

The management of adrenocortical cancer is challenging because of limited therapies, heterogeneous clinical presentations, and varying degree of aggressiveness at diagnosis. This article evaluates the use of mitotane for palliation in patients with advanced adrenocortical carcinomas, assessing response to treatment and treatment‐related side effects.
Keywords: Mitotane, Adrenocortical carcinoma, Adrenal insufficiency
Abstract
Purpose.
Based largely on reports that predate modern reporting standards, mitotane has been considered a systemic treatment option for both hormone control and antitumor control of metastatic adrenocortical cancer (ACC), although the therapeutic window is narrow.
Methods.
We searched electronic medical records to identify patients with metastatic ACC treated and prescribed single‐agent mitotane at Memorial Sloan Kettering Cancer Center from March 15, 1989–September 18, 2015. Reference radiologists reviewed all imaging and determined efficacy according to Response Evaluation Criteria in Solid Tumors 1.1. Patient demographics, toxicities, and treatment outcomes were reviewed. Next‐generation sequencing was performed in selected cases.
Results.
Thirty‐six patients were identified. The mean age was 54 and 50% had functional tumors. Grade 3 or greater toxicities were documented in 16 out of 36 patients (44%) and 17% had documented long term adrenal insufficiency. Progression of the disease as the best response occurred in 30 out of 36 patients (83%) and one patient (3%) experienced clinical progression. Three patients achieved a complete response (CR) (8%), one patient achieved a partial response (3%), and one patient (3%) had stable disease after slow disease progression prior to initiation of therapy (durable for 6 months). All responders had nonfunctional tumors. Next‐generation sequencing in two of the three CR patients was performed and failed to identify any novel alterations.
Conclusion.
In this retrospective series, mitotane had a low response rate and low tumor control rate; however, a disproportionately high complete response rate suggested it should be used in selected individuals. Adrenal insufficiency is common with mitotane use and aggressive treatment with steroid supplementation should be considered when appropriate to avoid excess toxicities. Biomarkers are desperately needed to further define this disease.
Implications for Practice.
This is the first objective report of single‐agent mitotane using modern objective criteria. Although the vast majority of patients did not respond (and toxicity was high), we identified a remarkable 8% complete response rate (i.e. cure) in biopsy proven stage IV adrenocortical cancer patients. Biomarkers are desperately needed for this rare disease.
Introduction
The management of adrenocortical cancer (ACC) poses a significant challenge because of limited therapies, heterogeneous clinical presentations, and varying degree of aggressiveness at diagnosis. There are very few randomized prospective trials, and as a result, physicians rely heavily on anecdotal evidence, uncontrolled trials, and retrospective studies [1], [2], [3].
Mitotane, or p′DDD, is a derivative of the insecticide dichlorodiphenyltrichloroethane and is the only drug approved by the U.S. Food and Drug Administration for ACC. Interest in mitotane and reports of its efficacy date back to the 1960s, when in vitro studies suggested that mitotane inhibited adrenocortical steroid biosynthesis while also increasing hepatic clearance of cortisol [4], [5]. At that time, several retrospective studies reported biochemical and tumor regression rates as high as 85% [1], [2], [3]. This older literature included unreliable study techniques and lacked modern standards of response and efficacy assessment. A pivotal New England Journal of Medicine paper included 105 patients, of which two thirds were functional and 30% (n = 31) had metastatic disease. Only eight patients (8%) in the entire cohort sustained a partial response, which did not affect overall survival. The authors concluded that mitotane may offer transient benefit, but mostly in endocrine symptoms [6]. Other studies also reported a biochemical response (lowering of hormone level) as a “response,” and as such implied a higher degree of tumor regression than was actually encountered. Mitotane can improve symptoms of hormone excess even in the setting of increasing tumor burden, which likely influenced early efficacy assessments [1], [2], [3].
As such, any standard recommendations based on these older retrospective studies must be made with extreme caution. More recent data, albeit also retrospective, suggested that these high response rates were not substantiated, although again an accurate assessment of true response rate remains unavailable [7], [8].
Equally if not more important than the question of efficacy is the degree of toxicity associated with the drug. Any therapy that is noncurative would, by definition, be palliative. The narrow therapeutic window can lead to chronic and often irreversible toxicity in the form of adrenal insufficiency, leading to nausea, vomiting, fatigue, and depression if left untreated. As a result of these issues, mitotane has been at the center of many controversies because of its questionable efficacy and associated toxicities. For this reason, we attempted to re‐evaluate the utility of mitotane for palliation in patients with advanced adrenocortical carcinomas, assessing response to treatment and treatment‐related side effects.
Methods
An institutional review board waiver of authorization was obtained to review electronic medical records of all patients with metastatic adrenocortical carcinoma who received single‐agent mitotane therapy at Memorial Sloan Kettering Cancer Center (MSKCC) from March 1989 through September 2015. Clinical histories and pathology were reviewed. Toxicities were categorized according to Common Terminology Criterion for Adverse Events (CTCAE) Version 4.0 (Bethesda, Maryland). Two reference radiologists reviewed all scans according to Response Evaluation Criteria in Solid Tumors 1.1 criteria. Progression‐free survival was calculated from the date of mitotane to the date of progression, the date of off study due to toxicity, or the date of death, whichever occurred first. All eligible patients had cross‐sectional images in our computerized system, and most patients had scans approximately every 2–6 months while on therapy. All patients were treated and prescribed mitotane at MSKCC and all patients were mitotane naïve prior to the initiation of therapy. Next‐generation sequencing was performed in selected “exceptional responders” (i.e., complete responders) using a platform developed by MSKCC known as IMPACT. MSK‐IMPACT has full coverage of 410 cancer‐related genes and detects base substitutions, small indels, copy number changes, and select gene rearrangements.
Results
Patient Characteristics
We identified 36 patients with a mean age of 54 (range, 23–88), 58% female. Eighteen of 36 tumors (50%) were functional: estradiol (n = 1), cortisol (n = 12), testosterone (n = 3), or both testosterone and cortisol (n = 2). Patient characteristics are outlined in Table 1. All patients had radiographic disease progression prior to the initiation of mitotane therapy. The median time on therapy was 13.5 weeks (range 3–702 weeks). Median follow‐up among survivors was 188 months. Median progression‐free survival was 3 months (95% confidence interval [CI] 2–6 months) and 1‐year survival was 55% [95% CI: 38%–70%] (Figs. 1, 2). The average maximum sustained daily dose of mitotane was 6.3 g (range 2–12 g). Therapeutic mitotane levels were available on 10 patients (see Table 1), and of those, 30% reached the target “therapeutic level” of 14–20 mcg/mL. Mitotane levels were available in one of the four responders; the patient achieved a therapeutic blood level of 19 mcg/mL (therapeutic range in that laboratory 14–20 mcg/mL) but required hospitalization for grade 3 nausea and dehydration (found to have a toxic level of 29 mcg/mL on 4500 mg daily) and was subsequently reduced to what are regarded as nontherapeutic doses.
Table 1. Patient characteristics.
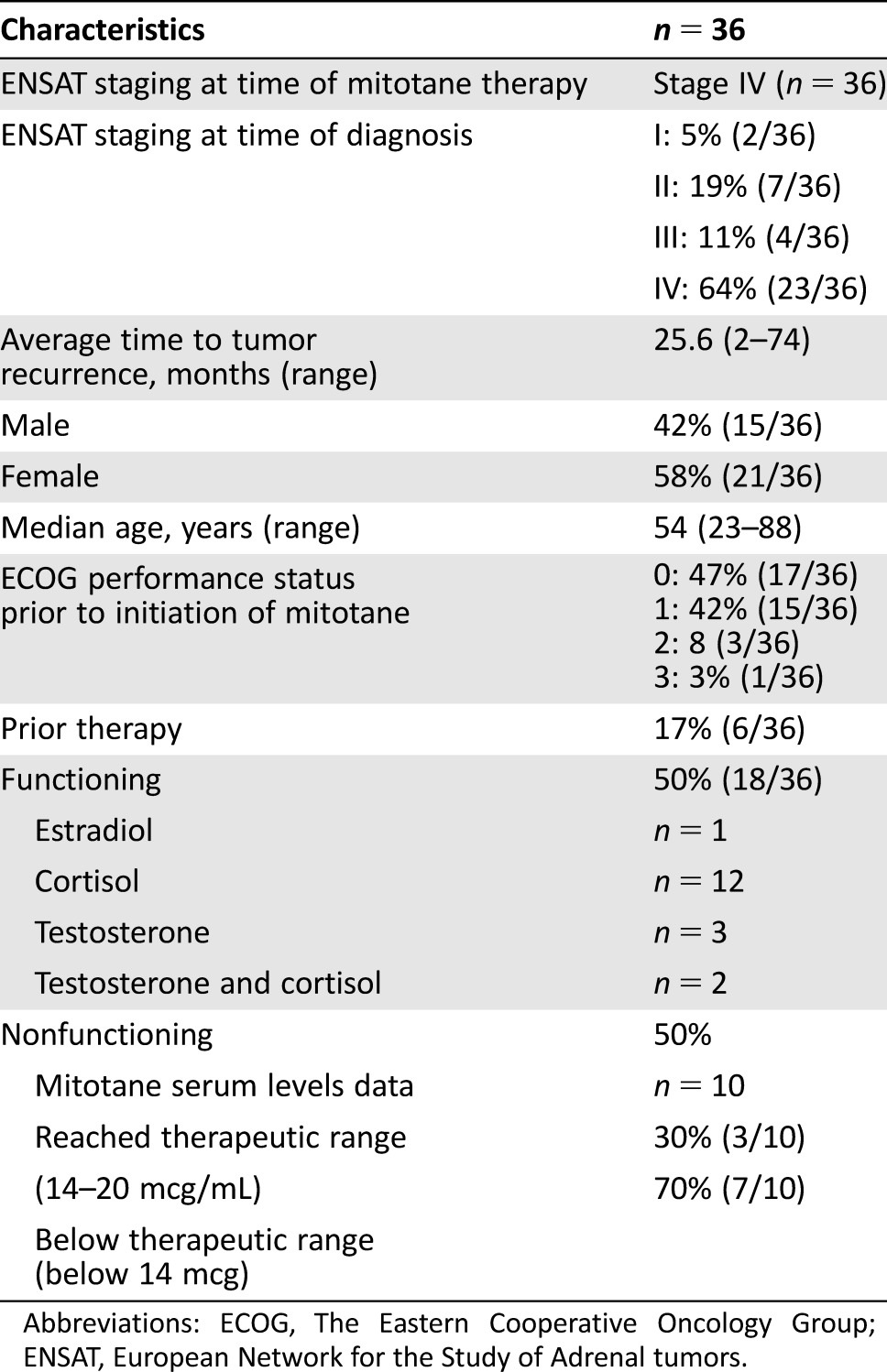
Abbreviations: ECOG, The Eastern Cooperative Oncology Group; ENSAT, European Network for the Study of Adrenal tumors.
Figure 1.
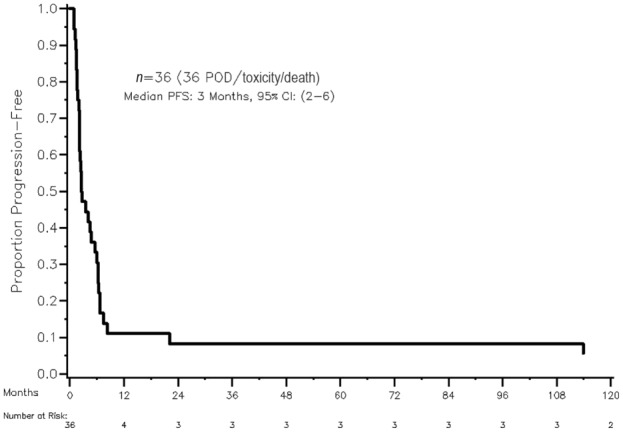
Progression‐free survival estimated using Kaplan‐Meier methods. Abbrevations: CI, confidence interval; PFS, progression‐free survival; POD, progression of disease.
Figure 2.
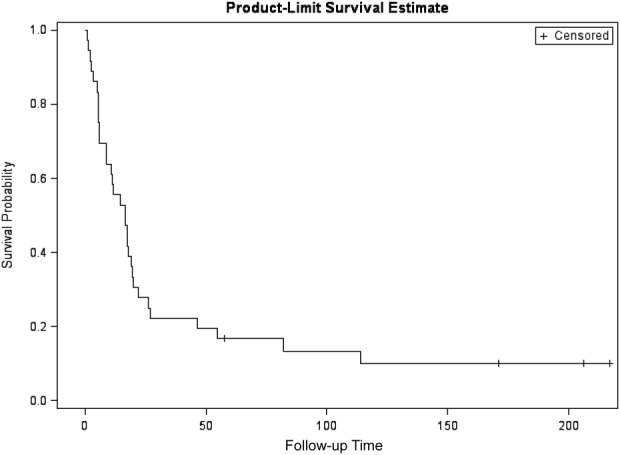
Overall survival. Patients who didn't experience the event at the end of the follow‐up were censored on June 13, 2016 (n = 4); median follow‐up among survivors was 188 months. Overall survival at 1 year was 55% [95% confidence interval: 38%–70%].
Toxicity
All 36 patients were assessable for toxicity. Table 2 lists the toxicities of mitotane that were possibly, probably, or definitely attributable to the drug. Grade 3 toxicities were observed in 16 of the 36 patients (44%). Severe toxicities requiring hospitalization in some patients included anorexia (8%), depression (6%), fatigue (19%), nausea (11%), vomiting (11%), electrolyte abnormalities (13%), abdominal pain (6%), bone marrow toxicity (20%), and rash (6%). Adrenal insufficiency, defined by use of steroid supplementation identified in the patient's pharmacy profile, occurred in 17% of patients. Those patients with documented adrenal insufficiency were recorded as such; associated symptoms related to the adrenal insufficiency (i.e., nausea, fatigue) were not separately recorded as toxicities. Given the retrospective nature of the review, we were unable to quantify the duration of these toxicities or if the symptoms improved with steroid replacement. The majority of the patients had their symptoms improve when the drug was temporarily held or discontinued. Additionally, 16% of the patients had labs done locally, which were not available for grading toxicity. All patients had the drug either interrupted, discontinued, or dose reduced. All four responders (all were nonfunctional had their mitotane treatment) interrupted and/or discontinued therapy because of toxicities. The three complete responders all remain adrenal insufficient, requiring lifelong hormone supplementation.
Table 2. Reported toxicities possibly, probably, or definitely related to mitotane.
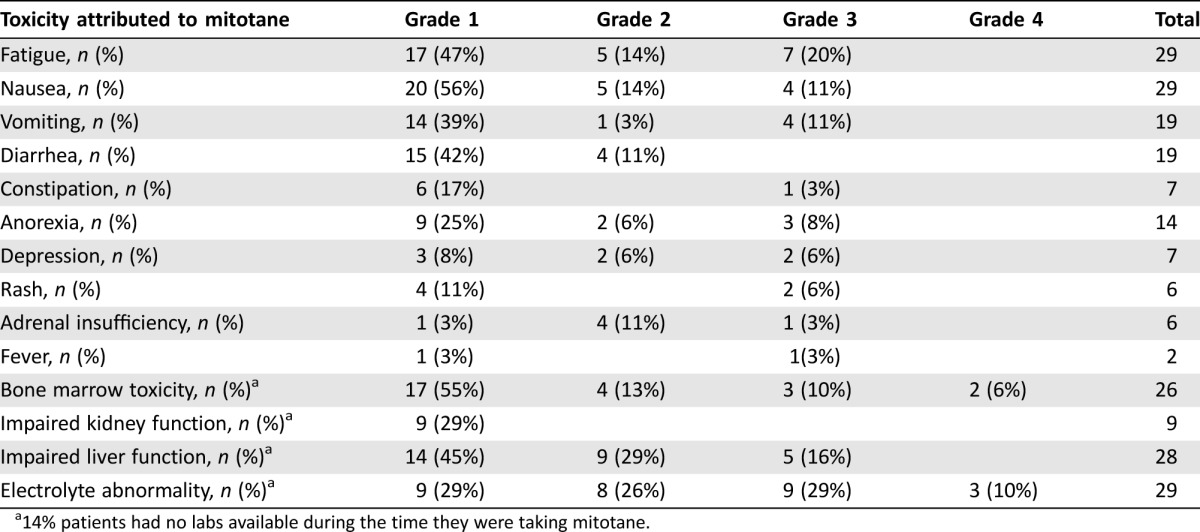
14% patients had no labs available during the time they were taking mitotane.
Efficacy
Radiographic progression of the disease occurred in 30 of 36 patients (83%) and one patient (3%) experienced clinical progression without imaging being obtained. Three patients had a complete response (8%), one patient had a partial response (3%), and one patient (3%) had stable disease (the latter of which had disease growth that stabilized on mitotane). The four patients (11%) with response were nonfunctional tumors, asymptomatic prior to initiating mitotane, and the disease was limited to one site (two patients liver only; one patient retroperitoneal lymphadenopathy only; one patient lung only); the three complete responders had low volume disease (<3 cm). Table 3 shows the cause of death for the 27 patients who died.
Table 3. Data on cause of death.
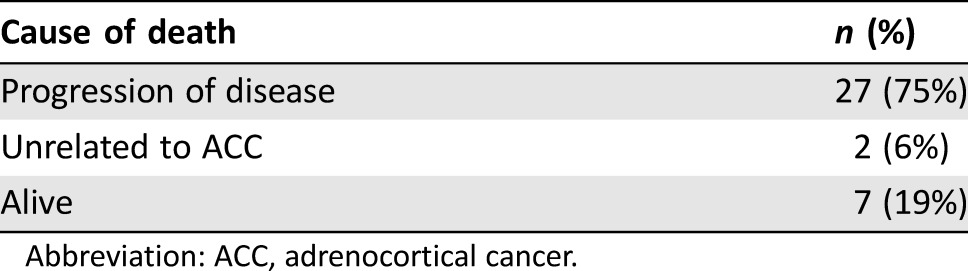
Abbreviation: ACC, adrenocortical cancer.
Germline Testing and Next‐Generation Sequencing
Only one patient underwent genetic germline testing and was found to have a deleterious inherited mutation (MLH1). Mismatch repair proteins on that tumor showed weak MLH1 staining, absent staining of the PMS2 protein, and normal/intact staining pattern of the MSH2 and MSH6 proteins. That patient did not respond to mitotane.
Next‐generation sequencing to better characterize these tumors was available on two of the four responders and one tumor harbored a frameshift indel in PTEN (p.K223fs) with amplification of several genes on chromosome 5, including TERT, IL7R, PDGFRB, CSF1R, and FGFR4. No genes were found to be deleted. A second tumor harbored a TP53 mutation and had several amplifications (CCDN1, FGF19, FGF3, FGF4, GLI1, SDHA) and deletions (ARID5B, CDKN1A, DAXX, EP300, ERBB2, HRAS, MCL1, PDCD1, PIM1, RARA). No germline mutations were identified in these two patients.
Discussion
In this retrospective study, 86% of our patients had disease progression on single‐agent mitotane and 44% experienced at least grade 3 toxicities thought to be possibly related to mitotane. There was, however, a remarkable 8% complete response rate.
What made the complete responders different? Two CR patients had clinical and radiographic asymptomatic and indolent disease prior to initiating mitotane therapy. The third CR had biopsy‐proven liver‐only disease growing quite rapidly prior to mitotane treatment. All patients had nonfunctional tumors and an excellent performance status prior to initiating mitotane (ECOG 0–1). It is possible that these patients may have achieved higher serum concentrations of mitotane, allowing for treatment response. The only CR patient with an available mitotane level achieved a therapeutic level once and was then hospitalized requiring dose reduction to subtherapeutic levels going against such a hypothesis. There are three studies suggesting that antitumor activity of mitotane requires a serum mitotane value greater than 14–20 mcg/mL [9], [10], [11]. In those studies, however, increase survival times were found in the group with higher mitotane levels when compared with patients not treated to such high levels irrespective of treatment duration. Although one possibility is that the higher therapeutic levels led to a higher response, a second and equally plausible hypothesis is that those patients selected to achieve higher levels were healthier, resulting in an undetected selection bias in patients who were going to have a better prognosis because of the biologically favorable tumors. This is an important consideration, because in general, clinicians are willing to aggressively use mitotane in patients who are higher risk and/or have more aggressive disease; in our series, however, none of the higher tumor burden and/or poor performance patients responded to single‐agent mitotane, bringing such an approach into question.
Nevertheless, many believe that mitotane is the therapeutic backbone of all metastatic ACC treatments, even in nonfunctional tumors, despite the absence of a randomized trial with mitotane as a variable. Indeed, the pivotal FIRM‐ACT study used mitotane in both the experimental and control arms despite the controversy regarding its efficacy. In that study, grade 3–4 adverse events were a remarkable 58% in the cisplatin, etoposide, doxorubicin, mitotane arm versus 41% in the streptozocin, mitotane arm. In addition, there is no plausible scientific explanation as to why mitotane would enhance the antitumor efficacy of cytotoxic chemotherapies [12]. Mitotane had previously been shown to be a putative inhibitor of P‐glycoprotein, and so was thought to potentiate the efficacy of other p‐glycoprotein substrates such as doxorubicin, leading to its initial incorporation into doxorubicin‐containing regimens; however, the hypothesis that p‐glycoprotein modulation would meaningfully increase antitumor efficacy has been repeatedly refuted by clinical experimental data and has now been abandoned.
Next‐generation sequencing of the tumors from the two complete responders failed to identify any novel mutations that could explain the differences. Both patients harbored the typical mutations, amplifications, and deletions reported in prior studies. A review of 17 ACC patients in our own MSKCC database identified similar alterations. It is noteworthy, however, that the effects of mitotane are thought to be based on metabolism of steroid biosynthesis, so it is certainly possible that mutations in these pathways are responsible for sensitivities or dependencies and such alterations would not have been identified in our next‐generation sequencing platform. Indeed, Zheng et al. recently reported a comprehensive genomic characterization of ACC [13]. Their study used integrated subtype analysis and identified three ACC subtypes with distinct clinical outcome and molecular alterations [13]. Such promising work potentially allows for a strategy to clinically stratify patients based on molecular markers. Moving forward, we hope to consider such classifications.
Our retrospective study has limitations. First, our study population has a low number of patients but would be the sample size of a typical phase II study. Second, it is hard to quantify the quality of life in a retrospective review except by a review of all progress notes and symptoms as best defined by CTCAE. Third, mitotane levels were not available on all patients, although as noted above, the importance or relevance of achieving that which is reported to be a therapeutic range is not validated and is based on retrospective analyses of small studies. Fourth, the median duration of treatment was 4 months, a short time to assess the efficacy of a drug that takes many weeks to get therapeutic levels; this series, therefore, likely reflects an “intention to treat” sample. Nevertheless, a median overall survival of 12 months compares quite similarly to the FIRM‐ACT trial, in which mitotane was used with other agents, and as noted, serious adverse events were 58%. Additionally, this retrospective series evaluated mitotane as an antitumor agent and did not evaluate the drug for control of hormone symptoms. Lastly, it is possible that many of the side effects described in this cohort were secondary to undetected adrenal insufficiency. Aggressive steroid replacement could have potentially improved their quality of life and reduced toxicities. Today we are much more aggressive in doing cortisol/ACTH hormone levels and treating accordingly. We recommend glucocorticoids and possibly mineralocorticoid supplementation in any patient with suspicion of adrenal insufficiency.
Conclusion
In summary, despite anecdotal and retrospective historical data suggesting antitumor activity of mitotane, our study failed to show that mitotane was clinically helpful in the vast majority of patients. There were, however, a few patients with excellent performance status that achieved durable complete responses, which is highlighted. The retrospective data presented might suggest that patients with ECOG 0–1 performance status and with low disease burden be considered for mitotane, whereas patients with high disease burden might benefit from cytotoxic chemotherapy. Future work needs to be done to evaluate this consideration further.
Acknowledgment
This work was supported by the philanthropic funds from The Drew O'Donoghue Fund. Presented at the ASCO 2015 General Meeting, Chicago (General Poster Session).
Author Contributions
Conception/Design: Diane L. Reidy‐Lagunes, Betty Lung, Anastasia Hrabovsky, Brian R. Untch, Nitya Raj, Ciara Kelly, Seth Katz, Joanne Chou, Anu Gopalan, Leonard B. Saltz, Scott Gerst
Provision of study material or patients: Diane L. Reidy‐Lagunes, Betty Lung, Anastasia Hrabovsky, Ciara Kelly
Collection and/or assembly of data: Diane L. Reidy‐Lagunes, Betty Lung, Anastasia Hrabovsky, Nitya Raj, Ciara Kelly, Lewis Kampel
Data analysis and interpretation: Diane L. Reidy‐Lagunes, Betty Lung, Anastasia Hrabovsky, Brian R. Untch, Nitya Raj, Ciara Kelly, Seth Katz, Joanne Chou, Anu Gopalan, Leonard B. Saltz, Scott Gerst
Manuscript writing: Diane L. Reidy‐Lagunes, Betty Lung, Anastasia Hrabovsky, Brian R. Untch, Nitya Raj, Ciara Kelly, Seth Katz, Lewis Kampel, Joanne Chou, Anu Gopalan, Leonard B. Saltz, Scott Gerst
Final approval of manuscript: Diane L. Reidy‐Lagunes, Betty Lung, Brian R. Untch, Nitya Raj, Seth Katz, Lewis Kampel, Joanne Chou, Anu Gopalan, Leonard B. Saltz, Scott Gerst
Disclosures
The authors indicated no financial relationships.
References
- 1. Bergenstal DM, Hertz R, Lipsett MB et al. Chemotherapy of adrenocortical cancer with o, p'DDD. Ann Intern Med 1960;53:672–682. [Google Scholar]
- 2. Hutter AM, Kayhoe DE. Adrenal cortical carcinoma. Clinical features of 138 patients. Am J Med 1966;41:572–580. [DOI] [PubMed] [Google Scholar]
- 3. Lubitz JA, Freeman L, Okun R. Mitotane use in inoperable adrenal cortical carcinoma. JAMA 1973;223:1109–1112. [PubMed] [Google Scholar]
- 4. Young RB, Bryson MJ, Sweat ML et al. Complexing of DDT and o,p'DDD with adrenal cytochrome P‐450 hydroxylating systems. J Steroid Biochem 1973;4:585–591. [DOI] [PubMed] [Google Scholar]
- 5. Hogan TF, Citrin DL, Johnson BM et al. o,p'–DDD (mitotane) therapy of adrenal cortical carcinoma: Observations on drug dosage, toxicity, and steroid replacement. Cancer 1978;42:2177–2181. [DOI] [PubMed] [Google Scholar]
- 6. Luton JP, Cerdas S, Billaud L et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med 1990;322:1195–1201. [DOI] [PubMed] [Google Scholar]
- 7. Haak HR, Hermans J, van de Velde CJ et al. Optimal treatment of adrenocortical carcinoma with mitotane: Results in a consecutive series of 96 patients. Br J Cancer 1994;69:947–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzalez RJ, Tamm EP, Ng C et al. Response to mitotane predicts outcome in patients with recurrent adrenal cortical carcinoma. Surgery 2007;142: 867–875; discussion 867–875. [DOI] [PubMed] [Google Scholar]
- 9. Baudin E, Pellegriti G, Bonnay M et al. Impact of monitoring plasma 1,1‐dichlorodiphenildichloroethane (o,p'DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer 2001;92:1385–1392. [DOI] [PubMed] [Google Scholar]
- 10. van Slooten H, Moolenaar AJ, van Seters AP et al. The treatment of adrenocortical carcinoma with o,p'‐DDD: Prognostic implications of serum level monitoring. Eur J Cancer Clin Oncol 1984;20:47–53. [DOI] [PubMed] [Google Scholar]
- 11. Terzolo M, Pia A, Berruti A et al. Low‐dose monitored mitotane treatment achieves the therapeutic range with manageable side effects in patients with adrenocortical cancer. J Clin Endocrinol Metab. 2000;85:2234–2238. [DOI] [PubMed] [Google Scholar]
- 12. Fassnacht M, Terzolo M, Allolio B et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 2012;366:2189–2197. [DOI] [PubMed] [Google Scholar]
- 13. Zheng S, Cherniack AD, Dewal N et al. Comprehensive pan‐genomic characterization of adrenocortical carcinoma. Cancer Cell 2016;29:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]