (Invitrogen). Mutations were validated by sequencing the PCR amplification products of the regions flanking the targeting sites.
Virus Production and Infection of hESCs
Lentiviral particles encoding full-length human HEBCan were commercially synthesized using the EX-CAG-GFP-Lv234 vector system (GeneCopoeia, Rockville, MD, USA). hESCs were transduced with HEBCan or control lentivirus (negative control particles, LP-NEG-GFP-LV234, GeneCopoeia), and the next day the medium was replaced by fresh TeSR-E8 medium, followed by an additional 72 hr of culture. GFP+ cells were sorted and expanded.
Flow Cytometry
The following antibodies were used for these studies: CD4-Alexa Fluor 700 (catalog #56-0048-82), CD5-PE-Cy7 (#25-0059-42), CD7-Alexa Fluor 700 (#56-0079-42), CD31-PerCP/eFluor710 (#46-0319-42), CD45-APC-eFluor780 (#47-0459-42), and CD144-PE (#12-1449-82) from eBioScience; CD8-PE (#555367), CD34-PE (#550761), and CD34-APC (#555824) from BD Pharmingen; KDR-PE (#359904) from BioLegend; and KDR-PE (#FAB357P100) from R&D Systems. Stained cells were analyzed using an LSRII (BD Biosciences) flow cytometer at the indicated time points. Data analysis was performed using FlowJo software. For T-lymphoid studies, analyses were carried out by gating on live cells, as indicated by lack of DAPI uptake, followed by gating on CD45+ cells. Gates were set using appropriate isotype controls.
Immunostaining
hESCs were passaged on Matrigel-coated plates, and cells were cultured in TeSR-E8 medium. Cells were fixed in 4% paraformaldehyde, and permeabilization and blocking were performed in 5% BSA and 1% Triton X-100 in PBS for 30 min. Cells were stained with anti-NANOG (Abcam, #AB80892), SOX2 (EMD Millipore, #AB5603), TRA-1-60 (EMD Millipore, #MAB4360), TRA-1-81 (EMD Millipore, #MAB4381), SSEA-4 (EMD Millipore, #MAB4304), or OCT4 (Santa Cruz Biotechnology, #SC-5279), all at 1:100. Secondary antibody was applied for 1 hr at room temperature. Nuclei were stained with DAPI (Life Technologies). Images were acquired using a Zeiss IX71 microscope and MetaMorph Advanced software.
Teratoma Formation
Mice were maintained at the SRI Comparative Research Facility, and all protocols were approved by the SRI Animal Care Committee. Six- to 8-week-old non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice were injected subcutaneously with 1 × 106 hESCs resuspended in Matrigel to allow teratoma formation for 8 weeks. The teratomas were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 10–12 μm thickness, and stained with H&E for examination. Histology examination was performed by the SRI histology core facility.
RNA-Sequencing
Total RNA was isolated from cells using TRIzol (Invitrogen) according to the manufacturer's protocol. Libraries were prepared using TruSeq Stranded Total RNA kit (Illumina), and rRNA depleted using Ribo-zero Gold rRNA beads. First-strand cDNA was generated using random primers, followed by second-strand cDNA synthesis, and adapter-ligated PCR-enriched products were used to create cDNA libraries. A single “A” base was added and adapter ligated, followed by purification and enrichment with PCR, to create cDNA libraries, which were sequenced for paired ends using the Illumina platform. Reads for each sample were aligned to the hg19 assembly of the human genome using the TopHat v2.1.0 (Kim et al., 2013) and Bowtie v2.2.6 (Langmead and Salzberg, 2012) software packages. The significant gene list was uploaded to DAVID (https://david.ncifcrf.gov/) and “Gene Functional Classification” was performed with GOTERM_BP_FAT, GOTERM_CC_FAT, and GOTERM_MF_FAT databases. RNA-seq was performed in the Princess Margaret Genomics Center at the University Health Network (Toronto, ON). Data have been deposited in the GEO with accession number GEO: GSE100417.
ChIP-Sequencing Data Analysis
ChIP-seq files from the ENCODE project (ENCODE Project Consortium, 2012) were loaded into the Integrated Genome Viewer browser (Robinson et al., 2011) for visualization of direct binding sites of p300, NANOG, and TCF12 (HEB) to the loci indicated in H1-hESCs. Input ChIP-seq files (control) are shown for background comparison with anti-transcription factor antibodies. NANOG, TCF12, p300, and control ChIP-seq data are all found within the GEO dataset GEO: GSE32465. Specifically, the accession numbers are GEO: GSM803437 (NANOG), GSM803427 (TCF12), GSM803542 (p300), and control (GSM1010818).
Differentiation of hESCs via EB Formation
To generate EBs, we treated hESCs with collagenase B (1 mg/mL; Roche) for 20 min. Cells were gently scraped with a cell scraper to form small aggregates (10–20 cells). Aggregates were resuspended in StemPro-34 (Invitrogen), supplemented with L-glutamine (2 mM), ascorbic acid (1 mM), monothioglycerol (4 × 10−4 M; Sigma-Aldrich), and transferrin (150 mg/mL; Roche). BMP4 (10 ng/mL; R&D), basic fibroblast growth factor (5 ng/mL; Peprotech), SB431542 (6 μM; Tocris), vascular endothelial growth factor (15 ng/mL; R&D), interleukin-6 (IL-6) (10 ng/mL; R&D), insulin-like growth factor 1 (25 ng/mL; R&D), IL-11 (5 ng/mL; R&D), stem cell factor (SCF) (50 ng/mL; Miltenyi), erythropoietin (2 U/mL), thrombopoietin (30 ng/mL; R&D), IL-3 (30 ng/mL; R&D), and FMS-like tyrosine kinase 3 ligand (FLT3L) (10 ng/mL; Miltenyi) were added as indicated (see Figure 3A). Cultures were maintained in a 5% CO2/5% O2/90% N2 environment. On the day of assay, EBs were harvested and dissociated to single cells by a 40-min treatment with 0.2% collagenase IV. Afterward, 1 mL of medium with serum was added and the EBs were dissociated to single cells by passaging six times through a 20-gauge needle.
Isolation and Reaggregation of KDR+ Cells
On day 4, EBs were trypsinized for 5 min at 37°C and triturated using a pipet. Single cells were stained with anti-human CD309/KDR-PE (BioLegend, #359904) for 30 min on ice and washed. KDR+ cells were enriched by magnetic-activated cell sorting using anti-PE-MicroBeads (Miltenyi) and LS columns following the manufacturer's protocol. KDR+-enriched cells were then sorted to ensure purity, and reaggregated in Aggrewells at 6.5 × 105 cells/mL per well. Cells were maintained as described above, and portions were analyzed at days 2, 4, and 6 after reaggregation with anti-human CD309/KDR-PE (BioLegend, #359904), CD34-APC (BD Pharmingen, #555824), and CD45-APC/eFluor780 (eBioscience, #47-0459-42).
OP9-DL4 Co-culture for T-Lineage Differentiation
OP9 cells expressing Delta-like 4 (OP9-DL4) were maintained in α-minimum essential medium supplemented with 20% fetal bovine serum (OP9 medium) as previously described (Schmitt and Zuniga-Pflucker, 2002). A total of 3–5 × 104 sorted human EB-derived CD34+ was added to individual wells of a 6-well plate containing OP9-DL4 cells, and cultured in OP9 medium supplemented with recombinant human (rh)FLT3L (5 ng/mL), rhIL-7 (5 ng/mL), and rhSCF (100 ng/mL) (PeproTech). Cells were passaged onto fresh OP9-DL4 cultures every 6 days.
Hematopoietic Colony Assay
Analysis of myeloerythroid potential was performed by plating 1 × 105 unfractionated EB populations, or 1 × 103 sorted cells (CD34/CD45 subsets), from d18 EBs, in methylcellulose H4434 (STEMCELL). Colonies consisting of erythroid and myeloid cells were scored after 10–14 days based on morphologic characteristics.
In Vitro Endothelial Tube Formation
Matrigel (300 μL) was dispensed into wells of a 12-well tray and polymerized at room temperature for 3 hr. CD34+ cells (3 × 105) in 500 μL of EBM-2 medium (Lonza) were seeded onto polymerized Matrigel and incubated overnight in a humidified atmosphere of 5% CO2 at 37°C. Images of Matrigel cultures were captured using a microscope (Zeiss).
Real-Time qRT-PCR
Total RNA was prepared with TRIzol (Invitrogen) and treated with RNase-free DNase (Qiagen). RNA (1 μg) was transcribed into cDNA using Oligo(dT) with Superscript III Reverse Transcriptase (Invitrogen). Real-time qRT-PCR was performed on a 7500 Real-Time PCR System (Applied Biosystems). All experiments were carried out in triplicate using Power SYBR Green Master Mix (Applied Biosystems). Gene expression was evaluated as Delta Ct relative to GAPDH. Primer sequences are listed in Table S1.
Western Blot
Cells were harvested and lysed on ice using lysis buffer (Pierce). Proteins were separated by SDS-PAGE, transferred onto nitrocellulose membranes, and probed overnight with anti-HEB (Santa Cruz, #SC-357), SOX2 (Millipore, #AB5603), NANOG (Abcam, #AB21624), OCT4 (Santa Cruz, #SC-5279), and αTUBULIN (Santa Cruz, #SC-5286) antibodies. Membranes were scanned using the Fusion Fx imaging system (Vilber Lourmat).
Author Contributions
Y.L., P.M.B., M.K.A., and J.C.Z.-P. conceived the project, designed experiments, and analyzed and interpreted the results; Y.L. and P.M.B. performed most experiments; J.S. and S.X. performed some experiments; K.Y. assisted in experiments; and Y.L. and M.K.A. wrote the manuscript with all other authors providing editorial advice.
Acknowledgments
We are thankful to Geneve Awong for assistance with flow cytometry and cell sorting and to Mahmood Mohtashami and Sean Oh for technical support. We are grateful to Christina Lee for expert assistance in the mouse work and mouse husbandry. Y.L. was supported by the Ontario-China Stem Cell Research and Commercialization Partnership. P.M.B. was supported by a Canadian Institutes of Health Research (CIHR) fellowship award. This work was funded by grants to Y.L. (National Natural Science Foundation of China 31571517), to M.K.A. (CIHR MOP-82861 and NSERC RGPIN 05333-14), and to J.C.Z.-P. (CIHR MOP-119538 and MOP-42387, NIH-1P01AI102853-01 grant, and the Krembil Foundation). J.C.Z.-P. is supported by a Canada Research Chair in Developmental Immunology.
Published: August 10, 2017
Footnotes
Supplemental Information includes six figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.07.011.
Contributor Information
Juan Carlos Zúñiga-Pflücker, Email: jczp@sri.utoronto.ca.
Michele K. Anderson, Email: manderso@sri.utoronto.ca.
Supplemental Information
References
- Barndt R., Dai M.F., Zhuang Y. A novel role for HEB downstream or parallel to the pre-TCR signaling pathway during alpha beta thymopoiesis. J. Immunol. 1999;163:3331–3343. [PubMed] [Google Scholar]
- Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein M., Anderson M.K. HEB in the spotlight: transcriptional regulation of T-cell specification, commitment, and developmental plasticity. Clin. Dev. Immunol. 2012;2012 doi: 10.1155/2012/678705. 678705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S., Teo A., Pauklin S., Hannan N., Cho C.H., Lim B., Vardy L., Dunn N.R., Trotter M., Pedersen R. Activin/Nodal signaling controls divergent transcriptional networks in human embryonic stem cells and in endoderm progenitors. Stem Cells. 2011;29:1176–1185. doi: 10.1002/stem.666. [DOI] [PubMed] [Google Scholar]
- Burns C.E., Traver D., Mayhall E., Shepard J.L., Zon L.I. Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes Dev. 2005;19:2331–2342. doi: 10.1101/gad.1337005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butko E., Pouget C., Traver D. Complex regulation of HSC emergence by the Notch signaling pathway. Dev. Biol. 2016;409:129–138. doi: 10.1016/j.ydbio.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I., Silva J., Colby D., Nichols J., Nijmeijer B., Robertson M., Vrana J., Jones K., Grotewold L., Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Chan S.S., Shi X., Toyama A., Arpke R.W., Dandapat A., Iacovino M., Kang J., Le G., Hagen H.R., Garry D.J. Mesp1 patterns mesoderm into cardiac, hematopoietic, or skeletal myogenic progenitors in a context-dependent manner. Cell Stem Cell. 2013;12:587–601. doi: 10.1016/j.stem.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Ware S.M., Sato A., Houston-Hawkins D.E., Habas R., Matzuk M.M., Shen M.M., Brown C.W. The Vg1-related protein Gdf3 acts in a Nodal signaling pathway in the pre-gastrulation mouse embryo. Development. 2006;133:319–329. doi: 10.1242/dev.02210. [DOI] [PubMed] [Google Scholar]
- Chen M.J., Li Y., De Obaldia M.E., Yang Q., Yzaguirre A.D., Yamada-Inagawa T., Vink C.S., Bhandoola A., Dzierzak E., Speck N.A. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell. 2011;9:541–552. doi: 10.1016/j.stem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.K., Webber T.D., Carlyle J.R., Nakano T., Lewis S.M., Zuniga-Pflucker J.C. Functional characterization of B lymphocytes generated in vitro from embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1999;96:9797–9802. doi: 10.1073/pnas.96.17.9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditadi A., Sturgeon C.M., Tober J., Awong G., Kennedy M., Yzaguirre A.D., Azzola L., Ng E.S., Stanley E.G., French D.L. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat. Cell Biol. 2015;17:580–591. doi: 10.1038/ncb3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditadi A., Sturgeon C.M., Keller G. A view of human haematopoietic development from the Petri dish. Nat. Rev. Mol. Cell Biol. 2017;18:56–67. doi: 10.1038/nrm.2016.127. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frelin C., Herrington R., Janmohamed S., Barbara M., Tran G., Paige C.J., Benveniste P., Zuniga-Pflucker J.C., Souabni A., Busslinger M. GATA-3 regulates the self-renewal of long-term hematopoietic stem cells. Nat. Immunol. 2013;14:1037–1044. doi: 10.1038/ni.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritz E., Hirschi K.K. Specification and function of hemogenic endothelium during embryogenesis. Cell. Mol. Life Sci. 2016;73:1547–1567. doi: 10.1007/s00018-016-2134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjimichael C., Nikolaou C., Papamatheakis J., Kretsovali A. MicroRNAs for fine-tuning of mouse embryonic stem cell fate decision through regulation of TGF-beta signaling. Stem Cell Reports. 2016;6:292–301. doi: 10.1016/j.stemcr.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann B.G., Labeit S., Poustka A., King T.R., Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- Hong S.H., Lee J.H., Lee J.B., Ji J., Bhatia M. ID1 and ID3 represent conserved negative regulators of human embryonic and induced pluripotent stem cell hematopoiesis. J. Cell Sci. 2011;124:1445–1452. doi: 10.1242/jcs.077511. [DOI] [PubMed] [Google Scholar]
- Hu J.S., Olson E.N., Kingston R.E. HEB, a helix-loop-helix protein related to E2A and ITF2 that can modulate the DNA-binding ability of myogenic regulatory factors. Mol. Cell. Biol. 1992;12:1031–1042. doi: 10.1128/mcb.12.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Zhang P., Hirai H., Elf S., Yan X., Chen Z., Koschmieder S., Okuno Y., Dayaram T., Growney J.D. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat. Genet. 2008;40:51–60. doi: 10.1038/ng.2007.7. [DOI] [PubMed] [Google Scholar]
- James D., Levine A.J., Besser D., Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- Kennedy M., D'Souza S.L., Lynch-Kattman M., Schwantz S., Keller G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M., Awong G., Sturgeon C.M., Ditadi A., LaMotte-Mohs R., Zuniga-Pflucker J.C., Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konantz M., Alghisi E., Muller J.S., Lenard A., Esain V., Carroll K.J., Kanz L., North T.E., Lengerke C. Evi1 regulates Notch activation to induce zebrafish hematopoietic stem cell emergence. EMBO J. 2016;35:2315–2331. doi: 10.15252/embj.201593454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis R., Karrasch C.C., Poulos M.G., Kunar B., Redmond D., Duran J.G.B., Badwe C.R., Schachterle W., Ginsberg M., Xiang J. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature. 2017;545:439–445. doi: 10.1038/nature22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohtashami M., Shah D.K., Nakase H., Kianizad K., Petrie H.T., Zuniga-Pflucker J.C. Direct comparison of Dll1- and Dll4-mediated Notch activation levels shows differential lymphomyeloid lineage commitment outcomes. J. Immunol. 2010;185:867–876. doi: 10.4049/jimmunol.1000782. [DOI] [PubMed] [Google Scholar]
- Monteiro R., Pinheiro P., Joseph N., Peterkin T., Koth J., Repapi E., Bonkhofer F., Kirmizitas A., Patient R. Transforming growth factor beta drives hemogenic endothelium programming and the transition to hematopoietic stem cells. Dev. Cell. 2016;38:358–370. doi: 10.1016/j.devcel.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C., McCaw P.S., Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- Park S.W., Lim H.Y., Do H.J., Sung B., Huh S.H., Uhm S.J., Song H., Chung H.J., Kim J.H., Kim N.H. Regulation of human growth and differentiation factor 3 gene expression by NANOG in human embryonic carcinoma NCCIT cells. FEBS Lett. 2012;586:3529–3535. doi: 10.1016/j.febslet.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Parker M.H., Perry R.L., Fauteux M.C., Berkes C.A., Rudnicki M.A. MyoD synergizes with the E-protein HEB beta to induce myogenic differentiation. Mol. Cell. Biol. 2006;26:5771–5783. doi: 10.1128/MCB.02404-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanpay A.C., Olson J.M. E protein dosage influences brain development more than family member identity. J. Neurosci. Res. 2008;86:1472–1481. doi: 10.1002/jnr.21615. [DOI] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt T.M., Zuniga-Pflucker J.C. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17:749–756. doi: 10.1016/s1074-7613(02)00474-0. [DOI] [PubMed] [Google Scholar]
- Schulte-Merker S., van Eeden F.J., Halpern M.E., Kimmel C.B., Nusslein-Volhard C. No tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development. 1994;120:1009–1015. doi: 10.1242/dev.120.4.1009. [DOI] [PubMed] [Google Scholar]
- Schwarz B.A., Bar-Nur O., Silva J.C., Hochedlinger K. Nanog is dispensable for the generation of induced pluripotent stem cells. Curr. Biol. 2014;24:347–350. doi: 10.1016/j.cub.2013.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.C., Price B.M., Green J.B., Weigel D., Herrmann B.G. Expression of a Xenopus homolog of Brachyury (T) is an immediate-early response to mesoderm induction. Cell. 1991;67:79–87. doi: 10.1016/0092-8674(91)90573-h. [DOI] [PubMed] [Google Scholar]
- Sugimura R., Jha D.K., Han A., Soria-Valles C., da Rocha E.L., Lu Y.F., Goettel J.A., Serrao E., Rowe R.G., Malleshaiah M. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017;545:432–438. doi: 10.1038/nature22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Thambyrajah R., Mazan M., Patel R., Moignard V., Stefanska M., Marinopoulou E., Li Y., Lancrin C., Clapes T., Moroy T. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 2016;18:21–32. doi: 10.1038/ncb3276. [DOI] [PubMed] [Google Scholar]
- Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Vallier L., Alexander M., Pedersen R.A. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci. 2005;118:4495–4509. doi: 10.1242/jcs.02553. [DOI] [PubMed] [Google Scholar]
- Vallier L., Mendjan S., Brown S., Chng Z., Teo A., Smithers L.E., Trotter M.W., Cho C.H., Martinez A., Rugg-Gunn P. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development. 2009;136:1339–1349. doi: 10.1242/dev.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Li L., Shojaei F., Levac K., Cerdan C., Menendez P., Martin T., Rouleau A., Bhatia M. Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity. 2004;21:31–41. doi: 10.1016/j.immuni.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wang D., Claus C.L., Vaccarelli G., Braunstein M., Schmitt T.M., Zuniga-Pflucker J.C., Rothenberg E.V., Anderson M.K. The basic helix-loop-helix transcription factor HEBAlt is expressed in pro-T cells and enhances the generation of T cell precursors. J. Immunol. 2006;177:109–119. doi: 10.4049/jimmunol.177.1.109. [DOI] [PubMed] [Google Scholar]
- Woll P.S., Morris J.K., Painschab M.S., Marcus R.K., Kohn A.D., Biechele T.L., Moon R.T., Kaufman D.S. Wnt signaling promotes hematoendothelial cell development from human embryonic stem cells. Blood. 2008;111:122–131. doi: 10.1182/blood-2007-04-084186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R.H., Sampsell-Barron T.L., Gu F., Root S., Peck R.M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008;3:196–206. doi: 10.1016/j.stem.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S.J., Foley J.W., Baker J.C. HEB associates with PRC2 and SMAD2/3 to regulate developmental fates. Nat. Commun. 2015;6:6546. doi: 10.1038/ncomms7546. [DOI] [PubMed] [Google Scholar]
- Yzaguirre A.D., de Bruijn M.F., Speck N.A. The Role of Runx1 in embryonic blood cell formation. Adv. Exp. Med. Biol. 2017;962:47–64. doi: 10.1007/978-981-10-3233-2_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambidis E.T., Peault B., Park T.S., Bunz F., Civin C.I. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood. 2005;106:860–870. doi: 10.1182/blood-2004-11-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.