

Abstract
Tissue development and homeostasis are governed by the actions of stem cells. Multipotent cells are capable of self-renewal during the course of one's lifetime. The accurate and appropriate regulation of stem cell functions is absolutely critical for normal biological activity. Several key developmental or signaling pathways have been shown to play essential roles in this regulatory capacity. Specifically, the Janus-activated kinase/signal transducer and activator of transcription, Hedgehog, Wnt, Notch, phosphatidylinositol 3-kinase/phosphatase and tensin homolog, and nuclear factor-κB signaling pathways have all been shown experimentally to mediate various stem cell properties, such as self-renewal, cell fate decisions, survival, proliferation, and differentiation. Unsurprisingly, many of these crucial signaling pathways are dysregulated in cancer. Growing evidence suggests that overactive or abnormal signaling within and among these pathways may contribute to the survival of cancer stem cells (CSCs). CSCs are a relatively rare population of cancer cells capable of self-renewal, differentiation, and generation of serially transplantable heterogeneous tumors of several types of cancer.
Keywords: cross-talk, microenvironment, signaling, STAT, stemness, Wnt
1. Introduction
The proper functioning of normal stem cells is an absolute necessity to sustain life. The specific properties that define a stem cell include the ability to self-renew and to generate the multitude of differentiated cellular progeny required to populate a tissue.[1] These hallmarks have been well established through >50 years of research in hematopoiesis. While continually maintaining their own stem cell pool for the entirety of adult life, hematopoietic stem cells in the bone marrow give rise to increasingly lineage-restricted progenitor cells, which ultimately produce 10 distinct types of daughter cells.[2] Established cellular differentiation pathways may not be exclusive and alternate pathways between various multipotent progenitors and differentiated progeny may also exist.[2]
In solid tissues, lineage-tracing studies in mouse models have identified cells of origin in numerous organs including the skin, mammary glands, and intestine, and have allowed for the visualization of stem cell self-renewal and the differentiation of their progeny within the de novo tissue architecture.[3–6] The small intestine is one such well-studied system. The small intestine is comprised of villi and crypts, or peaks and valleys of epithelial cells, which proliferate rapidly to produce completely new tissue over several days.[7] Two intestinal stem cell types have been identified: the crypt base columnar cells, which are marked by their expression of the orphan leucine-rich repeat-containing G protein-coupled receptor (Lgr) 5; and cells that reside at the +4 location from the bottom of the crypt, which express other biomarkers including Bmi1.[3,7–9]
Given the replicative and regenerative potential of a normal organ stem cell, it has been postulated that cancer may develop from an analogous small population of cells with self-renewal properties. This is the rationale for the cancer stem cell (CSC) hypothesis, that is, a small subpopulation of CSCs, or tumor-initiating cells, is responsible for propagating a tumor through their ability to self-renew and to create the heterogeneous milieu of nontumorigenic cells that make up the bulk of a tumor.[10] These 2 traits define the characteristic “stemness” of CSCs thought to support their function.[11–13] Mounting data have identified CSCs in many solid tumors, as well as some hematopoietic cancers.[12,14] Several models have been proposed to describe how CSC stemness may help identify the cancer cell of origin and account for the molecular and phenotypic differences within and between these diverse types of cancer.[15] (See “Cancer Stem Cells: Understanding Tumor Hierarchy and Heterogeneity” for a broader review.) Although these models are still under study, growing evidence suggests that CSCs may contribute to or outright underlie important disease milestones ranging from tumorigenesis to metastasis.[12]
Mechanistic studies have indicated that dysfunction of several developmental and homeostatic stemness signaling pathways may support unregulated self-renewal and differentiation that drive CSC functions.[11,16–18] The growing list of dysfunctional signaling pathways in CSCs include the Janus-activated kinase/signal transducer and activator of transcription (JAK/STAT), Hedgehog, Wnt, Notch, phosphatidylinositol 3-kinase/phosphatase and tensin homolog (PI3K/PTEN), and nuclear factor-κB (NF-κB).[12] As yet the precise mechanisms underlying dysfunctional CSC signaling pathways have not been elucidated. This article highlights current understanding of the function of signaling pathways in both normal stem cells and CSCs.
2. Signaling pathways in normal stem cells and CSCs
The molecular signaling pathways that govern normal stem cell homeostasis are highly regulated. Not surprisingly, many of these pathways are abnormally activated or repressed in human cancers and in experimental models of tumorigenesis. Such abnormalities contribute to the self-renewal, proliferative, survival, and differentiation properties of CSCs. In general, these pathways are intricate, with many extrinsic and intrinsic molecular signals and regulatory elements. Many of these “pathways” are not linear, but rather interwoven networks of signaling mediators that feed into one another, facilitating inter-pathway cross talk. Thus, several molecular methodologies, including the expression of cell surface proteins (e.g., CD44 and CD133), intracellular markers (e.g., aldehyde dehydrogenase [ALDH]), stemness genes (e.g., OCT4 and SOX2), and phenotypic assays (e.g., tumorsphere formation and serially transplantable in vivo tumors) are used to test stemness.[11,19–21] This review will highlight some of these methods to describe how signaling pathways support CSC function.
2.1. JAK/STAT pathway
The JAK/STAT signaling pathway is activated through the binding of diverse ligands, such as interleukins, interferons, hormones, and growth factors, to their respective receptors.[22] Ligand binding stimulates receptor oligomerization and aggregation of JAK proteins (JAK1-3 and TYK2) around the cytoplasmic domain of the receptor, prompting phosphorylation and activation of the JAK proteins (Fig. 1).[22,23] Activated JAK proteins promote the phosphorylation of the cytoplasmic domain of the receptor and recruitment of the STAT protein family. Once associated with JAKs, STATs become phosphorylated, resulting in dimerization and translocation to the nucleus to initiate transcription of target genes.[22,23] A wide range of cytoplasmic proteins function as negative regulators of this pathway, including those that inhibit binding of STATs to JAKs at the membrane (SOCS proteins), those that promote dephosphorylation of STATs (protein phosphatases), and/or those that sequester STATs away from the nucleus (PIAS proteins).[23]
Figure 1.
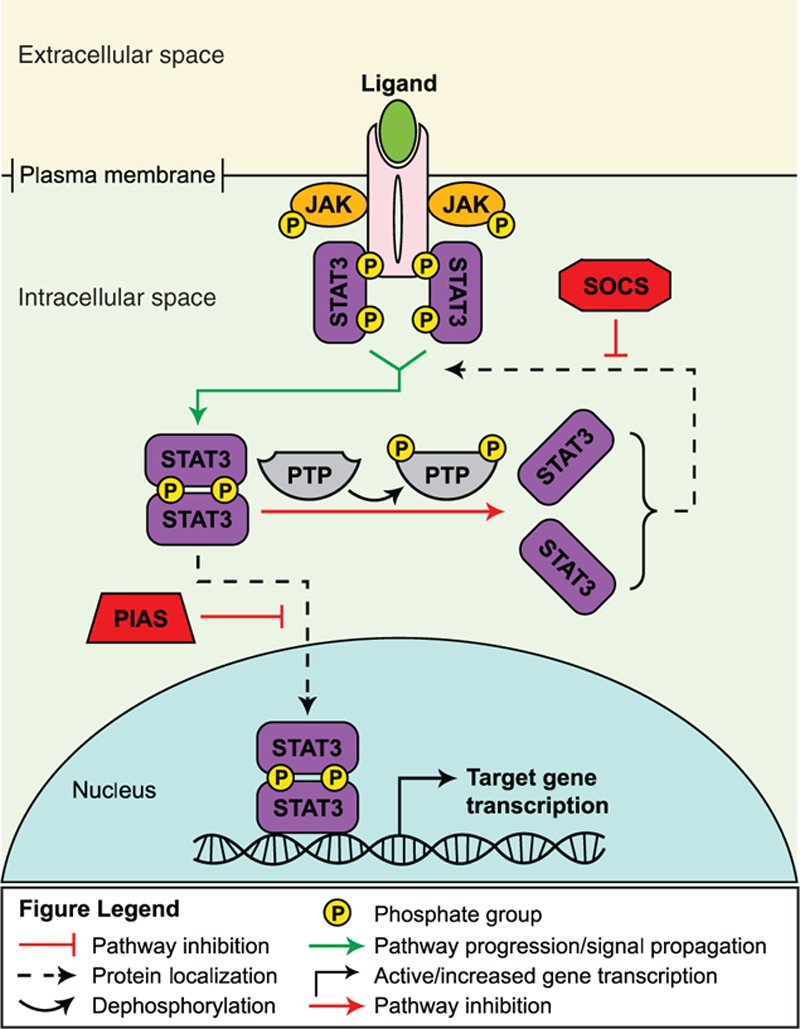
JAK/STAT signaling pathway.[23]
The kinases and transcription factors that constitute the JAK/STAT pathway are evolutionarily conserved signaling molecules that regulate numerous cellular processes across many tissue types, including those of the immune and nervous systems.[23] JAK/STAT signaling in stem cells has been shown to be involved in maintaining embryonic stem cell self-renewal properties, hematopoiesis, and neurogenesis.[23,24]
Evidence that this pathway is activated aberrantly in CSCs has been found in stem-like cells isolated from tumors of the breast, prostate, blood, and glia. For example, stem-like cells isolated from prostate cancer cells were shown to overexpress several genes involved in JAK/STAT signaling including IFNK, IFNGR, IL6, CSF2, and STAT1,[25] and the activated form of STAT3 was demonstrated to be the most significantly upregulated JAK/STAT signaling protein in breast CSC-like cells.[26]
Modulation of the JAK/STAT pathway in CSCs has been shown to enhance or repress the expansion of these cancer-forming cells in solid tumor model systems.[23] In a study of glioblastoma, tumor growth factor-beta (TGF-β) was shown to regulate the self-renewal and differentiation properties of glioma-initiating cells derived from patient samples of glioblastoma multiforme. The actions of TGF-β were mediated through leukemia inhibitory factor (LIF) activation of the JAK/STAT pathway.[27] Another study of glioblastoma demonstrated that chemical inhibition of STAT3 in CSCs prevents proliferation and tumorsphere formation, decreases the expression of the neural stem cell genes Olig2 and nestin, and increases the expression of the neuronal differentiation gene βIII-tubulin.[28] These data suggest that JAK/STAT signaling is important for glioblastoma CSC proliferation and positively regulates glioblastoma stemness. Similar experiments in breast CSCs have indicated that chemical inhibition of STAT3 reduced CSC abundance, proliferation, and clonogenicity, further supporting the role of JAK/STAT signaling in promoting cancer stemness.[26]
JAK/STAT signaling has been implicated in CSC-mediated metastasis. Stage IV clinical tumor sample-derived colon cancer CSCs were unresponsive to TGF-β stimulation or inhibition as the genes for TGF-β receptors were found to be mutated. However, the CSCs expressed high levels of the TGF-β gene and formed in vivo tumors that expressed high levels of the activated form of SMAD2, a TGF-β signal transducer, exclusively in the stromal compartment. Importantly, TGF-β inhibitor-fed mice showed a significantly reduced incidence of liver metastasis. TGF-β signaling pathway analysis of the activated stromal cells indicated that the cells secreted significant levels of interleukin-11 (IL-11), a known ligand of JAK/STAT signaling, which corresponded to the high levels of the activated form of the STAT3 protein observed in adjacent tumor cells. Accordingly, expression of the IL11 gene was found to be significantly reduced in activated stromal cells isolated from primary tumor samples of TGF-β inhibitor-feeding mice bearing colon cancer xenografts. Furthermore, mouse colon cancer xenografts derived from a TGF-β-secreting cell line deficient in the JAK/STAT receptor GP130, resulted in an increase in apoptosis of tumor cells isolated from early liver metastases, indicating a potential requirement of JAK/STAT signaling for the survival of early metastatic colonies. Collectively, these data suggest that CSC-dependent, tumor microenvironment-mediated JAK/STAT signaling may be important for initial phases of colon cancer metastasis.[29]
Aberrant JAK/STAT signaling has also been observed in myeloproliferative malignancies.[30] Isolation and analysis of CSCs from patients with acute myeloid leukemia (AML) identified constitutive activation of JAK/STAT signaling. In vitro studies indicated that the growth and survival of these CSCs were reduced when treated with a JAK1/2 inhibitor. Moreover, the CSCs lost their ability to engraft immunodeficient mice or to form AML upon secondary transplantation.[31]
2.2. Hedgehog pathway
The major players in the Hedgehog pathway include 3 secreted Hedgehog ligands—Sonic, Desert, and Indian—their cognate receptor Patched, the transmembrane protein Smoothened, and 3 Gli transcription factors (Glis1–3; Gli was named as such because of the identification and isolation of Gli1 from a glioma cell line) that modulate activation or repression of the pathway.[18,32] The Patched receptor functions as a constitutive inhibitor of Smoothened when it is unoccupied by ligand. In this state, target gene transcription is repressed by Gli3 and Gli2-R (Gli2 in its repressor form). Upon ligand binding to Patched, the repression upon Smoothened is released, which allows the transcriptional activators Gli1 and Gli2-A (Gli2 in its activator form) to facilitate transcription of target genes (Fig. 2).[18]
Figure 2.
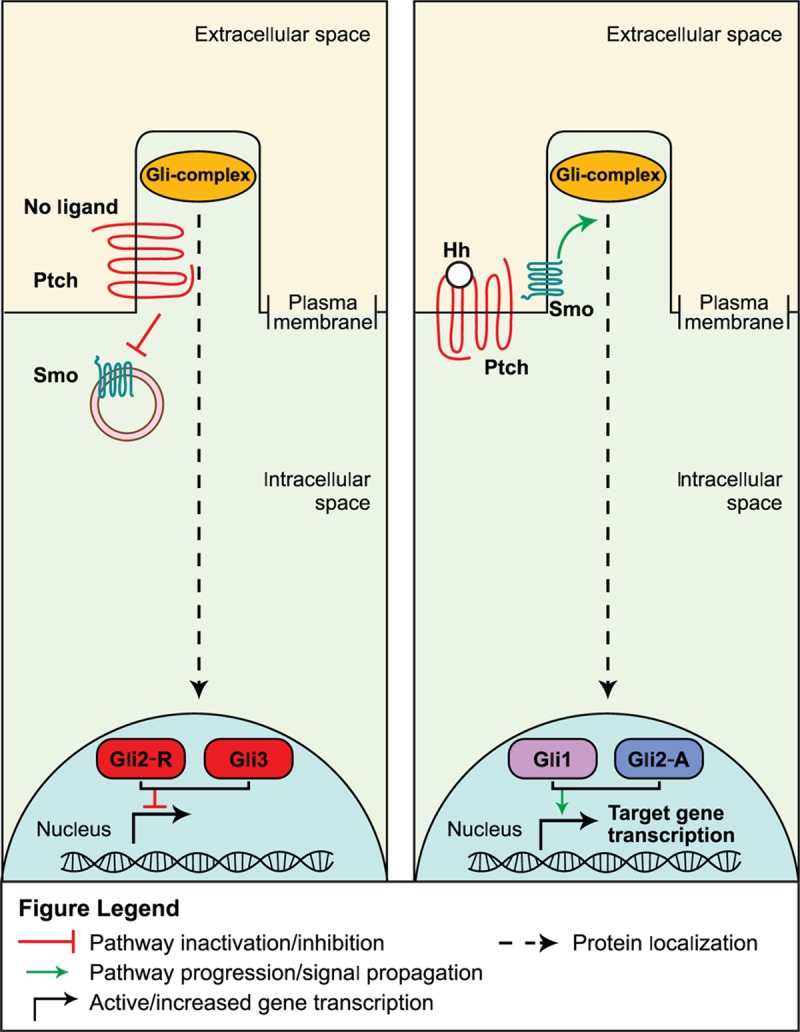
Hedgehog inhibition (left panel) and activation (right panel) signaling pathways.[18]
The Hedgehog pathway is essential for the development and proper patterning of many organs during embryogenesis, including the nervous system, skeleton, limbs, lung, heart, and gut, by controlling cellular proliferation, differentiation, and migration.[18,33] Unlike many other pathways described here, the Hedgehog pathway is largely inactive in most postnatal tissues except the adult central nervous system, skin, hair, and teeth. Recently, Hedgehog activity has been shown to regulate the resident stem and/or progenitor cell populations.[18,33]
Mouse studies and in vitro analyses of cancer cell lines and patient samples have confirmed the presence of aberrant Hedgehog signaling in more than a dozen types of cancers.[18] The role for Hedgehog signaling in CSC function has been documented in various cancers, including basal cell carcinoma (BCC), multiple myeloma, glioblastoma, chronic myeloid leukemia (CML), and colon cancer.[18,34]
Humans with mutations in the patched 1 (PTCH1) gene are predisposed to developing medulloblastomas. Additionally, analysis of the corresponding mouse gene, Ptch1, indicated that mice bearing a heterozygous mutation in the gene had a similar predisposition.[35] Furthermore, mutations in the PTCH1 gene cause Gorlin syndrome, a disease predisposing patients to advanced BCC.[36] A recent analysis deleting Ptch1 in various cellular compartments in murine skin identified that stem cells located within several different areas of the hair follicle can form BCC upon loss of Ptch1. Loss of Ptch1 in the stem cells of the interfollicular epidermis, however, had no effect. Interestingly, the ability of aberrant Hedgehog signaling to induce BCC depended on the presence of sensory nerves in the stem cell niche.[36] These findings support previous studies indicating that BCCs may arise from overactive Hedgehog signaling (via Gli2) in small populations of residual, long-term cancer-initiating cells in skin and hair follicles.[34]
The Hedgehog pathway has also been shown to regulate the properties of CSCs in multiple myeloma, glioma, and CML.[37–39] In an analysis of progenitor cells isolated from a human multiple myeloma cell line, the SMO gene, which encodes the Smoothened protein, was overexpressed and corresponded with high Gli1 transcriptional activity, in comparison to nonstem cancer cells.[38] Chemical inhibition of the Smoothened protein attenuated proliferation, stemness maintenance, and self-renewal in CSCs, suggesting strongly that Hedgehog signaling promotes these CSC functions in multiple myeloma.[38] In human glioma, CSCs were observed to overexpress the Hedgehog signaling genes Gli1, SHH, and PATCHED1.[37] Treatment of glioma CSCs with a Hedgehog signaling inhibitor resulted in a decrease in proliferation, survival, self-renewal, and clonogenicity along with reduced expression of the stemness genes NANOG, SOX2, and OCT4.[37] Moreover, Hedgehog signaling was demonstrated to support tumor growth in nude mice using gliomasphere cells expressing a conditionally suppressible form of SMO.[37] These data indicate that glioma CSCs may require Hedgehog signaling for functional support.[37] In CML, a murine model of BCR-Abl1–induced CML was used to demonstrate that deletion of SMO significantly reduced the frequency of CML CSCs.[39] Overexpression of SMO in a SMO-deficient mouse model of CML increased CSC frequency 4-fold and significantly increased CML progression.[39] In vitro expression analysis of Numb, a prodifferentiation gene, indicated the existence of an inverse relationship between SMO and Numb in which SMO-deficient CML CSCs expressed high levels of Numb. These results were complimentary to those from experiments demonstrating that overexpression of Numb in SMO-expressing CML CSCs reduces colony numbers.[39] Finally, chemical inhibition of SMO in CSCs was found to prevent the propagation of CML in CSC transplantation assays.[39] Collectively, these data suggest that Hedgehog signaling controls frequency and maintenance of stemness in CSCs, and may be important in CSC-driven propagation of CML.[39]
A similar role for CSCs in colon cancer was observed in experiments in which genetic knockdown of SMO in CSCs resulted in complete ablation of the malignant stem cells over only a single in vivo serial tumor passage.[40] Moreover, genetic knockdown of PTCH1 in CSCs resulted in an increase in their numbers during serial tumor passaging.[40] These results suggest that Hedgehog signaling may be required for colon CSC stemness and survival, which may promote tumor maintenance and growth.[40] Additionally, patient-derived CSCs isolated from colon cancer liver metastases were found to express high levels of Hedgehog signaling genes Gli1, Gli2, and HIP compared with nonmetastatic controls.[40]Gli1, however, was exclusively expressed in CSCs.[40] Coupled with results from experiments demonstrating that the frequency of CSCs increases with disease progression,[40] these data indicate that Hedgehog signaling activity may increase as colon cancer advances.[40] Indeed, CSCs isolated from liver metastases expressed higher levels of SNAIL1, a Gli1 target, compared with nonmetastatic controls.[40] Given that SNAIL1 facilitates epithelial–mesenchymal transition,[41] a process strongly implicated in promoting metastasis,[41] the gene transcriptional data suggest that Hedgehog signaling is important in the prometastatic role of CSCs implied in this study.[40]
2.3. Wnt pathway
The Wnt pathway is a highly complex, evolutionarily conserved signaling pathway encompassing 19 Wnt ligands and >15 receptors.[42] Conventionally, the Wnt pathway is considered to encompass 2 signaling pathways: canonical (mediated through the transcriptional regulator β-catenin) and noncanonical (β-catenin-independent).[42]
The canonical Wnt signaling pathway is activated when Wnt ligands secreted by 1 cell bind to Frizzled receptors and/or the low-density lipoprotein-related protein (LRP) 5 and LRP 6 co-receptors on a neighboring cell (Fig. 3).[7] Recent data have described enhancement of the Wnt pathway through the actions of R-spondin ligands bound to Lgrs, as well.[7,8,43]
Figure 3.
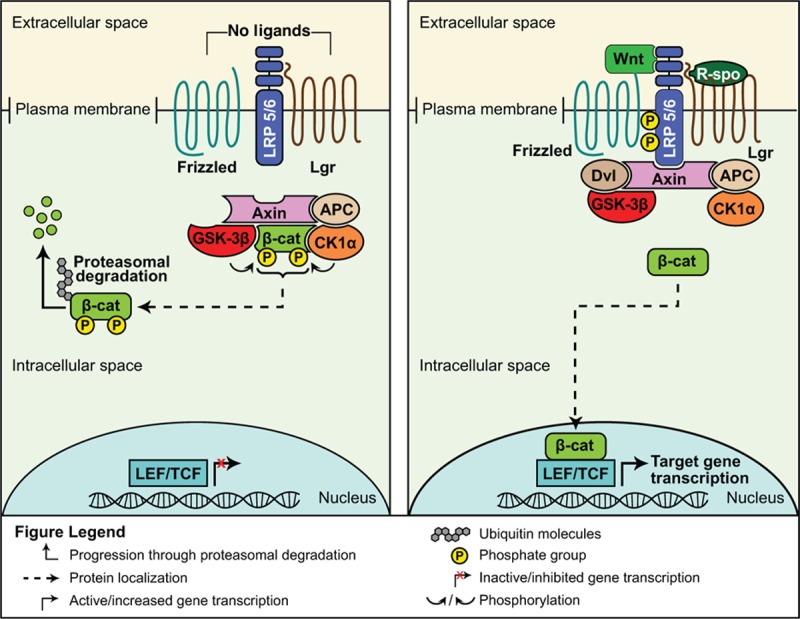
Inactive (left panel) and active (right panel) Wnt signaling pathways.[8]
In the absence of Wnt ligand binding, transcriptional regulator β-catenin protein intracellular levels are kept low through the actions of a destruction complex. The complex includes the scaffolding proteins Axin and adenomatous polyposis coli (APC), and the kinase proteins glycogen synthase kinase-3β (GSK-3β) and casein kinase 1α (Ck1α). GSK-3β and Ck1α, phosphorylate β-catenin tagging it for ubiquitination and subsequent proteosomal degradation.[7,42] When Wnt binds to Frizzled/Lrp, the cytoplasmic domain of Lrp becomes phosphorylated, sequestering GSK-3β and Axin and recruiting the scaffolding protein Disheveled (Dvl). This disassembles the destruction complex. Free β-catenin translocates into the nucleus, binds to lymphoid enhancer factor (LEF)/T-cell factor (TCF) transcription factors, and activates transcription of numerous target genes.[7,44]
Signaling through the canonical and noncanonical Wnt pathways is essential for embryonic development and homeostasis of a wide range of tissues.[8,42] In general, the canonical Wnt pathway regulates proliferation, survival, and cell fate decisions; the noncanonical Wnt pathway regulates asymmetrical cell division, cell polarity, and migration. However, these pathways are not mutually exclusive and cross-talk may occur between the 2 arms.[42] Lineage tracing has identified numerous postnatal tissues, including the epidermis, hair follicle, intestine, mammary gland, central nervous system, and kidney, from which normal stem cells are regulated by the canonical Wnt signaling pathway.[7]
Mutations in genes encoding Wnt pathway mediators are commonplace in many cancers including medulloblastoma, lymphoma, and leukemia, as well as breast, gastric, and colorectal cancer (CRC).[45] Moreover, APC tumor suppressor mutations cause familial adenomatous polyposis and the majority of sporadic CRCs.[45]
Experimental evidence supports a role for Wnt activation in regulating CSCs of blood, intestine, lung, mammary gland, nervous system, skin, and urinary tract cancers.[8] A role for activated Wnt signaling in the development of skin cancer was uncovered through the use of a reporter mouse strain. Wnt was activated in bulge stem cells (identified by their expression of cell surface molecule CD34) of the hair follicle.[46] To establish an initiating role for β-catenin in skin tumorigenesis, genetic deletion of β-catenin in chemically induced skin tumors resulted in complete tumor regression preceded by a marked depletion in the pool of CD34+ stem cells. Furthermore, these tumors lost the ability to propagate secondary tumors upon transplantation.[46] Conversely, expression of a nondegradable β-catenin in the skin expanded the stem cell population.[46]
Support for Wnt signaling playing a causal role in mammary tumorigenesis comes from numerous transgenic mouse models of pathway activation, which invariably result in hyperplastic and tumorigenic phenotypes.[47] In human breast cancers, many activators of the Wnt pathway and downstream target genes are amplified or upregulated while inhibitors of the pathway are inactivated or epigenetically silenced.[47] Normal stem cells capable of reconstituting the entire mammary epithelia have been identified in mice.[48,49] Within an established mouse model of mammary tumorigenesis caused by constitutive overexpression of Wnt1 in the mammary glands, mammary stem cells were >6 times more abundant than control glands.[48] In a murine mammary tumor virus (MMTV)-Wnt1 mouse model study, deletion of the Lrp5 receptor inhibited hyperplastic growth and tumorigenesis and decreased the number of progenitor cells within the glands.[50]
As mentioned above, loss of the tumor suppressor APC gene results in familial and sporadic CRCs in humans.[45] In mice, deletion of the Apc gene results in rapid formation of adenomas in the intestine. By genetically crossing this mouse model with a reporter mouse model, which allows for tracking of the Lgr5-positive crypt stem cells, the generation of intestinal cancer caused by loss of Apc was found to be driven by a crypt stem cell.[51]
In addition to tumorigenesis, Wnt signaling has been associated with CSC-mediated metastasis and maintenance of CSC stemness.[52] Flow cytometry analysis indicated that breast CSCs expressed significantly higher levels of the Wnt signaling proteins LEF1, cyclin D1, β-catenin, and TCF-4, compared with nonstem cancer cell controls.[52] Wnt ligand treatment resulted in a significant increase in Wnt-responsive gene transcription in breast CSCs compared with nonstem cancer cell controls.[52] Similarly, knockdown of Wnt1 in the CSCs decreased expression of the stemness genes CD44, ALDH1, and Sca-1, tumorsphere formation, and a reduction in the CSC subpopulation of breast cancer cells.[52] These data suggest that Wnt signaling is required for maintenance of stemness in breast cancer.[52] Finally, expression analysis of Wnt genes in metastatic and nonmetastic breast CSCs indicated that TCF-4 expression levels are significantly higher in the former.[52] These data are consistent with others that demonstrate a high percentage of CSCs exist among breast cancer cells that overexpress Dvl and display a propensity for lymphatic metastasis.[53] These data suggest that amplified Wnt signaling in CSCs may be important for breast cancer metastasis.[52]
Although less studied than the canonical pathway, noncanonical Wnt signaling may also play a role in tumor initiation through the actions of the noncanonical Wnt ligand Wnt5a. Within a mouse model of ErbB2-driven mammary tumorigenesis, Wnt5a can function as a tumor suppressor. Its production by luminal cells functions in a paracrine manner to limit the expansion of basally located tumor-initiating cells.[54] In a glioma study, depletion of Disheveled2 (Dvl2) inhibited cell proliferation, promoted differentiation, and inhibited 3-dimensional neurosphere formation, as well as tumorigenesis. These regulatory actions of Dvl2 may depend on canonical and noncanonical Wnt signaling pathways.[44]
2.4. Notch pathway
Notch ligands (DLL1, DLL3, DLL4, JAG1, and JAG2) and receptors (Notch1-4) are transmembrane proteins. The pathway is activated when a ligand expressed on 1 cell binds to a receptor on an adjacent cell (Fig. 4).[55] This interaction initiates proteolytic cleavage of the cytoplasmic domain of the receptor, first by a disintegrin and metalloproteinases (ADAMs), then by γ-secretase. This dual cleavage releases the Notch intracellular domain (NICD) into the cytoplasm where it translocates into the nucleus and activates transcription of target genes via the CBF1, Suppressor of Hairless, LAG-1/recombination signal binding protein for immunoglobulin κ J region (CSL/RBPJ) transcription factor.[55,56]
Figure 4.
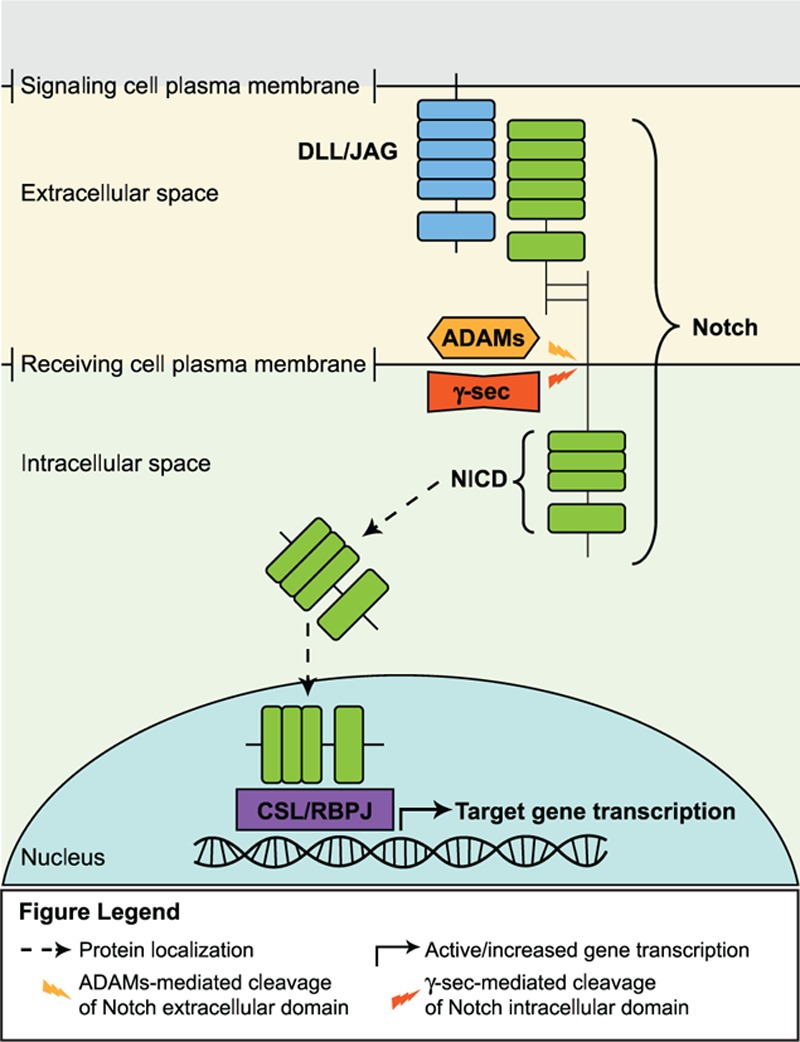
Notch signaling pathway.[55]
As with many of the aforementioned signaling pathways, the Notch pathway is highly conserved and critical in the regulation of cell fate specification and differentiation of stem and progenitor cells in the development of the embryonic central nervous and hematopoietic systems, vasculature, heart, eyes, pancreas, intestine, and other organs.[55–57] In adults, Notch signaling has been shown to influence stem and progenitor cell functions in the skin, hematopoietic system, intestine, and skeletal muscle system.[57]
The Notch pathway has been demonstrated to regulate the properties of tumor cells in many cancers, including leukemia, glioblastoma, and those of the breast, colon, pancreas, and lung, among others.[58] Evidence for Notch signaling in regulation of tumor-initiating cells in cancer is, however, just recently beginning to be understood.[58] The uncertainty of the potential role for Notch signaling in CSCs is, in part, because activated Notch signaling can act as a tumor promoter or suppressor in different tissues, depending on the context.[58] For example, like APC mutations in CRC, activating mutations in NOTCH1 are causal for the development of T cell acute lymphoblastic leukemia.[58,59] Furthermore, Notch signaling has been shown to regulate cell fate in the intestinal crypts. Inhibition of Notch signaling resulted in complete conversion of transit amplifying cells into differentiated goblet cells. In adenomas caused by Apc mutations, inhibition of Notch forced the adenoma cells to differentiate into goblet cells, suggesting that both Notch and Wnt activation may be necessary to maintain the undifferentiated CSC state.[60] In another study investigating the role of Notch in maintenance of CSC stemness, patient-derived pancreatic CSCs were shown to express higher levels of the Notch signaling genes Notch1, Notch3, Jag1, Jag2, and the Notch target gene Hes1.[61] Treatment of the CSCs with a gamma secretase (γ-sec) inhibitor decreased the CSC subpopulation and tumorsphere formation frequency.[61] Similarly, knockdown of Hes1 or treatment of the CSCs with delta/Serrate/Lag-2 peptide, a Notch receptor agonist, respectively decreased and increased CSC tumorsphere formation, suggesting that Notch signaling activity is required for CSC stemness.[61] Cell death assays indicated that γ-secretase inhibitor treatment impaired cell cycle progression and increased apoptosis, indicating that Notch signaling may promote cell survival to support pancreatic CSC stemness.[61]
In another example, the Notch pathway may be activated in nearly three-quarters of primary esophageal adenocarcinoma samples as compared with normal esophageal mucosa; interestingly, tumors that were partly or fully refractory to treatment exhibited higher Notch signaling activity.[62] In xenograft models of tumorigenesis using esophageal adenocarcinoma cells, inhibition of the Notch pathway via treatment with a γ-secretase inhibitor greatly reduced primary tumor growth. The subsequent failure of secondary tumor growth upon retransplantation suggested that loss of Notch resulted in depletion of the cancer-initiating cell population.[62] Finally, analysis of CSC survival, gene expression, and growth properties confirmed that Notch signaling was enriched in this subpopulation of cells and its inhibition resulted in a reduction of this population by half.[62]
Conversely, Notch may serve as a tumor suppressor in murine skin.[63] Deletion of Notch1 in the epidermis resulted in the development of spontaneous BCC-like skin tumors. Moreover, loss of Notch1 in the skin increased the susceptibility of these mice to develop tumors induced by treatment with carcinogenic chemicals. In this scenario, 4 different types of tumors developed (papillomas, benign dysplastic lesions, BCC-like, and squamous cell carcinoma), suggesting the potential contribution of additional mutational hits to the development of the various tumor types. When further investigating possible mechanisms for the ensuing tumorigenesis, it was discovered that Gli2 and free β-catenin were aberrantly upregulated in the epidermis of Notch1-null mice, indicating that activated Hedgehog and Wnt pathways were important mediators of tumorigenesis in the absence of Notch signaling.[63]
2.5. PI3K and PTEN pathway
In response to ligand binding to receptor tyrosine kinases, intracellular PI3K phosphorylates the membrane lipid phosphatidylinositol (3,4)-bis-phosphate (PIP2) to become phosphatidylinositol (3,4,5)-tris-phosphate (PIP3); in doing so, PIP3 becomes a docking site for protein kinase B (PKB) (Fig. 5).[64] Once bound to PIP3, PKB becomes phosphorylated and activated by various kinases including mammalian target of rapamycin (mTOR) and DNA-dependent protein kinase, which promotes PKB-mediated phosphorylation and activation or repression of downstream mediators; a negative regulator of the process is the phosphatase PTEN, which dephosphorylates PIP3 to PIP2.[64]
Figure 5.
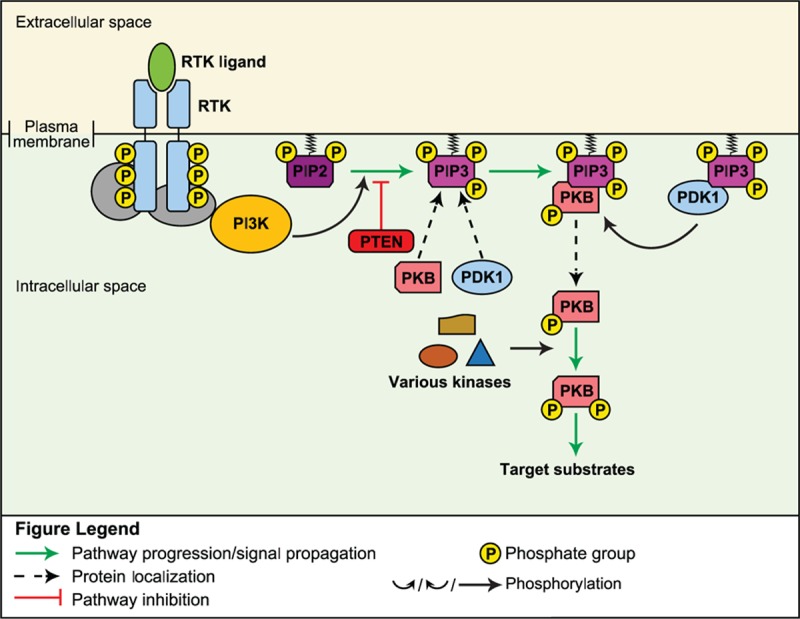
PI3K/PTEN signaling pathway.[64]
PI3K signaling is highly conserved and is involved in regulation of numerous cellular processes including cell cycle progression, growth, and survival, in response to activation by diverse ligands including growth factors, integrins, and cytokines, among others.[64] PI3K signaling has been shown to be an important mediator of self-renewal in mouse embryonic stem cells, as well as of the expansion and lineage specification of hematopoietic stem cells.[65–68] Furthermore, PI3K signaling may play a role in the regulation of intestinal and neural stem cells.[69]
Inactivating mutations in PTEN are found commonly in glioblastoma multiforme.[70] A recent analysis investigated the initiating role of PTEN in the transformation of neural stem cells into glioblastoma multiforme; while retaining the ability to express key stemness regulators OCT4, SOX2, and NANOG and to differentiate into multiple lineages, deletion of PTEN in neural stem cells leads to a neoplastic phenotype, including enhanced growth abilities, resistance to stress-induced cell death, and increased migratory and invasive properties in vivo. In contrast, loss of the PTEN gene in mesenchymal stem cells resulted in cellular senescence, suggesting that this neoplastic phenotype is cell context-specific.[70]
Activation of PKB or inactivation of PTEN has been observed in other solid tumors, leukemias, and myeloproliferative neoplasias.[71] In mouse studies, loss of Pten in hematopoietic stem cells has been shown to play a causal role in the initiation of myeloproliferative disease and leukemia, and in 1 recent study, the development of such neoplasias in the absence of Pten was shown to be dependent upon expression of the class IA PI3K p110β.[67,68,71] Another recent analysis established a role for the microRNA miR-126 in the maintenance of a CSC state through PI3K signaling in AML. At high levels, miR-126 maintained leukemia stem cells in a quiescent, primitive state, restricting entry into the cell cycle, increasing self-renewal, and reducing differentiation by repressing PI3K signaling. In contrast, loss of miR-126 activity resulted in proliferation and differentiation of progenitors through activation of PI3K.[72] Similar results were reported in a study in which miR-10b was shown to inhibit PTEN expression in support of breast CSC self-renewal.[73] Genetic ablation of PTEN in the CSCs resulted in an increase in mammosphere formation and increased the expression of the stemness genes OCT4 and SNAIL1.[73] Overexpression of miR-10b-resistant mutant PTEN in the CSCs decreased the expression of the stemness gene SNAIL1.[73] These results collectively indicate that PTEN signaling plays a suppressive role in the maintenance of breast CSC stemness.[73]
2.6. NF-κB pathway
NF-κB transcription factors consist of 5 different proteins that function as dimers; they are normally inactivated in the cytoplasm through binding to IκB proteins.[74] In the canonical NF-κB signal transduction cascade, activation occurs through binding of ligands, such as tumor necrosis factor alpha (TNF-α), IL-1β, or bacterial cell wall components, to their respective receptors, for example, TNF receptor, IL-1 receptor, or toll-like receptors (TLRs).[75] Adapter proteins are recruited, facilitating the phosphorylation and activation of IκB kinase (IKK) proteins (mainly IKKβ in canonical signaling), which subsequently initiate the phosphorylation of IκB proteins, marking them for ubiquitination and degradation.[74,75] Degradation of IκB releases NF-κB, allowing it to translocate into the nucleus to activate transcription of target genes (Fig. 6 left panel).[74] In addition, a separate noncanonical NF-κB pathway exists that is activated by different receptors, such as receptor activator of NF-κB (RANK) and CD40, signaling through NF-κB–inducing kinase and IKKα; in this alternate pathway, p100/RelB dimers are processed into p52/RelB dimers that translocate into the nucleus and activate transcription (Fig. 6 right panel).[74,75]
Figure 6.
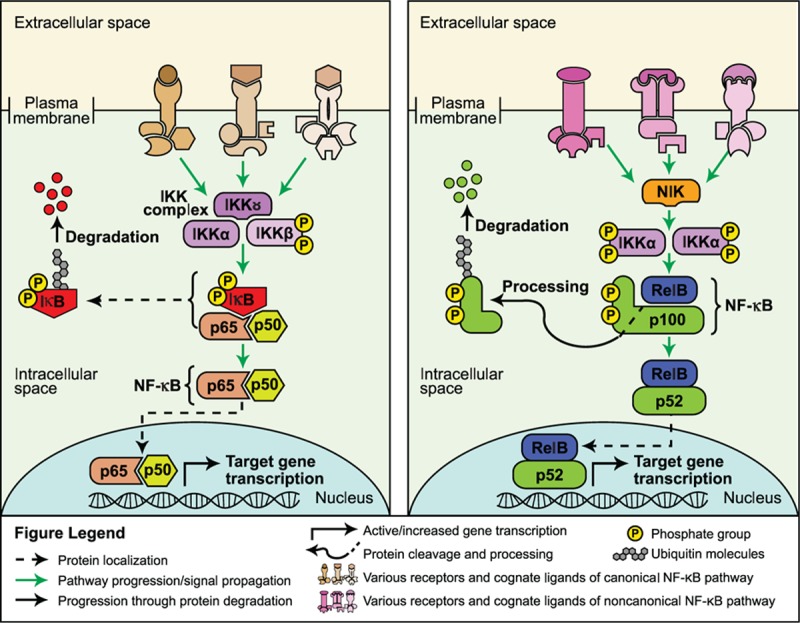
Canonical (left panel) and noncanonical (right panel) NF-κB signaling pathways.[74]
The NF-κB pathway is a highly complex and critical signaling pathway extensively studied for its role in regulating inflammatory and immune responses; within and beyond the immune system, the NF-κB pathway is involved in cellular proliferation, survival, and differentiation.[74] In contrast with the other pathways discussed here, a role for NF-κB in the regulation of embryonic or adult normal stem cell functions has not been fully established.[76,77] Mouse studies have found that loss of p65 or p52/RelB results in a significant reduction in the number of hematopoietic stem cells, the ability of these cells to self-renew, and an abnormal differentiation profile of committed hematopoietic progeny.[77,78] In adult murine neurogenesis, loss or inhibition of TLR2 and 4 in neural stem cells affected their self-renewal and cell fate determination characteristics; this effect was modulated through NF-κB activation.[79]
Mutations causing overactivation of the NF-κB pathway have been reported in a number of blood cancers including B-cell chronic lymphocytic leukemia, B- and T-cell lymphomas, mucosa-associated lymphoid tissue lymphomas, diffused large B cell lymphoma, and multiple myeloma.[80] In addition to hematologic malignancies, constitutive activation of the NF-κB pathway has been documented in gastrointestinal, genitourinary, gynecological, thoracic, head and neck, and breast tumors and cell lines, among others.[81]
Much scientific evidence supporting a role for NF-κB in cancer has identified an essential connection between its regulation of inflammation and the development or maintenance of the cancerous state.[80] Outside of this inflammatory contribution, whether NF-κB signaling constitutes an important pathway controlling the self-renewal and multipotency properties of CSCs across tissue types, however, is unclear. Evidence for NF-κB activity in regulating a breast CSC population comes from transgenic mouse studies. In one analysis, constitutive expression of RANK resulted in expansion of mammary stem and progenitor cell populations and disruption of the luminal and basal cell compartments. The disruption in the 2 cellular compartments was associated with a failure to establish the luminal subpopulation responsible for alveolar development, resulting in defective lactation upon parity. Furthermore, the mammary glands were hyperplastic and spontaneously formed tumors that were heterogeneous in cellular composition, each with distinct morphologies, suggesting that each tumor arose from different progenitor cell types.[82]
Another transgenic mouse model used to investigate NF-κB signaling in CSCs is the MMTV-ErbB2/neu model of mammary tumorigenesis. Constitutive activation of ErbB2 in the mammary gland and NF-κB inhibition via use of a mutated inactive IKKα protein resulted in a decreased incidence and delayed rate of tumor formation; the self-renewal ability of tumor-initiating cells was compromised in this model, as demonstrated through secondary transplantation studies.[83] In addition, NF-κB signaling is essential for maintaining a basal progenitor cell population in these tumors, rather than a luminal one, and that this cell type may be the predominant tumor-initiating cell in ErbB2-driven tumorigenesis.[84] In another preclinical study using a mouse mammary tumor cell line and a mouse mammary tumor model harboring active ErbB2, inhibition of NF-κB signaling reduced cell proliferation and colony formation in soft agar and greatly reduced the rate of tumorigenesis and resulting tumor number.[85] The authors proposed that NF-κB is regulating the mammary tumor-initiating cell population since inhibition of NF-κB reduced the number of progenitor cells and the ability of these cells to form 3-dimensional mammospheres in culture, and decreased the promoter activity levels of the embryonic stem cell regulators SOX2 and NANOG genes by approximately 90% and 50%, respectively.[85]
NF-κB signaling has also been implicated in enabling CSCs to mediate metastasis. A recent study reported that breast CSCs may promote metastasis to lungs via LIN28, a stemness factor and downstream effector of IKKβ.[86] Gene expression analysis of patient-derived Stage I, II, and III breast cancer cells indicated that expression levels of LIN28 increased with disease progression. In vitro analysis of breast cancer cell lines indicated that breast CSCs expressed higher protein levels of LIN28 and the activated form of IKKβ than nonstem cancer cell counterparts. Genetic silencing or chemical inhibition of IKKβ reduced the expression of the stemness proteins LIN28, OCT4, SOX2, and NANOG. Finally, treatment of breast cancer xenograft-bearing mice with an IKKβ inhibitor almost completely eliminated the incidence of lung metastasis, and significantly reduced the frequency of CSCs in the metastatic foci, compared with vehicle control.[86] However, treatment with a conventional taxane resulted in a significantly higher incidence of lung metastasis while increasing the frequency of CSCs in the metastatic foci, compared with vehicle control.[86] The ability of IKKβ inhibition to limit the incidence of metastatic foci in mice was preserved when combined with taxane treatment in the mouse model of metastasis.[86] These data collectively demonstrate that NF-κB signaling may support CSC stemness and activate LIN28 to promote tumor metastasis in breast cancer.[86]
2.7. Interaction between signaling pathways
As mentioned previously, these complex signal transduction pathways are not linear and in some cases, cross-talk between and among various pathways occurs in the regulation of the tumor-initiating cell phenotype. Some examples of convergence between pathways were discussed earlier. For example, in one study, cooperation between the Notch and Wnt signaling pathways was necessary to maintain an undifferentiated intestinal CSC state.[60] In another study, the spontaneous development of skin tumors after deletion of Notch1 in murine epidermis was suggested to occur through the activation of the Hedgehog and Wnt pathways.[63]
In some cases, cross-talk between pathways may promote resistance to cancer therapeutics through maintenance of the CSC population; PI3K and Notch signaling pathways may contribute to CSC expansion and the resistance to PI3K inhibitors that often results in treatment failure for patients with triple-negative breast cancer.[87] Although treating triple-negative breast cancer cells with a PI3K/mTOR inhibitor resulted in growth inhibition for some cells, there remained a surviving subpopulation of resistant cells with increased stem cell properties that exhibited increased expression and activation of Notch1.[87] Breast cancer cells that were resistant to treatment with the PI3K/mTOR inhibitor retained the ability to generate 3-dimensional mammospheres in cell culture and to form tumors in xenograft mouse models; however, concomitant reduction in Notch1 levels resulted in failure of these cells to form mammospheres, a reduction in primary tumor growth in xenografts, and a significant decrease in the ability of CSCs from primary xenograft tumors to generate new tumors upon retransplantation.[87] In another study analyzing mouse models of medulloblastoma caused by overactivation of Hedgehog signaling, activation of the PI3K pathway was shown to be essential in the survival of CSCs in the perivascular niche after irradiation; importantly, these cells were responsible for tumor recurrence.[88]
Other studies have found a potential for cooperation between the Hedgehog and PI3K pathways in the regulation of CSCs in biliary tract and pancreatic cancers.[89,90] Treatment of biliary tract CSCs with Hedgehog and mTOR/PI3K inhibitors limited the number and size of tumor spheres, decreased expression of the stemness genes NANOG and OCT4, and reduced tumorigenicity in xenograft mouse models. These inhibitory effects were greater than those observed with either individual drug treatment.[90] Similar results were found in pancreatic CSCs. Concomitant treatment of pancreatic CSCs with inhibitors of Smoothened (the Hedgehog pathway) and a dual inhibitor of PI3K and mTOR reduced the expression of OCT4, SOX2, and NANOG, decreased the ability of these cells to self-renew upon serial 3-dimensional tissue culturing, and reduced tumor growth in xenografts to a much greater extent than either agent used alone.[89]
2.8. CSC microenvironment associations
A seminal role for the microenvironment or niche in the maintenance of the stem cell state, whether under normal or cancerous circumstances, can be inferred when considering that the key signaling pathways believed to be involved in this process are activated through intercellular interactions. In other words, ligands secreted by one cell activate cognate receptors on another cell.[8,18,23,55,64,74] Not only are multiple cell types involved, but interactions with extracellular matrix and vasculature contribute to microenvironment dynamics.[91,92]
In normal intestinal homeostasis, it has been suggested that the stem cells in the crypt are constrained to the positions that they are in through their close proximity with Wnt-producing Paneth cells.[93] Wnt signaling has been shown to be activated in colon CSCs via the actions of myofibroblasts in the microenvironment.[94] Using a reporter mouse model of Wnt signaling activity, CSCs harboring the greatest level of Wnt signaling activation were found to have the greatest tumorigenic potential in xenograft mouse models[94]; of interest, these cells were preferentially localized close to myofibroblasts. The authors established that signals from the myofibroblasts further potentiated Wnt signaling activity in these CSCs. The impact of myofibroblasts on the maintenance of a tumorigenic microenvironment was highlighted by the fact that CSCs with low Wnt signaling activity were extremely limited in their ability to form tumors upon injection unless myofibroblasts were coinjected with them.[94] Furthermore, myofibroblasts were likely stimulating stabilization of β-catenin in colon CSCs through secretion of hepatocyte growth factor, which was hypothesized to bind to the c-Met receptor leading to downstream signaling through PKB and GSK-3β, suggesting cross-talk between the 2 pathways.[94]
Although the NF-κB pathway has been known to influence tumor growth through the microenvironment via its regulation of inflammation, its influence on tumor initiation may be through both cell autonomous and noncell autonomous mechanisms.[80,95] In a mouse model of colon cancer, inhibition of the NF-κB pathway through deletion of Ikkβ in enterocytes or myeloid cells resulted in a 75% and 50% decrease in tumor incidence, respectively, and a decrease in tumor size in the myeloid-specific deletion.[95] Significantly, the authors found that this decreased tumor burden was the result of different mechanisms of action in each cell type; loss of NF-κB signaling in enterocytes led to apoptosis with no change in inflammatory response, whereas loss of NF-κB signaling in myeloid cells greatly decreased inflammation.[95]
In breast cancer, numerous preclinical studies have led to a generalized model of breast CSC regulation through the microenvironment in which ILs, chemokines, growth factors, Wnts, and other proteins secreted by fibroblasts, macrophages, endothelial cells, immune cells, and mesenchymal stem cells activate PI3K/PKB, STAT3, Wnt, and the NF-κB pathways in breast CSCs, leading to self-renewal and the reinforcement of positive feedback loops between these cells and the cells within the microenvironment.[91]
Examples of cellular interactions within the breast tumor niche modulating CSC functions and outcomes include studies of NF-κB. In a mouse model of ErbB2-driven mammary tumorigenesis, inhibition of NF-κB activity reduced the pool of CSCs as well as the development of tumors. In tumors that did develop, there was a significant reduction in vasculature accompanied by a decrease in the levels of vascular endothelial growth factor and the endothelial receptor platelet and endothelial cell adhesion molecule 1/CD31. Furthermore, mammary tumors harbored approximately half the quantity of tumor-associated macrophages as control tumors, supporting a role for NF-κB in influencing the microenvironment within a tumor.[85] In another study of human basal-like breast cancer cell lines, NF-κB activity levels correlated with the CSC population; reduction of activity decreased numbers of CSCs and reduced tumorigenicity, whereas forced activation of NF-κB increased the number of CSCs and enhanced tumorigenicity.[96] However, upon closer examination, it was discovered that this effect was driven by a noncell autonomous interaction between nonstem cancer cells and CSCs; NF-κB activation in nonstem cancer cells drove expression of the Notch ligand JAG1, which lead to expansion of the CSC population in trans.[96]
In neural cancers, studies have demonstrated that CSCs congregate in hypoxic areas of the tumor and in the perivascular niche.[92] In one murine study, nitric oxide produced by endothelial cells within gliomas stimulated adjacent glioma stem-like cells that expressed Notch1; Notch signaling was important for tumor initiation and growth and correlated with enhancement of a stem cell population.[97] Another study analyzing human gliomas and utilizing glioma tumor sphere tissue culture methods identified that Sonic Hedgehog secreted by endothelial cells regulated glioma stem cell functions.[98]
3. Conclusions
Mounting evidence supports CSCs as the possible cells of origin in the development of many cancers. Although no one CSC model will be sufficient to explain how all cancers develop, or even to explain how different cancers develop within the same tissue, common to many of these hypothetical models is the dysregulation of key signaling pathways. Further understanding how CSCs and their signaling pathways function may lead to the development of new therapeutic approaches.
Acknowledgment
This supplement was supported by Boston Biomedical Pharma, Inc., Cambridge, MA. Editorial support was provided by Jacqueline Egan, Allison England, PhD, and Brij Patel, PhD, of Guidemark Health, Stamford, CT.
Footnotes
Abbreviations: β-cat = β-catenin, γ-sec = gamma secretase, ADAM = a disintegrin and metalloproteinases, ALDH = aldehyde dehydrogenase, AML = acute myeloid leukemia, APC = adenomatous polyposis coli protein, APC = human adenomatous polyposis coli gene, Apc = mouse adenomatous polyposis coli gene, BCC = basal cell carcinoma, CK1α = casein kinase 1α, CML = chronic myeloid leukemia, CRC = colorectal cancer, CSC = cancer stem cell, CSL/RBPJ = CBF1, Suppressor of Hairless, LAG-1/recombination signal binding protein for immunoglobulin κ J region, DLL = Delta-like proteins, Dvl = Disheveled, Dvl2 = Disheveled2, GSK-3β = glycogen synthase kinase-3β, Hh = Hedgehog, IκB = IκB proteins, IKK = IκB kinase, IL11 = interleukin 11 gene, IL-11 = interleukin-11 protein, IL6 = interleukin 6, JAG = Jagged proteins, JAK/STAT = Janus-activated kinase/signal transducer and activator of transcription, LEF = lymphoid enhancer factor, Lgr = leucine-rich repeat-containing G protein-coupled receptor, LIF = leukemia inhibitory factor, LRP = low-density lipoprotein-related protein, MMTV = murine mammary tumor virus, mTOR = mammalian target of rapamycin, NF-κB = nuclear factor-κB, NICD = Notch intracellular domain, NIK = NF-κB-inducing kinase, PI3K = phosphatidylinositol 3-kinase, PIAS = Protein Inhibitor of Activated STAT, PIP2 = phosphatidylinositol (3,4)-bis-phosphate, PIP3 = phosphatidylinositol (3,4,5)-tris-phosphate, PKB = protein kinase B, Ptch = patched protein, PTCH1 = human patched 1 gene, Ptch1 = mouse patched 1 gene, PTEN = human phosphatase and tensin homolog gene, Pten = mouse phosphatase and tensin homolog gene, PTEN = phosphatase and tensin homolog protein, PTP = protein tyrosine phosphatase, R-spo = R-spondin, RANK = receptor activator of NF-κB, Smo = Smoothened protein, SOCS = Suppressors of Cytokine Signaling, TCF = T-cell factor, TGF-β = tumor growth factor-beta, TLR = toll-like receptor, TNF-α = tumor necrosis factor alpha.
The author reports no conflicts of interest.
References
- 1.Cahan P, Daley GQ. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat Rev Mol Cell Biol 2013; 14:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rieger MA, Schroeder T. Hematopoiesis. Cold Spring Harb Perspect Biol 2012; 4:a008250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449:1003–1007. [DOI] [PubMed] [Google Scholar]
- 4.Inman JL, Robertson C, Mott JD, et al. Mammary gland development: cell fate specification, stem cells and the microenvironment. Development 2015; 142:1028–1042. [DOI] [PubMed] [Google Scholar]
- 5.Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005; 437:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mascre G, Dekoninck S, Drogat B, et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012; 489:257–262. [DOI] [PubMed] [Google Scholar]
- 7.Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014; 346:1248012. [DOI] [PubMed] [Google Scholar]
- 8.Holland JD, Klaus A, Garratt AN, et al. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol 2013; 25:254–264. [DOI] [PubMed] [Google Scholar]
- 9.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet 2008; 40:915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66:9339–9344. [DOI] [PubMed] [Google Scholar]
- 11.Ajani JA, Song S, Hochster HS, et al. Cancer stem cells: the promise and the potential. Semin Oncol 2015; 42 (suppl 1):S3–S17. [DOI] [PubMed] [Google Scholar]
- 12.Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin 2013; 34:732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao B, Ahmad A, Azmi AS, et al. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol 2013; Chapter 14:Unit 14 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zagozdzon R, Golab J. Cancer stem cells in haematological malignancies. Contemp Oncol (Pozn) 2015; 19 (1A):A1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015; 16:225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroon P, Berry PA, Stower MJ, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res 2013; 73:5288–5298. [DOI] [PubMed] [Google Scholar]
- 17.Lin L, Liu A, Peng Z, et al. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res 2011; 71:7226–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merchant AA, Matsui W. Targeting Hedgehog—a cancer stem cell pathway. Clin Cancer Res 2010; 16:3130–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012; 22:457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boman BM, Wicha MS. Cancer stem cells: a step toward the cure. J Clin Oncol 2008; 26:2795–2799. [DOI] [PubMed] [Google Scholar]
- 21.Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009; 69:3382–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene 2000; 19:5662–5679. [DOI] [PubMed] [Google Scholar]
- 23.Stine RR, Matunis EL. JAK-STAT signaling in stem cells. Adv Exp Med Biol 2013; 786:247–267. [DOI] [PubMed] [Google Scholar]
- 24.Chambers I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells 2004; 6:386–391. [DOI] [PubMed] [Google Scholar]
- 25.Birnie R, Bryce SD, Roome C, et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol 2008; 9:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Wulfkuhle J, Zhang H, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A 2007; 104:16158–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penuelas S, Anido J, Prieto-Sanchez RM, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009; 15:315–327. [DOI] [PubMed] [Google Scholar]
- 28.Sherry MM, Reeves A, Wu JK, et al. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009; 27:2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calon A, Espinet E, Palomo-Ponce S, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012; 22:571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Freitas RM, da Costa Maranduba CM. Myeloproliferative neoplasms and the JAK/STAT signaling pathway: an overview. Rev Bras Hematol Hemoter 2015; 37:348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cook AM, Li L, Ho Y, et al. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood 2014; 123:2826–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinzler KW, Bigner SH, Bigner DD, et al. Identification of an amplified, highly expressed gene in a human glioma. Science 1987; 236:70–73. [DOI] [PubMed] [Google Scholar]
- 33.Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014; 141:3445–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutchin ME, Kariapper MS, Grachtchouk M, et al. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev 2005; 19:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JY, Nelson AL, Algon SA, et al. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev Biol 2003; 263:50–66. [DOI] [PubMed] [Google Scholar]
- 36.Peterson SC, Eberl M, Vagnozzi AN, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015; 16:400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement V, Sanchez P, de Tribolet N, et al. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 2007; 17:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peacock CD, Wang Q, Gesell GS, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A 2007; 104:4048–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009; 458:776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varnat F, Duquet A, Malerba M, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med 2009; 1:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013; 27:2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov 2014; 13:513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Lau W, Barker N, Low TY, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011; 476:293–297. [DOI] [PubMed] [Google Scholar]
- 44.Pulvirenti T, Van Der Heijden M, Droms LA, et al. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res 2011; 71:7280–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol 2012; 4:a0008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malanchi I, Peinado H, Kassen D, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008; 452:650–653. [DOI] [PubMed] [Google Scholar]
- 47.Incassati A, Chandramouli A, Eelkema R, et al. Key signaling nodes in mammary gland development and cancer: beta-catenin. Breast Cancer Res 2010; 12:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature 2006; 439:84–88. [DOI] [PubMed] [Google Scholar]
- 49.Stingl J, Eirew P, Ricketson I, et al. Purification and unique properties of mammary epithelial stem cells. Nature 2006; 439:993–997. [DOI] [PubMed] [Google Scholar]
- 50.Lindvall C, Evans NC, Zylstra CR, et al. The Wnt signaling receptor Lrp5 is required for mammary ductal stem cell activity and Wnt1-induced tumorigenesis. J Biol Chem 2006; 281:35081–35087. [DOI] [PubMed] [Google Scholar]
- 51.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009; 457:608–611. [DOI] [PubMed] [Google Scholar]
- 52.Jang GB, Kim JY, Cho SD, et al. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep 2015; 5:12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pandit TS, Kennette W, Mackenzie L, et al. Lymphatic metastasis of breast cancer cells is associated with differential gene expression profiles that predict cancer stem cell-like properties and the ability to survive, establish and grow in a foreign environment. Int J Oncol 2009; 35:297–308. [PubMed] [Google Scholar]
- 54.Borcherding N, Kusner D, Kolb R, et al. Paracrine WNT5A signaling inhibits expansion of tumor-initiating cells. Cancer Res 2015; 75:1972–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta 2013; 1830:2481–2495. [DOI] [PubMed] [Google Scholar]
- 56.Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015; 12:445–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiba S. Notch signaling in stem cell systems. Stem Cells 2006; 24:2437–2447. [DOI] [PubMed] [Google Scholar]
- 58.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 2011; 11:338–351. [DOI] [PubMed] [Google Scholar]
- 59.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306:269–271. [DOI] [PubMed] [Google Scholar]
- 60.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435:959–963. [DOI] [PubMed] [Google Scholar]
- 61.Abel EV, Kim EJ, Wu J, et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One 2014; 9:e91983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Da Silva TG, Jin K, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res 2014; 74:6364–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicolas M, Wolfer A, Raj K, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 2003; 33:416–421. [DOI] [PubMed] [Google Scholar]
- 64.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 2012; 4:a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paling NR, Wheadon H, Bone HK, et al. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem 2004; 279:48063–48070. [DOI] [PubMed] [Google Scholar]
- 66.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet 2008; 9:115–128. [DOI] [PubMed] [Google Scholar]
- 67.Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006; 441:475–482. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, Grindley JC, Yin T, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006; 441:518–522. [DOI] [PubMed] [Google Scholar]
- 69.Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol 2005; 21:605–631. [DOI] [PubMed] [Google Scholar]
- 70.Duan S, Yuan G, Liu X, et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat Commun 2015; 6:10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuzugullu H, Baitsch L, Von T, et al. A PI3K p110beta-Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat Commun 2015; 6:8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lechman ER, Gentner B, Ng SW, et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell 2016; 29:214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bahena-Ocampo I, Espinosa M, Ceballos-Cancino G, et al. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep 2016; 17:648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell 2008; 132:344–362. [DOI] [PubMed] [Google Scholar]
- 75.Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 2013; 12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev 2007; 3:7–17. [DOI] [PubMed] [Google Scholar]
- 77.Stein SJ, Baldwin AS. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood 2013; 121:5015–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao C, Xiu Y, Ashton J, et al. Noncanonical NF-kappaB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells 2012; 30:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rolls A, Shechter R, London A, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol 2007; 9:1081–1088. [DOI] [PubMed] [Google Scholar]
- 80.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 2009; 1:a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem 2010; 336:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pellegrini P, Cordero A, Gallego MI, et al. Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem Cells 2013; 31:1954–1965. [DOI] [PubMed] [Google Scholar]
- 83.Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A 2007; 104:15852–15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang W, Tan W, Wu X, et al. A NIK-IKKalpha module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013; 23:647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu M, Sakamaki T, Casimiro MC, et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res 2010; 70:10464–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen C, Cao F, Bai L, et al. IKKbeta enforces a LIN28B/TCF7L2 positive feedback loop that promotes cancer cell stemness and metastasis. Cancer Res 2015; 75:1725–1735. [DOI] [PubMed] [Google Scholar]
- 87.Bhola NE, Jansen VM, Koch JP, et al. Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and notch-dependent cancer stem cell population. Cancer Res 2016; 76:440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hambardzumyan D, Becher OJ, Rosenblum MK, et al. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008; 22:436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sharma N, Nanta R, Sharma J, et al. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015; 6:32039–32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zuo M, Rashid A, Churi C, et al. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. Br J Cancer 2015; 112:1042–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 2011; 121:3804–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lathia JD, Heddleston JM, Venere M, et al. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell 2011; 8:482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011; 469:415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vermeulen L, De Sousa EMF, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12:468–476. [DOI] [PubMed] [Google Scholar]
- 95.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118:285–296. [DOI] [PubMed] [Google Scholar]
- 96.Yamamoto M, Taguchi Y, Ito-Kureha T, et al. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun 2013; 4:2299. [DOI] [PubMed] [Google Scholar]
- 97.Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010; 6:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yan GN, Yang L, Lv YF, et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol 2014; 234:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]