

Abstract
Cancer stem cells (CSCs) are a class of pluripotent cells that have been observed in most types of solid and hematologic cancers. CSCs have been shown in numerous cancer models to be involved in tumor development, cell proliferation, and metastatic dissemination, while possessing a capacity for sustained self-renewal. CSCs, which typically represent a small proportion of total cells of a given tumor, also exhibit resistance to chemotherapy and radiotherapy. Indeed, exposure to these treatments may promote “stemness” in nonstem cancer cells, which may explain why successful therapeutic reduction of tumor bulk will often fail to produce clinical improvement. Acquisition of stemness involves epithelial-mesenchymal transition (EMT), in which epithelial cells are transformed into a mesenchymal phenotype characterized by increased capacities for migration, invasiveness, and resistance to apoptosis. EMT may also contribute to metastasis by driving dissemination of mesenchymal CSCs to distant locations, whereupon the CSCs revert to an epithelial phenotype to support metastatic tumor growth. Several different approaches to treatment aimed at overcoming the intrinsic resistance of CSCs to conventional therapies are currently being developed. These include agents targeting tumorigenic pathways, such as JAK2/STAT3 and PI3K/mTOR, and immunotherapies, including vaccines and natural killer cells employed to induce a T cell response.
Keywords: cancer stem cell, CSC-targeting, epithelial-mesenchymal transition, metastasis, recurrence, resistance, tumor microenvironment
1. Introduction
Cancer stem cells (CSCs) describe a class of pluripotent cancer cells that behave analogously to normal stem cells in their ability to differentiate into the spectrum of cell types observed in tumors.[1,2] These characteristics of what is known as “stemness” allow for the growth of the primary cancer tumor as well as the development of new tumors.[1,2] CSCs may be identified via phenotypic surface markers (such as CD34+/CD38− in leukemia stem cells), which has allowed for the observation of CSCs in most types of human cancer, including breast, brain, liver, lung, gastric, colon, prostate, pancreatic, and head and neck cancers, as well as multiple myeloma, leukemia, and melanoma.[2–6] In addition to their pluripotency, CSCs have been shown to be involved in fundamental processes of tumor development, cell proliferation, and metastatic dissemination, while possessing a capacity for self-renewal that make CSCs immortal.[2,7]
The qualities of stemness that define CSCs appear to be confined to a relatively small proportion of cancer cells, which are able to produce the diversity and heterogeneity of cancer cells that compose most of the tumor bulk. CSCs of several types of cancer may be markedly more tumorigenic than nonstem cancer cells.[1,2] CSCs are, however, generally resistant to chemotherapy and radiotherapy, meaning that despite these therapies successfully destroying a large proportion of the tumor bulk, the result may not be significant clinical improvement.[1] Taken together, the available data regarding CSCs indicate that they represent a rich source for understanding the development, and potential treatment, of a spectrum of cancer types. This article reviews the role of CSCs, as they are currently understood, with a particular focus on the role of CSCs in tumor growth, recurrence, metastasis, and treatment resistance.
1.1. Epithelial–mesenchymal transition and the acquisition of stemness
Epithelial-mesenchymal transition (EMT) refers to the process by which epithelial cells undergo several changes, including loss of polarity, which transitions the cells into a mesenchymal phenotype (Fig. 1).[8,9] The mesenchymal phenotype is associated with an increased capacity for migration, resistance to apoptosis, increased production of extracellular matrix (ECM) degrading enzymes, and increased proclivity for invasiveness.[9,10] The acquisition of the properties of stemness by cancer cells—that is, the transition from a nonstem cancer cell to a CSC—occurs via the process of EMT and the concurrent acquisition of an invasive mesenchymal phenotype.[3] Epithelial cells, which normally interact with the basement membrane, once transitioned into mesenchymal cells, migrate from the epithelial layer at the same time that the basement membrane undergoes degradation.[8,9]
Figure 1.
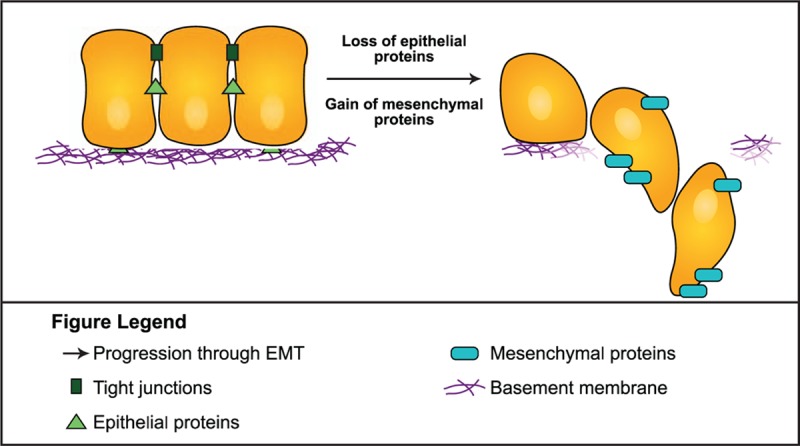
In addition to its role in the development of stemness in cancer, EMT is involved in a variety of physiologic processes, including tissue repair and embryogenesis.[9,11] In embryogenesis, EMT operates at multiple stages, including the conversion of trophectoderm cells to promote invasion of the endometrium to anchor the placenta, and is subsequently involved in the differentiation of the mesendoderm into the mesoderm and the endoderm.[9] In tissue regeneration, a different type of EMT, type 2, responds to inflammation associated with tissue damage, and produces fibroblasts and other repair-related cells to reconstruct tissue.[9] In fact, there are 3 EMT subtypes categorized by their biological context: type 1 refers to EMTs related to embryo formation, implantation, and organ development; type 2 describes EMTs related to tissue regeneration, wound healing, and organ fibrosis; and type 3 are EMTs occurring in tumor cells that have already undergone genetic and/or epigenetic alterations. Although all 3 EMT types involve similar underlying biological processes, type 3 EMTs are those primarily relevant to CSCs and cancer progression, and produce outcomes significantly different from those observed in type 1 and 2 EMTs.[9]
Either exogenous expression of transcription factors that promote EMT induction or exposure to particular cytokines can spontaneously initiate the process in which cancer cells acquire CSC and mesenchymal phenotype properties.[12] With regard to breast cancer cells, ectopic expression of Twist, Snail, or FoxC2 transcription factors have been shown to induce EMT in human mammary epithelial cells, with spontaneous transformation into a mesenchymal phenotype, although without robust plasticity.[3] Interestingly, exposure of nonstem breast cancer cells to transforming growth factor beta (TGF-β) has been shown to produce mesenchymal/CSC-like cells with a high degree of plasticity, whereas removal or inhibition of TGF-β causes the cells to lose their mesenchymal/CSC-like characteristics, and to regain an epithelial and nonstem cell phenotype.[3]
EMT is, in fact, 1 side of a 2-way process, because epithelial and mesenchymal cells can move in either direction as a part of EMT or its reverse, mesenchymal-epithelial transition (MET).[9] Other pathways associated with the regulations of CSCs include Notch, Hedgehog, TGF-β, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and Wnt, which are associated with EMT induction, whereas human epidermal growth factor receptor (HER) and pathways involving bone morphogenetic proteins are associated with MET induction.[8] By undergoing EMT, and acquisition of a mesenchymal phenotype, CSCs are produced possessing characteristics that promote tumor progression, recurrence, metastasis, and resistance to therapy.[8]
1.2. Tumor growth
Although normal stem cells follow strict signaling pathways that direct differentiation into mature cells and govern self-renewal, dysregulation of such signaling can result in the formation of tumors.[13] Oncogenic hits, such as alterations occurring in the tumor microenvironment, or intrinsic alterations, such as APC/KRAS mutations, can also promote the initiation, expansion, and progression of malignancies.[1,14] Additionally, stromal fibroblasts secrete growth factors and inflammatory cytokines, including interleukin-8, an activator of the Janus-activated kinase/signal transducer and activator of transcription (JAK/STAT) 3 pathway, which is known to promote tumor initiation and cancer progression.[15]
The CSC model of development and differentiation can, to a large extent, account for the extraordinary heterogeneity, both of function and phenotype, present in many types of solid tumors and hematologic cancers.[7,16] The potential for multiple variations in manifestations of tumor propagation contribute to the observed heterogeneity of tumors (Fig. 2).[16] In its simplest manifestation, a single CSC may expand and differentiate into a tumor. Alternatively, multiple CSC pools may be present, each of which has the potential to develop into independent tumors. Activation of dormant CSCs—potentially dormant for years—may result in cancer recurrence at a later time. A secondary distinct CSC may also arise during tumor progression, possibly due to a mutation or epigenetic modification, and where the new CSC exhibits more aggressive growth, it may become the driver of tumor formation thereafter. Last, intrinsic or microenvironmental influences may cause nonstem cancer cells to revert to CSC-like phenotypic states.[16]
Figure 2.
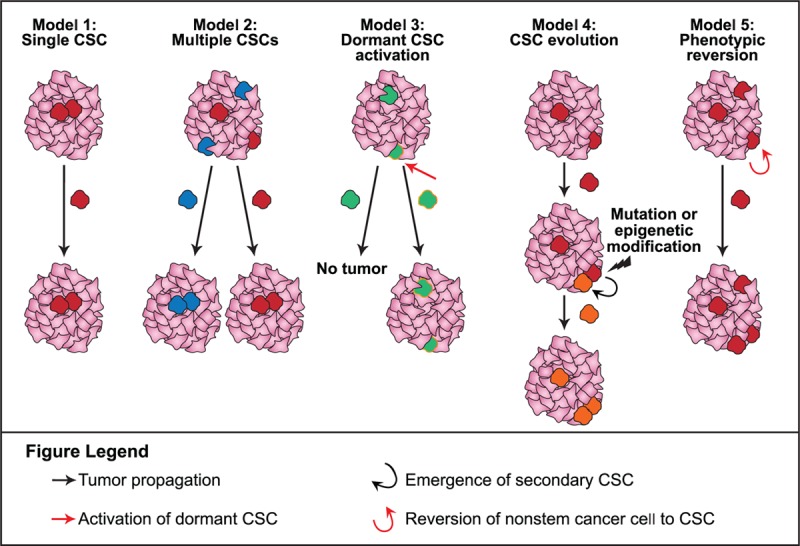
Models of CSC-driven tumor heterogeneity.[16] CSC = cancer stem cell.
The importance of the microenvironment, or niche, in which CSCs exist is underscored in a study in which 2 metastatic breast cancer cell lines (MDA-MB231 and BCM2) were exposed to repeated hypoxia/reoxygenation cycles, in replication of microenvironments frequently observed in solid tumors.[17] After a single round of hypoxia and reoxygenation, a small subset of cells survived the hypoxia, forming spherical clusters and proliferating once reoxygenation had taken place.[17] After 3 rounds of hypoxia and reoxygenation, a novel subpopulation of cells was established, with a high degree of tumorigenicity and surface marker expression (i.e., CD44++/CD24−/ESA+) associated with breast CSCs.[17] Moreover, compared with the parental breast cancer cell lines from which the subpopulations were produced, the subpopulation exhibited a far higher proportion of cells with surface markers indicative of CSCs.[17] Consistent with previous observations regarding CSCs, the cycling hypoxia-selected-cancer-cell subpopulation also exhibited a high propensity for metastasis and an increasing mesenchymal phenotype.[17]
1.3. Metastasis
The precise role of EMT/CSCs in metastatic processes remains an area of investigation. In 2002, a reversible EMT model was proposed to explain metastasis. In this model, primary epithelial tumor cells initiate EMT, which enables dissemination throughout the body. Once settled at a distant site, tumor cells undergo reversal via MET, thereby producing epithelial metastases.[18] More recently, other investigators have described 3 functional categories for the complex molecular signaling pathways and regulators involved in EMT: EMT effectors, which execute the EMT program; EMT core regulators, which are the transcription factors that mediate EMT; and EMT inducers, the extracellular triggers for the activation of EMT.[10]
The metastatic process in cancer is generally described as occurring in 5 steps: invasion, intravasation, transport, extravasation, and colonization.[10] In the first step, invasion of surrounding tissue, EMT has been proposed to have a role at various levels of the process, including the initial degradation of the ECM, loss of cellular junctions, and induction of proteases to facilitate local invasion.[10] In the second step, intravasation, tumor cells enter the vascular system and are disseminated to distant locations in the body. This process is not entirely understood, although several hypotheses have been proposed to explain the potential role of EMT.[10] Evidence for EMT's role in intravasation is diverse and compelling, if only partial at present. In prostate cancer, migration through the endothelial cell barrier has been found to be contingent on the expression of ZEB1 in PC-3 human prostate cells that exhibit EMT.[19] In breast cancer, it was found that MCF-7 breast cancer cells expressing Snail1 can transmigrate through basement membrane via mobilization of the membrane-type matrix metalloproteinases (MT1-MMP and MT2-MMP). These cells were, furthermore, shown to promote angiogenesis and intravasation into the host vasculature.[20]
Transportation via systemic circulation is challenging due to the obstacles preventing tumor cells surviving long enough to relocate to a blood vessel wall and undergo extravasation.[10] The role of EMT in transportation was addressed in a study of premalignant circulating pancreatic cancer cells in a murine model. In many cases, these cells possessed a mesenchymal phenotype. In addition, these cells expressed Zeb1, which is indicative of EMT occurring before tumor formation and were ultimately observed to seed the liver.[21] Another study found a preferential association of circulating tumor cells (CTCs) with platelet cells, which are a source of TGF-β, and which, in subsequent direct platelet-cell contact, activate the TGF-β and NF-κB pathways. This resulted in transition to a mesenchymal phenotype and increased metastasis in vivo.[22]
To explore the role of EMT in extravasation, investigators employed a model using a zebrafish, a visually transparent species, allowing for live imaging of cells. They observed that vascular endothelial growth factor A expression increased extravasation through the blood vessel wall and Twist1 expression increased both extravasation and intravascular migration.[23] The reversal of EMT to MET and the process of colonization at the metastatic site is suggested by the fact that most macrometastases are epithelial, and is supported by experimental data showing that, whereas EMT is necessary to achieve intravasation and extravasation, a loss of signaling to induce EMT at the metastatic site is necessary to achieve proliferation of cancer cells and formation of metastases.[24]
1.4. Limitations of current therapies
Although chemotherapy and radiotherapy have undergone significant refinements and improvements in efficacy and administration in recent years, conventional cancer treatments remain inadequate for many patients, particularly those whose cancer is diagnosed at a late stage.[25] Delays in cancer diagnosis reduce overall treatment efficacy in part as a result of the increased likelihood of the occurrence of metastatic disease, but also partly because more advanced disease may require more intensive treatment, which may, itself, result in treatment intolerance.
Resistance to chemotherapy and radiotherapy has been observed repeatedly in CSCs.[1] If CSCs are resistant to treatment, and treatment to destroy cancer cells succeeds primarily in killing only nonstem cancer cells, it may explain the phenomenon of successful therapeutic tumor shrinkage without a corresponding improvement in patient survival.[26] This phenomenon indicates a need to modify therapeutic approaches so that tumor shrinkage is not itself the determinant of therapeutic success.
1.5. Resistance to chemotherapy and radiotherapy
As noted, although chemotherapy and/or radiotherapy resistance is common, recent observations are showing that CSCs are particularly resistant to these modalities.[27–30] Numerous factors, including the central role of the tumor microenvironment, have been hypothesized to contribute to CSC treatment resistance.[31]
The tumor microenvironment is typically rich in a diversity of proteins, including growth factors (e.g., TGF-β) and cytokines, which likely activate pathways that impact the survival of CSCs.[3] Elevated levels of the cytokine Oncostatin M in the tumor microenvironment of breast cancer were associated with aggressive metastatic disease and chemotherapy resistance. Moreover, chemotherapy was shown to induce additional Oncostatin M secretion, which may exacerbate the aggressive characteristics and treatment resistance of CSCs.[3] Based on these results, the investigators concluded that the plentiful cytokines and growth factors present in the CSC niche promote CSC plasticity and EMT properties that ultimately result in poor treatment outcomes.[3]
Apart from the role of the tumor niche, chemotherapeutic resistance of CSCs has been hypothesized to derive from a mismatch between the relatively slow cell cycle of CSCs in contrast with the relatively rapidly dividing cancer cells that many chemotherapy agents are intended target.[1,13] With regard to resistance to radiotherapy, limited data point to resistance being, in some cases, a consequence of lower levels of reactive oxygen species (ROS) in CSCs. Researchers have observed that ROS occurs at reduced levels in CSCs compared with nonstem cancer cells in some human breast tumors as well as head and neck tumors.[32] They also showed that CSC-enriched tumors sustained less DNA damage compared with the nonstem cancer cells after exposure to radiotherapy.[32] The authors additionally observed increased expression of genes involved in ROS defenses among a CSC population enriched with cells with low ROS levels. Inhibition of ROS scavenger production resulted in increased sensitization to radiotherapy and a reduction in clonogenicity in this population.[32]
1.6. Enrichment and acquisition of stemness due to treatment
The problem of resistance of CSCs to conventional cancer therapies is not simply a matter of an inability of chemotherapy and radiation to destroy the CSCs but rather that the treatment itself has been shown to increase CSC characteristics in nonstem cancer cells, and can even convert nonstem cancer cells to CSCs.[33–36] Studies in breast cancer found that irradiation of breast cancer cells resulted in an increase in the number of CSCs and, in some cases, converted nontumorigenic cancer cells into CSCs.[33,35] The potential for chemotherapy to induce stemness was shown when hepatocellular carcinoma cells were exposed to carboplatin-gained stemness characteristics, including self-renewal and the expression of pluripotency genes (SOX2 and OCT3/4).[34] In addition, carboplatin treatment of nonstem hepatocellular carcinoma cells resulted in increased self-renewal capacity.[34] Similarly, other investigators found that human gastric cancer cell lines, after exposure to the chemotherapy agent 5-fluorouracil (5-FU), exhibited both resistance to 5-FU and features consistent with stemness, including tumorigenicity and capacity for self-renewal.[36]
1.7. Recurrence
The role of CSCs in promoting cancer recurrence may be viewed from several different perspectives. One is derived from the propensity of CSCs to resist chemotherapy and radiotherapy, thereby maintaining a pool of tumorigenic cells from which recurrence can develop.[37] A separate perspective on recurrence relates to the importance of EMT and the conversion of epithelial cells to a mesenchymal phenotype. Good treatment responses among a small population of patients with breast cancer were observed to be coincident with a reduction in the proportion of mesenchymal CTCs, compared with that of epithelial CTCs.[38] Furthermore, poor treatment responses and progressive disease observed in a subpopulation of patients with breast cancer were found to be coincident with an increase in the proportion of mesenchymal CTCs compared with that of epithelial CTCs.[38] Relatedly, hepatocellular CTCs expressing the CSC biomarker epithelial cell adhesion molecule have been shown to express EMT biomarkers, occur in higher numbers among patients with metastatic or recurrent disease, and correlate with increased probability of pre- and postoperative tumor recurrence, suggesting that stem cell-like subpopulations of mesenchymal CTCs may be indicators of micrometastatic status and prognostic of tumor recurrence risk.[12]
EMT must be reversed to MET at the metastatic site to achieve proliferation and differentiation.[3] Similarly, recurrence requires a preservation of proliferative and differentiating qualities to thrive.[3] In recurrence, exposure to chemotherapy or radiotherapy reduces the overall tumor bulk but largely at the expense of nonstem cancer cells, thereby preserving and expanding the relative ratio of CSCs to tumor as a whole.[3] For example, data from breast cancer biopsy studies show that after endocrine therapy or chemotherapy, tumors are enriched in cells with gene signatures indicative of mammosphere-forming cancer cells expressing CSC surface markers compared with pretreatment.[39]
The potential for treatment to enrich refractory cancer cells, and promote stemness in CSCs or nonstem cancer cells, represents still another avenue for disease recurrence and progression. This relation between enrichment and recurrence was well illustrated in a recent in-vitro study in which a stemness-inhibiting molecule (BBI608) blocked cancer relapse in a xenografted human pancreatic cancer model.[40]
1.8. Rationale and potential for treatment
Although the activity of CSCs in various forms of cancer is complex and yet to be fully elucidated, opportunities for therapies targeting CSC, EMT, and CSC pathways, among other areas, exist. As was suggested by the pancreatic cancer model study, inhibiting stemness has the potential to reduce the risk posed by CSCs by limiting or eliminating their capacity for tumorigenesis, proliferation, metastasis, and recurrence. Treatments targeting pathways of tumorigenesis, such as JAK2/STAT3 and PI3K/mTOR, offer a potentially viable therapeutic approach in several cancer types.[41,42] For example, treatment of patient-derived ovarian cancer cells from recurrent tumors with paclitaxel activated the JAK2/STAT3 pathway and increased in CSC marker protein and gene expression among surviving cells.[41] Additionally, combination treatment of patient-derived ovarian cancer cells with paclitaxel and the JAK2-specific small molecule inhibitor CYT387 attenuated paclitaxel-induced JAK2/STAT3 activation and CSC and embryonic stem cell marker protein expression and expression of CSC genes of the residual cells.[41] When cells surviving treatment with paclitaxel plus CYT387 were transplanted into mice, the tumor burden was significantly lower than that seen in mice transplanted with cells surviving treatment with only paclitaxel or CYT387.[41] Such preclinical studies have informed the development of therapies targeting CSCs. Table 1[43–48] lists several CSC pathways under clinical study in various solid and hematological cancers.
Table 1.
Snapshot of CSC signaling pathways under phase I/II through phase III clinical study∗.

Immunotherapies offer an additional potential approach to CSC treatment. Natural killer cells, for example, have been shown to offer some degree of efficacy in glioma CSCs and oral squamous carcinoma CSCs, although less promising results have been observed in brain and breast CSCs.[49] Separately, anti-CSC vaccines using dendritic cells to induce tumor-specific T cell responses in murine models have shown therapeutic potential, particularly in treating microscopic tumors or as combinatorial agents in treating macroscopic tumors with conventional therapy.[49] Antigens associated with particular CSCs, such as ALDH, CD44, CD133, and HER2, constitute potential targets for CSC-specific immunotherapeutic approaches.[49]
2. Conclusions
The recognition of CSCs as a major component and driver of key processes in cancer progression, such as tumor growth, recurrence, metastasis, and treatment resistance, constitutes a landmark discovery in cancer research. The evolving CSC model is helping to explain a variety of unresolved questions, such as why the destruction of nonstem cancer cells of the tumor bulk may be associated with little or no improvement in patient outcomes. Recent discoveries concerning the centrality of CSC in cancer offer researchers a conceptual framework that is vital to decoding previously unexplained processes in cancer pathophysiology, and offers new approaches and new targets for cancer treatment.
Acknowledgment
This supplement was supported by Boston Biomedical Pharma, Inc., Cambridge, MA. Editorial support was provided by Jacqueline Egan, Allison England, PhD, and Brij Patel, PhD, of Guidemark Health, Stamford, CT.
Footnotes
Abbreviations: 5-FU = 5-fluorouracil, CSC = cancer stem cell, CTC = circulating tumor cell, ECM = extracellular matrix, EMT = epithelial-mesenchymal transition, EphA3 = ephrin type-A receptor 3, FAK = focal adhesion kinase, HER = human epidermal growth factor receptor, IL-3R = interleukin-3 receptor, JAK/STAT = Janus-activated kinase/signal transducer and activator of transcription, MET = mesenchymal-epithelial transition, NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells, ROS = reactive oxygen species, TGF-β = transforming growth factor beta.
The author reports no conflicts of interest.
References
- 1.Ajani JA, Song S, Hochster HS, et al. Cancer stem cells: the promise and the potential. Semin Oncol 2015; 42 (suppl 1):S3–S17. [DOI] [PubMed] [Google Scholar]
- 2.Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin 2013; 34:732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doherty MR, Smigiel JM, Junk DJ, et al. Cancer stem cell plasticity drives therapeutic resistance. Cancers (Basel) 2016; 8:pii: E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takaishi S, Okumura T, Tu S, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009; 27:1006–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith LM, Nesterova A, Ryan MC, et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer 2008; 99:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730–737. [DOI] [PubMed] [Google Scholar]
- 7.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012; 22:457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, Cong Y, Wang D, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep 2014; 2:78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013; 27:2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radisky DC, LaBarge MA. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008; 2:511–512. [DOI] [PubMed] [Google Scholar]
- 12.Sun YF, Xu Y, Yang XR, et al. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology 2013; 57:1458–1468. [DOI] [PubMed] [Google Scholar]
- 13.Han LSS, Shi S, Gong T, et al. Cancer stem cells: therapeutic implications and perspectives in cancer therapy. Acta Pharm Sin B 2013; 3:65–75. [Google Scholar]
- 14.Visvader JE. Cells of origin in cancer. Nature 2011; 469:314–322. [DOI] [PubMed] [Google Scholar]
- 15.Chen S, Huang EH. The colon cancer stem cell microenvironment holds keys to future cancer therapy. J Gastrointest Surg 2014; 18:1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10:717–728. [DOI] [PubMed] [Google Scholar]
- 17.Louie E, Nik S, Chen JS, et al. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res 2010; 12:R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2:442–454. [DOI] [PubMed] [Google Scholar]
- 19.Drake JM, Strohbehn G, Bair TB, et al. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell 2009; 20:2207–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ota I, Li XY, Hu Y, et al. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc Natl Acad Sci U S A 2009; 106:20318–20323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011; 20:576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoletov K, Kato H, Zardouzian E, et al. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci 2010; 123 (pt 13):2332–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai JH, Donaher JL, Murphy DA, et al. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012; 22:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Centers for Disease Control and Prevention Cancers Diagnosed at Late Stages Despite Available Screening Tests. Atlanta, GA:Centers for Disease Control and Prevention; 2014. [Google Scholar]
- 26.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Res 2006; 66:1883–1890. [DOI] [PubMed] [Google Scholar]
- 27.Touil Y, Igoudjil W, Corvaisier M, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res 2014; 20:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duru N, Fan M, Candas D, et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin Cancer Res 2012; 18:6634–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips TM, McBride WH, Pajonk F. The response of CD24 (−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 2006; 98:1777–1785. [DOI] [PubMed] [Google Scholar]
- 30.Sims-Mourtada J, Izzo JG, Apisarnthanarax S, et al. Hedgehog: an attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin Cancer Res 2006; 12:6565–6572. [DOI] [PubMed] [Google Scholar]
- 31.Borovski T, De Sousa EMF, Vermeulen L, et al. Cancer stem cell niche: the place to be. Cancer Res 2011; 71:634–639. [DOI] [PubMed] [Google Scholar]
- 32.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458:780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Li W, Patel SS, et al. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget 2014; 5:3743–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu X, Ghisolfi L, Keates AC, et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012; 11:2691–2698. [DOI] [PubMed] [Google Scholar]
- 35.Lagadec C, Vlashi E, Della Donna L, et al. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012; 30:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu ZY, Tang JN, Xie HX, et al. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int J Biol Sci 2015; 11:284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dave B, Mittal V, Tan NM, et al. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res 2012; 14:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu M, Bardia A, Wittner BS, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013; 339:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 2009; 106:13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Rogoff HA, Keates S, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A 2015; 112:1839–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abubaker K, Luwor RB, Zhu H, et al. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014; 14:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolev VN, Wright QG, Vidal CM, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res 2015; 75:446–455. [DOI] [PubMed] [Google Scholar]
- 43.Boston Biomedical Pharmaceuticals. Current Clinical Trials in Oncology. 2016; Available at: http://www.bostonbiomedical.com/research-and-development/pipeline/ Accessed July 25, 2016. [Google Scholar]
- 44.KaloBios Pharmaceuticals. KBB004 (ANTI-EPHA3). 2016; Available at: http://www.kalobios.com/our-programs/kb004/ Accessed August 2, 2016. [Google Scholar]
- 45.OncoMed Pharmaceuticals. List of OncoMed Trials. 2016; Available at: http://www.oncomed.com/Pipeline.html Accessed July 25, 2016. [Google Scholar]
- 46.Verastem Pharmaceuticals. Products Overview. 2016; Available at: http://www.verastem.com/products/ Accessed July 25, 2016. [Google Scholar]
- 47.Stemcentrix. Clinical Trials: Small Lung Cell Cancer. 2016; Available at: http://www.stemcentrx.com/ct-small-cell-lung-cancer.html Accessed August 8, 2016. [Google Scholar]
- 48.Stemline Therapeutics. Clinical Trials. 2016; Available at: http://www.stemline.com/clinical-trials.asp Accessed August 4, 2016. [Google Scholar]
- 49.Pan Q, Li Q, Liu S, et al. Concise review: targeting cancer stem cells using immunologic approaches. Stem Cells 2015; 33:2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]