

Summary
The roles of histone demethylases (HDMs) for the establishment and maintenance of pluripotency are incompletely characterized. Here, we show that JmjC-domain-containing protein 1c (JMJD1C), an H3K9 demethylase, is required for mouse embryonic stem cell (ESC) self-renewal. Depletion of Jmjd1c leads to the activation of ERK/MAPK signaling and epithelial-to-mesenchymal transition (EMT) to induce differentiation of ESCs. Inhibition of ERK/MAPK signaling rescues the differentiation phenotype caused by Jmjd1c depletion. Mechanistically, JMJD1C, with the help of pluripotency factor KLF4, maintains ESC identity at least in part by regulating the expression of the miR-200 family and miR-290/295 cluster to suppress the ERK/MAPK signaling and EMT. Additionally, we uncover that JMJD1C ensures efficient generation and maintenance of induced pluripotent stem cells, at least partially through controlling the expression of microRNAs. Collectively, we propose an integrated model of epigenetic and transcriptional control mediated by the H3K9 demethylase for ESC self-renewal and somatic cell reprogramming.
Keywords: JMJD1C, H3K9 demethylase, KLF4, embryonic stem cells, self-renewal, reprogramming, microRNAs, ERK/MAPK signaling, EMT
Graphical Abstract
Highlights
-
•
JMJD1C is required for the maintenance of ESC identity
-
•
Depletion of Jmjd1c leads to the activation of ERK/MAPK signaling and EMT
-
•
JMJD1C interplays with KLF4 to activate the expression of miR-200 family
-
•
JMJD1C ensures efficient induction of pluripotency partially via miR-200 family
In this article, Jin and colleagues show that JMJD1C is required for mouse ESC self-renewal and efficient somatic cell reprogramming. In ESCs, JMJD1C cooperates with KLF4 to suppress the ERK/MAPK signaling and EMT at least in part through promoting miR-200 family and miR-290/295 cluster expression. Additionally, JMJD1C ensures efficient somatic cell reprogramming partially via the miR-200 family.
Introduction
Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are both pluripotent stem cells, self-renewing indefinitely in vitro and differentiating into all cell types of an organism (De Los Angeles et al., 2015, Evans and Kaufman, 1981, Takahashi and Yamanaka, 2006). Thus, they have provided new prospects for basic research and regenerative medicine (Cell Stem Cell Editorial Team, 2016, Cherry and Daley, 2012). The unique properties of ESC/iPSCs are controlled by multiple regulatory mechanisms (Hackett and Surani, 2014, Niwa, 2007). First, OCT4, SOX2, and NANOG are core transcription factors for the establishment and maintenance of pluripotency (Ng and Surani, 2011). Recently, more transcriptional factors have been shown to involve in the regulation of ESC self-renewal (Ding et al., 2015, Ivanova et al., 2006, Ng and Surani, 2011). Second, leukemia inhibitor factor, together with bone morphogenetic protein 4 or serum, can maintain mouse ESC self-renewal, whereas fibroblast growth factor (FGF)/ERK1/2 signaling and calcineurin-nuclear factor of activated T cells (NFAT) pathways are both essential and sufficient to induce ESC differentiation and early mouse embryonic development (Huang et al., 2015, Kunath et al., 2007, Li et al., 2011). Additionally, accumulating lines of evidence have indicated that epigenetic modulators also play critical roles in the ESC fate control (Liang and Zhang, 2013). To fulfill the full potential of ESCs, it is necessary to elucidate how these regulatory mechanisms are precisely orchestrated to safeguard ESCs at an undifferentiated state. The interplay between transcriptional factors and epigenetic regulators in ESCs has been intensively investigated (Ang et al., 2011, Costa et al., 2013, Ding et al., 2015, He et al., 2013, Loh et al., 2007). However, how epigenetic regulators have crosstalks with signaling pathways is poorly understood.
Epigenetic regulation, mediated by DNA/RNA modifications, histone modifications/variants, and non-coding RNAs, provides ESCs with a unique chromatin conformation, allowing for the activation of pluripotency genes and the suppression of developmental genes (Boland et al., 2014, Zhao and He, 2015). Histone modifications are the important part of epigenetics and play critical roles in the regulation of ESC identity (Lessard and Crabtree, 2010). Generally H3K4 methylation correlates with gene activation, while H3K27 and H3K9 methylation are associated with gene repression (Kouzarides, 2007). The Trithorax group (trxG)-mediated H3K4 methylation and the Polycomb group (PcG)-mediated H3K27 methylation have been widely investigated in the maintenance and establishment of the pluripotent state (Gu and Lee, 2013, Lessard and Crabtree, 2010, Liang and Zhang, 2013). In contrast, the distribution and functions of H3K9 methylation are more complicated and less studied in ESCs and somatic cell reprogramming. It has been reported that H3K9 dimethylation (H3K9me2) has a broad distribution and covers more than one-third of the genome (Liu et al., 2015b). Interestingly, H3K9me2 is maintained at a low level in ESCs and undergoes drastic changes during ESC differentiation (Wen et al., 2009), indicating a key role of H3K9 demethylases in the maintenance of ESC identity.
H3K9 demethylation is mainly catalyzed by Fe(II)- and α-ketoglutarate-dependent JmjC-domain-containing proteins, including the JMJD1/KDM3 family, JMJD2/KDM4 family, and PHF8/KDM7B (Mosammaparast and Shi, 2010, Qiu et al., 2010). In addition, flavin adenine dinucleotide (FAD)-dependent amine oxidase LSD1/KDM1A has also been demonstrated to remove H3K9me1/2 (Laurent et al., 2015, Mosammaparast and Shi, 2010). Among these H3K9 demethylases, a few have been shown to be required for the maintenance of ESC identity, including JMJD1A, JMJD2B, and JMJD2C (Das et al., 2014, Loh et al., 2007). Besides, the regulatory mechanisms underlying the role of the three H3K9 demethylases in ESCs are not fully addressed, particularly from the perspective of interaction between histone modifications and signaling pathways. Hence, we asked whether there would be other H3K9 demethylases required for the induction and maintenance of pluripotency, and if so, how the function is executed.
In this study, to address the above questions we performed a functional RNAi screen against H3K9 demethylases in mouse ESCs and found that JMJD1C is required for ESC self-renewal and efficient somatic cell reprogramming. Mechanistically, JMJD1C could antagonize the ERK/MAPK pathway and the epithelial-to-mesenchymal transition (EMT) process to maintain ESCs in the undifferentiated state. Moreover, our results reveal a direct regulatory role of JMJD1C and pluripotency factor KLF4 for the expression of the miR-200 family and miR-290/295 cluster, which is, at least in part, responsible for JMJD1C's function in ESCs. Therefore, this study reports an important function of JMJD1C, revealing a crosstalk between an epigenetic regulator and one of the key signaling pathways in ESC fate determination.
Results
JMJD1C Is Required for the Maintenance of ESC Self-Renewal
We began with comparing the expression level of various H3K9 demethylases between mouse ESCs and mouse embryonic fibroblasts (MEFs) and found substantially higher transcript levels of most examined H3K9 demethylases in ESCs than in MEFs (Figure S1A), suggesting their potential association with the ESC state. To find H3K9 demethylases required for the maintenance of ESC identity, we performed a small-scale functional RNAi screen against H3K9 demethylases (Figure S1B) and found that, in addition to JMJD1A and JMJD2C (Das et al., 2014, Loh et al., 2007), JMJD1B and JMJD1C were also required to sustain ESCs in the undifferentiated state (Figure S1C). In contrast, knockdown (KD) of Jmjd2a, Jmjd2b, Jmjd2d, or Phf8 did not change the ESC morphology markedly. JMJD1C was previously identified to associate with OCT4, NANOG, TFCP2L1, and ESRRB (Costa et al., 2013, Ding et al., 2012, van den Berg et al., 2010). Moreover, our real-time qRT-PCR results showed that Jmjd1c was highly expressed in undifferentiated ESCs and downregulated during embryoid body (EB) formation, paralleling with levels of Oct4 and Nanog (Figure 1A). However, its role in regulating mouse ESC self-renewal is as yet unreported. Therefore, we decided to focus on Jmjd1c in the subsequent study.
Figure 1.

JMJD1C Is Required for ESC Self-Renewal
(A) qRT-PCR analysis of Jmjd1c, Oct4, and Nanog expression levels during embryoid body (EB) formation. Data are presented as mean ± SD of three independent experiments.
(B) Bright-field images and alkaline phosphatase (AP) staining of ESCs after Jmjd1c knockdown (KD) for 4 days. See also Figure S1E.
(C) qRT-PCR analysis of pluripotency and primordial germ cell (PGC) gene expression after 4 days of Jmjd1c KD. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D) Western blot analysis of JMJD1C, OCT4, SOX2, and KLF4 after 4 days of Jmjd1c KD. α-TUBULIN was used as a loading control. See also Figure S1F.
(E) qRT-PCR analysis of lineage-specific gene expression after 4 days of Jmjd1c KD. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(F and G) Colony-formation assays for Jmjd1c KD (shJmjd1c#3, shJmjd1c#4) and control (shNT) ESCs (F) and their quantification (G). Data are presented as mean ± SD of four independent experiments. ∗∗∗p < 0.001.
(H and I) Schematic illustration (H) and results (I) of competition assays for mixed populations of Jmjd1c KD and control ESCs. Data are presented as mean ± SD of three independent experiments.
To silence Jmjd1c expression efficiently and specifically, we designed four short hairpin RNAs (shRNAs) targeting its mRNA sequences and found that shJmjd1c#3 and shJmjd1c#4 had higher efficiencies to KD Jmjd1c expression than the other two shRNAs (Figure S1D). Jmjd1c KD resulted in the loss of typical ESC morphology and alkaline phosphatase (AP) activities, while transfection of ESCs with non-target control shRNA (shNT) did not have such effects (Figure 1B). Similar results were obtained in another ESC line, CGR8 (Figure S1E). In addition, Jmjd1c KD led to significantly decreased expression levels of pluripotency genes (Oct4, Sox2, Nanog, Klf2, Klf4, Klf5, Esrrb, Nr5a2, Tbx3, Tcl1, and Tfcp2l1) (Figures 1C, 1D, and S1F) and increased levels of lineage markers, including those of the endoderm (Bmp2, Dab2, and Gata4), the mesoderm (T, Bmp4, and Isl1), the ectoderm (Fgf5, Nestin, Pax6, and Cxcl12), and the trophectoderm (Cdx2 and Eomes) (Figure 1E). Primordial germ cell markers such as Stella (Dppa3), Prdm1, and Tcfap2c also were downregulated in Jmjd1c KD ESCs (Figure 1C), verifying the previous report that JMJD1C is required for the long-term maintenance of mouse male germ cells (Kuroki et al., 2013). To validate that JMJD1C was indeed required for ESC self-renewal, we performed ESC colony formation and competition assays and found that Jmjd1c KD drastically reduced the capacity of ESCs to self-renew, as evidenced by reduced undifferentiated ESC colony numbers and percentages of shJmjd1c-transfected GFP+ cells (Figures 1F–1I). Taken together, our results reveal that JMJD1C is required for the maintenance of ESC self-renewal.
Silencing of Jmjd1c Activates ERK/MAPK Signaling and the Epithelial-to-Mesenchymal Transition
Given that JMJD1C is known as an H3K9 demethylase responsible for the removal of H3K9me1/2 (Chen et al., 2015, Kim et al., 2010), we reasoned that JMJD1C might regulate gene expression through removing H3K9 methylation. We therefore examined the global H3K9 methylation level. As anticipated, Jmjd1c KD resulted in increased levels of H3K9 methylation, including H3K9me1, H3K9me2, and H3K9me3 (Figure 2A). To understand how Jmjd1c KD and resultant changes in H3K9 methylation would affect ESCs at a global gene expression level, we compared the whole-genome transcriptomes between the control and Jmjd1c KD ESCs (6 samples, including 2 shNT control samples and 4 shJmjd1c samples, 2 from #3 and 2 from #4 shRNA, respectively; Figure 2B) and identified 540 upregulated and 321 downregulated genes, respectively. Gene ontology (GO) analysis of these differentially expressed genes (DEGs) showed that the top GO terms were associated with cell differentiation and the EMT process (Figures 2C and S2A). Signaling pathway analysis indicated that these DEGs were enriched in the terms of focal adhesion, extracellular matrix-receptor interaction, MAPK signaling, and RAS signaling (Figures 2D and S2B). As the activated MAPK signaling and EMT have been demonstrated to be key events for mouse ESCs to exit from the self-renewal state (Kunath et al., 2007, Li et al., 2011), we first examined the phosphorylated ERK1/2 levels and found that Jmjd1c KD activated ERK1/2 signaling (Figure 2E). Next, we examined EMT markers in Jmjd1c KD cells and found that depletion of Jmjd1c led to the downregulation of epithelial markers, including Cdh1 (E-cadherin), Epcam, Cldn3, Crb3, and Ocln (Figure 2F), and the upregulation of mesenchymal markers, including Cdh2 (N-cadherin), Zeb2, Twist2, and Mmp9 (Figure 2G). These data suggest that JMJD1C may function to suppress the activation of ERK/MAPK signaling and EMT process in undifferentiated ESCs.
Figure 2.

Knockdown of Jmjd1c Activates the ERK/MAPK Signaling and EMT in ESCs
(A) Representative western blot analysis of global H3K9 methylation levels after 4 days of Jmjd1c KD in CGR8 and J1 ESC lines. Histone H3 and α-TUBULIN were used as loading controls.
(B) Hierarchical clustering of differentially expressed genes (DEGs) (fold changes >1.5) from Jmjd1c KD (shJmjd1c#3, shJmjd1c#4) and control (shNT) ESCs. Data from two independent experiments are shown.
(C) Biological process GO enrichments of DEGs (fold changes >1.5). See also Figure S2.
(D) Signaling pathway analysis of DEGs (fold changes >1.5). See also Figure S2.
(E) Representative western blot analysis of ERK1/2 and phospho-ERK1/2 after 4 days of Jmjd1c KD. α-TUBULIN was used as a loading control.
(F and G) qRT-PCR analysis of transcript levels for markers of the epithelial-to-mesenchymal transition (EMT) process after 4 days of Jmjd1c KD. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Inhibition of ERK/MAPK Signaling Abolishes ESC Differentiation Induced by Jmjd1c Depletion
To answer the question of whether activated ERK/MAPK signaling process could be the cause of ESC differentiation induced by Jmjd1c KD, we tested whether inhibition of the pathway could rescue Jmjd1c KD-induced phenotypes. Strikingly, shJmjd1c-treated ESCs exhibited largely normal compacted colonies in the presence of a MEK1/2 inhibitor PD0325901, whereas the same cells displayed an extensively differentiated morphology in the absence of the inhibitor (Figure 3A). Moreover, treatment of Jmjd1c KD cells with the inhibitor restored the pluripotency gene expression almost to normal levels (Figures 3B and S3A) and attenuated the upregulation of lineage markers (Figures 3C and S3B). The inhibition of ERK/MAPK signaling also suppressed the EMT process, as shown by restoring Jmjd1c deficiency-caused alterations in the expression levels of epithelial and mesenchymal markers (Figures 3D, 3E, S3C, and S3D). The latter finding is consistent with our previous finding that the enhanced EMT was downstream of the activated ERK/MAPK signaling (Li et al., 2011). Additionally, matrix metalloproteinases (MMPs) are key components of the EMT, and the broad-spectrum MPP inhibitor GM6001 is known to prevent the EMT process (Cavallaro and Christofori, 2004, Tan et al., 2010). We therefore treated the control and Jmjd1c-deficient cells with GM6001 and found that the inhibitor rescued the differentiation phenotypes partially, being less efficient than the ERK/MAPK inhibitor in terms of both cell morphology (Figure 3F) and expression levels of epithelial and mesenchymal markers (Figures 3G, 3H, S3E, and S3F). Collectively, these results indicate that JMJD1C suppresses ERK/MAPK signaling and the EMT process to maintain ESC identity.
Figure 3.

Inhibition of ERK/MAPK Signaling or EMT Rescues the Differentiation Phenotype Caused by Jmjd1c Depletion
(A) Bright-field images of Jmjd1c-depleted and control ESCs after treatment with DMSO or the MEK1/2 inhibitor PD0325901 (1 μM) for 4 days.
(B and C) qRT-PCR analysis of transcript levels for pluripotency (B) and lineage (C) genes in Jmjd1c-depleted and control ESCs after treatment with DMSO or the MEK1/2 inhibitor PD0325901 (1 μM) for 4 days. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
(D and E) Expression levels of EMT markers in Jmjd1c-depleted and control ESCs after treatment with DMSO or the MEK1/2 inhibitor PD0325901 (1 μM) for 4 days. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
(F) Bright-field images of Jmjd1c-depleted and control ESCs after treatment with DMSO or the MMPs inhibitor GM6001 (50 μM) for 4 days.
(G and H) Expression levels for EMT markers in Jmjd1c-depleted and control ESCs after treatment with DMSO or the MMPs inhibitor GM6001 (50 μM) for 4 days. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
The miR-200 Family and miR-290/295 Cluster Are Key Downstream Targets of JMJD1C
The next question we asked was how JMJD1C suppressed the ERK/MAPK signaling and EMT process in ESCs. Given that JMJD1C acts to reduce the levels of H3K9me1/2, repressive marks for gene expression (Chen et al., 2015, Kim et al., 2010), we hypothesized that JMJD1C might suppress the ERK/MAPK signaling and EMT process in an indirect manner. MicroRNAs generally play inhibitory roles in regulating gene expression at a post-transcriptional and/or translational level (Macfarlane and Murphy, 2010). We inferred that JMJD1C might antagonize the ERK/MAPK signaling and EMT process through controlling microRNA expression. To test this deduction, we examined whether expression levels of microRNAs highly expressed in pluripotent stem cells, including miR-290/295, miR-302/367, miR-17/92, miR-106a/363, miR-106b/25, miR-200b/429, miR-200c/141, and miR-183/182 (Greve et al., 2013), could be regulated by JMJD1C. As each microRNA cluster is composed of several microRNAs, we first examined primary transcripts of these microRNA clusters in Jmjd1c KD cells. Clusters of miR-183/182, miR-106b/25, miR-200b/429, miR-200c/141, and miR-290/295 were downregulated significantly by Jmjd1c KD, while there were no significant differences for clusters of miR-17/92, miR-106a/363, and miR-302/367 between control and Jmjd1c KD cells (Figure 4A). We then examined the mature microRNAs of clusters controlled by JMJD1C. Notably, all members of these clusters examined had significantly lower levels in Jmjd1c KD cells than in control cells (Figure 4B), suggesting an important role of JMJD1C for maintaining the expression of these microRNAs. To provide evidence for the direct regulation of these microRNA expressions, we conducted chromatin immunoprecipitation (ChIP)-qPCR assays using affinity-purified rabbit polyclonal JMJD1C antibodies made in our laboratory and ChIP-grade hemagglutinin (HA) antibodies in ESCs where an FLAG-HA tag was knocked into the C terminus of the endogenous Jmjd1c locus by the CRISPR/Cas9 system (Figure S4A), respectively. With multiple primers against promoter regions of miR-200b/429 cluster, miR-200c/141 cluster, and miR-290/295 cluster, we detected relatively high enrichments of JMJD1C at P1–P5 regions of the miR-200b/429 cluster, at P5–P8 regions of the miR-200c/141 cluster, at P1–P4 and P6–P8 regions of the miR-290/295 cluster (Figures 4C and 4D). In addition, we found JMJD1C enrichments at enhancer and promoter regions of Oct4 (Figure S4B). Specifically, these JMJD1C enrichments were significantly reduced when Jmjd1c expression was silenced (Figures S4C and S4D, compared with data in Figures S4B and 4D). Furthermore, we found that JMJD1C bound to the promoter regions of Klf4, an important ESC self-renewal-associated gene, particularly at P4–P7 regions (Figures S4E and S4F). Thus, JMJD1C might directly activate the expression of miR-200b/429, miR-200c/141, and miR-290/295 clusters and pluripotency factors (Oct4 and Klf4) to maintain ESC identity.
Figure 4.

JMJD1C and KLF4 Directly Regulate Expression of miR-200 Family and miR-290/295 Cluster to Sustain ESC Identity
(A) qRT-PCR analysis of primary transcripts of microRNA clusters after 4 days of Jmjd1c KD. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
(B) qRT-PCR analysis of mature transcripts of microRNA clusters after 4 days of Jmjd1c KD. Data are presented as mean ± SD of four independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C) Schematic illustration showing the location of amplicons used to evaluate ChIP-enriched genomic regions and transcription factor binding sites at loci of miR-200 family and miR-290/295 cluster.
(D) ChIP-qPCR assays using JMJD1C and HA antibodies in Jmjd1cFLAG-HA ESCs. Relative fold enrichments are shown. The value from control immunoglobulin G (IgG) ChIP-qPCR under the same condition was set as 1. Data are presented as mean ± SD of four independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) Bright-field images of control and Jmjd1c-depleted ESCs after 4 days of transfection with negative control (NC), microRNA mimics, or microRNA mutants. Mut4n and mut7n are mutants of miR-200 family, harboring 4- and 7-base mutations, respectively, in the seed region.
(F) ChIP-qPCR assays using KLF4 antibodies in Jmjd1cFLAG-HA ESCs. Relative fold enrichments are shown. The value from control IgG ChIP-qPCR under the same condition was set as 1. Data are presented as mean ± SD of three independent experiments; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(G) Western blot analysis of KLF4 and JMJD1CFLAG-HA after 3 days of Klf4 KD. α-TUBULIN was used as a loading control.
(H) ChIP-qPCR assays using HA antibodies in control and Klf4-depleted Jmjd1cFLAG-HA ESCs. Relative fold enrichments were normalized to HA ChIP-qPCR in control Jmjd1cFLAG-HA ESCs under the same condition. Data are presented as mean ± SD of three independent experiments ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To determine the causative link between Jmjd1c KD-induced ESC differentiation and altered expression levels of JMJD1C-regulated microRNAs and pluripotency factors, we examined their expression levels at 24 hr post infection of the Jmjd1c shRNA virus. As a result, we found that primary transcripts of miR-200 family and miR-290-295 cluster as well as Klf4 mRNA levels were downregulated at this early time point after Jmjd1c KD, while the expression levels of most lineage markers did not change (Figure S4G), suggesting that these microRNAs and Klf4 might be important downstream targets of JMJD1C. To test this assumption at a functional level, we overexpressed mimics of these microRNAs in Jmjd1c-deficient ESCs. Notably, miR-200s and miR-295, rather than their mutants, largely rescued the differentiation phenotype caused by Jmjd1c KD (Figures 4E and S4H–S4J). Thus, the miR-200 family and miR-290/295 cluster could be key effectors for JMJD1C to maintain ESCs at the undifferentiated state.
We finally asked how JMJD1C was localized to its target genes. It has been reported that JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors (Chen et al., 2015). We therefore analyzed the genomic regions specifically enriched for JMJD1C and predicted the transcription factor binding motifs by DIANA-miRGen v3.0 (Georgakilas et al., 2016). Specific DNA binding motifs of TFCP2L1, OCT4/SOX2, ESRRB, KLF4, and FOXP1 were found (Figure 4C). Among these factors, KLF4 binding motifs were present at promoters of both miR-200 family and miR-290/295 cluster. We conducted KLF4 ChIP-qPCR assays and validated the enrichment of KLF4 at these regions (Figure 4F). Importantly, Klf4 KD, without reducing JMJD1C protein levels, led to reduced JMJD1C binding to these microRNA loci (Figures 4G and 4H) and the downregulation of miR-200 family and miR-290/295 cluster (Figure S4K). Furthermore, we found protein interactions between endogenous KLF4 and JMJD1C in ESCs (Figure S4L). Altogether, these results lead to our conclusion that KLF4 recruits JMJD1C to the promoters of miR-200 family and miR-290/295 cluster to activate their expression.
JMJD1C Suppresses the ERK/MAPK Signaling and EMT through Controlling miR-200 Family and miR-290/295 Cluster Expression
To identify genes targeted by the miR-200 family and miR-290/295 cluster, we predicted their targets using TargetScan (Mouse 7.1) (Lewis et al., 2005) and overlapped these predicted targets with Jmjd1c KD-upregulated DEGs detected by our microarray analysis (Figures 2B, 5A, and 5B), uncovering 93 and 42 potential targets of miR-200 family and miR-290/295 cluster, respectively (Table S1). Among these potential targets, there were components of ERK/MAPK signaling, including integrin β-8 (Itgb8), insulin growth factor 2 (Igf2), c-Jun, Ets1, Tgfb2, and Tgfbr2 (Hazzalin and Mahadevan, 2002, Rauch et al., 2016, Stupack and Cheresh, 2002), as well as EMT inducers, including Zeb2, Lats2, c-Jun, Ets1, Tgfb2, Tgfbr2, and Igf2 (Lamouille et al., 2014, Liu et al., 2015a, Shaikhibrahim and Wernert, 2012, Zhang et al., 2012). To narrow down and find true functional targets, we then examined their expression patterns in control and Jmjd1c KD cells transfected with mimics of miR-200 family and miR-290/295 cluster, respectively. Overexpression of miR-200s or miR-295, in particular microRNAs sharing an AAUACUG seed sequence (miR-429, miR-200b and miR-200c), abrogated the upregulation of these identified genes in Jmjd1c KD cells (Figures 5C and S5A). In addition, miR-200s or miR-295 diminished the downregulation of epithelial markers and upregulation of mesenchymal markers caused by Jmjd1c KD (Figures 5D, 5E, S5B, and S5C). Mutations of these microRNAs in the seed sequence led to the loss of their ability to rescue Jmjd1c KD-mediated aberrant expression of these marker genes (Figures S5A–S5C), demonstrating the function specificity of the microRNAs. Notably, miR-200s could reverse the increased pERK1/2 level caused by Jmjd1c KD (Figure 5F). Collectively, these results support the notion that JMJD1C suppresses the ERK/MAPK signaling and the EMT process, at least in part, through controlling miR-200 family and miR-290/295 cluster expression.
Figure 5.

miR-200 Family and miR-290/295 Cluster Maintain ESC Identity through Suppressing ERK/MAPK Signaling and EMT Process
(A and B) Venn diagrams of overlapping genes between predicted microRNA targets (miR-200s [A] and miR-295 [B]) by TargetScan and Jmjd1c KD-caused upregulated genes (DEGs) detected by KD microarray analysis.
(C) qRT-PCR analysis of key targets of miR-200s and miR-295 in control and Jmjd1c-depleted ESCs 4 days post transfection with NC or microRNA mimics. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D and E) qRT-PCR analysis of EMT markers in control and Jmjd1c-depleted ESCs 4 days post transfection with NC or microRNA mimics. Data are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) Representative western blot analysis of ERK1/2 and phospho-ERK1/2 in control and Jmjd1c-depleted ESCs 3 days post transfection with NC or miR-200s (miR-200a, miR-200b, and miR-200c). α-TUBULIN was used as a loading control.
JMJD1C Ensures Efficient Somatic Cell Reprogramming
We next tested the role of JMJD1C in the re-establishment of pluripotency from MEFs mediated by Yamanaka factors (OCT4, SOX2, KLF4 and C-MYC, OSKM). First, Jmjd1c expression was gradually upregulated during MEF reprogramming, in parallel with the upregulation of Nanog and Esrrb (Figure 6A). Second, when Jmjd1c was depleted by its specific shRNA in MEFs carrying a transgenic Oct4 promoter-driven GFP cassette, the number of OCT4-GFP-positive colonies decreased significantly, with approximately 40% of that in control cells (Figure 6B). To further validate the role of JMJD1C, we derived Jmjd1c+/+ (wild-type [WT]), Jmjd1c+/− (heterogeneous [HET]), and Jmjd1c−/− (knockout [KO]) MEFs from corresponding mouse embryos and reprogrammed these MEFs to iPSCs. Strikingly, Jmjd1c KO almost abolished the ability of MEFs to reprogram, as indicated by AP staining results at reprogramming day 14 (Figures 6C and 6D). Intriguingly, Jmjd1c HET MEFs also displayed a significantly reduction in the AP-positive colony number. To determine whether overexpression of miR-200 family could rescue the defect of Jmjd1c-null MEFs in reprogramming, we infected Jmjd1c WT and Jmjd1c-null MEFs with retroviruses carrying sequences for the OSKM and miR-200s to generate iPSCs. Our results showed that miR-200s could restore the reprogramming efficiency of Jmjd1c-null MEFs to levels comparable with that of Jmjd1c HET MEFs (Figures 6C and 6D). Although miR-200b could enhance the reprogramming efficiency in both KO and WT MEFs, the effect was more robust in KO MEFs than in WT MEFs (Figures 6C and 6D). Therefore, it is likely that JMJD1C ensures efficient somatic cell reprogramming, partially if not mostly, by regulating microRNA expression.
Figure 6.

JMJD1C Is Indispensable for Efficient Somatic Cell Reprogramming
(A) qRT-PCR analysis of Jmjd1c, Nanog, and Esrrb expression levels during MEF reprogramming. Data are presented as mean ± SD of three independent experiments.
(B) Analysis of GFP+ colonies generated from control (shLuc) and Jmjd1c-depleted (shJmjd1c) Oct4-GFP MEFs. Data are presented as mean ± SD of three independent experiments. ∗∗p < 0.01.
(C and D) Analysis of AP colonies generated from Jmjd1c wild-type (WT), heterogeneous (HET), and knockout (KO) MEFs, as well as WT and KO MEFs ectopically overexpressing miR-200s. Data in (D) are presented as mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) RT-PCR analysis of the activation of endogenous (Endo) pluripotency genes and silencing of exogenously introduced (Retro) pluripotency genes in Jmjd1c WT and KO iPSCs. Gapdh was used as an internal control.
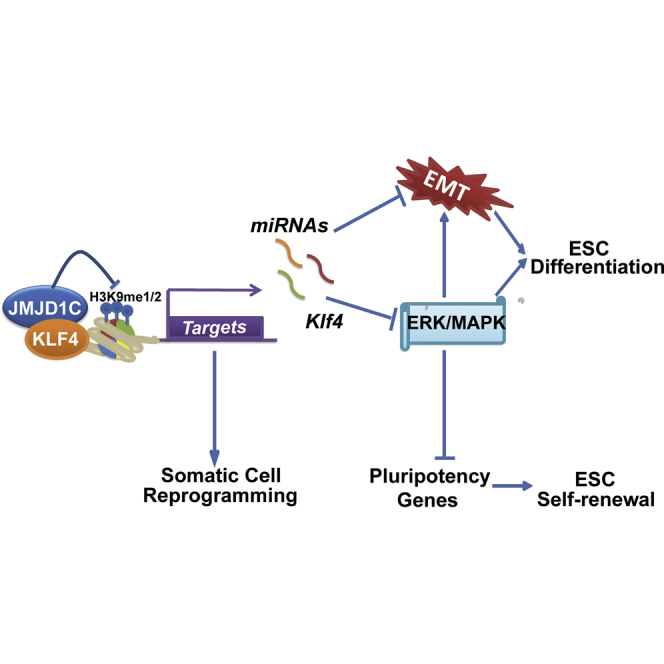
(F) A proposed working model of JMJD1C in mouse ESCs and for somatic cell reprogramming. JMJD1C is recruited to the regulatory regions of subsets of pluripotency genes and microRNAs (targets) with the help of KLF4 to activate their transcription. Members of the miR-200 family and miR-290/295 cluster suppress the ERK/MAPK signaling and EMT process. In addition, JMJD1C ensures efficient induction of pluripotency partially via microRNA expression.
Finally, we evaluated the role of JMJD1C in the derivation and maintenance of iPSC lines. We found that WT iPSCs showed the typical morphology of ESCs and were expandable, while KO iPSCs tended to differentiate and died gradually after several passages, suggesting that JMJD1C was required for the maintenance of iPSC identity. Furthermore, endogenous pluripotency genes and exogenously introduced pluripotency genes were efficiently activated and silenced, respectively, in WT iPSCs (Figure 6E). In contrast, exogenously introduced pluripotency genes were not fully silenced and endogenous pluripotency genes were not robustly activated in Jmjd1c KO iPSCs, indicative of partial reprogramming (Figure 6E). This observation points out that JMJD1C is indispensable for bona fide induction of pluripotency from somatic cells.
Discussion
ESCs have a more open and transcriptionally permissive chromatin state than differentiated cells (Meshorer and Misteli, 2006). Methylation of the repressive marker H3K9 is maintained at a low level in ESCs (Liu et al., 2015b). Some H3K9 demethylases have been indicated to be required for the maintenance of ESC identity (Das et al., 2014, Loh et al., 2007). Loh et al. (2007) showed that JMJD1A and JMJD2C maintain ESC self-renewal via the regulation of Tcl1 and Nanog expression, respectively. Recently, Das et al., (2014) proposed that JMJD2B and JMJD2C play distinct and combinational functions in the maintenance of mouse ESC identity. Their studies propose that JMJD2B and NANOG act through an interconnected regulatory loop, while JMJD2C assists PRC2 in transcriptional repression. More recently, Pedersen et al. (2016) revealed that JMJD2A and JMJD2C have redundant roles in mouse ESCs, and their simultaneous loss impairs ESC proliferation and induces ESC differentiation into primitive endoderm. These studies reveal the existence of distinct functions and complicated regulatory mechanisms for H3K9 demethylases.
Here, we report an essential role of JMJD1C, a member of the JMJD1/KDM3 demethylase family, for the maintenance of ESC self-renewal. Distinctly from previously reported roles of the JMJD2/KDM4 family H3K9 demethylases, JMJD1C cooperates with pluripotency transcription factor KLF4 to promote the expression of the miR-200 family and miR-290/295 cluster to suppress the ERK/MAPK signaling and EMT process, ensuring ESC self-renewal. In addition, we identify subsets of genes potentially responsible for JMJD1C-microRNA-mediated function in ESCs. These include genes associated with the ERK/MAPK signaling and EMT process. Thus, we establish a previously unrecognized JMJD1C/KLF4-microRNA-ERK/MAPK axis, providing important insights into the crosstalk between the epigenetic regulator and signaling in controlling ESC fate.
It has been recently demonstrated that H3K9 methylation is a barrier for reprogramming somatic cell into iPSCs (Chen et al., 2013). Consistent with this result, most H3K9 demethylases, including JMJD1A/B/C and JMJD2B/C, have been shown to be required for efficient somatic cell reprogramming. In line with our results, Shakya et al. (2015) showed that Jmjd1c KD reduced somatic cell reprogramming efficiency. However, Chen et al. (2013) showed that Jmjd1c KD in pre-iPSCs did not obviously affect vitamin C-induced further reprogramming. Their failure to detect JMJD1C's role in reprogramming could be explained as follows. First, they generated iPSCs in a medium containing vitamin C; different culture conditions may give rise to different results. Second, the conversion from pre-iPSCs to fully reprogrammed iPSCs represents the late stage of the reprogramming; the function of JMJD1C could not be detected if it would act in earlier stages of pluripotency induction.
The mechanism underlying the function of H3K9 demethylases in reprogramming remains largely unknown. JMJD1C was shown to induce endogenous Oct4 expression during reprograming of fibroblasts to pluripotency (Shakya et al., 2015). Here, we show that JMJD1C ensures efficient somatic cell reprogramming, at least in part, by regulating miR-200 family expression, providing a potential mechanistic explanation. This notion is also supported by the previous study that miR-200 family is critical for MET and iPSC generation (Hu et al., 2014, Wang et al., 2013). Moreover, characterization of WT and Jmjd1c KO iPSCs reveals that normal expression of JMJD1C is critical for the full activation of endogenous pluripotency genes and efficient silencing of exogenously introduced pluripotency genes. Therefore, this study provides crucial insights into how JMJD1C functions in maintaining ESC self-renewal and during somatic cell reprogramming.
Despite critical roles of JMJD1C in pluripotency regulation, Jmjd1c KO mice are viable (Kuroki et al., 2013). The JMJD1/KDM3 family has three members (JMJD1A/KDM3A, JMJD1B/KDM3B, and JMJD1C/KDM3C), which may have functional redundancy in vivo. In the absence of Jmjd1c, other family members may compensate its deficiency during embryonic development. Similar to JMJD1C, both JMJD1A and JMJD2C have been demonstrated to be required for ESC self-renewal (Das et al., 2014, Loh et al., 2007). However, both Jmjd1a and Jmjd2c KO mice are also viable (Inagaki et al., 2009, Pedersen et al., 2014, Tateishi et al., 2009), suggesting the possibility of redundancy for the JMJD1/KDM3 family in vivo.
In addition, studies have suggested that the expression of JMJD1C associates with normal development such as spermatogenesis (Kuroki et al., 2013, Nakajima et al., 2016), circulating vascular endothelial growth factor levels (Choi et al., 2016), and concentrations of liver enzymes (Chambers et al., 2011), and that its dysfunction results in various diseases such as leukemia (Chen et al., 2015, Sroczynska et al., 2014, Zhu et al., 2016), intracranial germ cell tumors (Wang et al., 2014a, Wang et al., 2014b), and Rett syndrome (Saez et al., 2016). Because the majority of these results were obtained from genome-wide association studies, the underlying mechanisms remain largely unknown. MicroRNAs of the miR-200 family play critical roles in the EMT process and development of cancers such as ovarian, breast, and prostate cancer (Feng et al., 2014). Discovery of the regulatory role for JMJD1C over the transcription of the miR-200 family opens new angles for the study of Jmjd1c deficiency-associated diseases and possible strategies for their treatment.
Experimental Procedures
Cell Culture
Mouse ESC lines (all experiments were performed in J1 ESC lines, unless indicated otherwise), MEFs, Plat-E cells, and HEK 293FT cells were cultured in the defined medium as described in Supplemental Experimental Procedures.
Lentiviral Production and RNAi Screen
Four different shRNAs were used for each H3K9 demethylase. Non-target shRNA (shNT) and shLuciferase (shLuc) were used as negative controls, and shRNAs against Oct4 and Nanog were used as positive controls. Viral particles for shRNAs were produced in 293FT cells. A detailed protocol of lentiviral production and infection of ESCs is available in Supplemental Experimental Procedures. The shRNA sequences used in this study are listed in Table S2.
RNA Isolation, Reverse Transcription, and Real-Time qPCR
Total RNA was isolated using the TRIzol Reagent (Invitrogen). Detailed procedures are provided in Supplemental Experimental Procedures. The primers are listed in the Table S3.
Competition Assays
Competition assays of ESCs were performed according to the protocol developed by the Ihor R Lemischka laboratory (Lee et al., 2012). In brief, 80% shRNA-expressing ESCs (GFP+ from shRNA-pLKO.pig) were mixed with 20% control shRNA-expressing ESCs (GFP− from shLuc-pLKO.1), and cultured under standard ESC self-renewal conditions. After six passages (12 days), FACS analysis was performed to determine the percentage of GFP+ cells. The percentage of GFP+ cells should remain ∼80% if an shRNA does not affect ESC self-renewal.
Colony-Formation Assays
Colony-formation assays were conducted as previously described (Ang et al., 2011). In brief, Jmjd1c or control shRNAs were introduced into J1 ESCs by lentivirus-mediated infection. Three days later, cells were trypsinized and seeded at a density of 2,000 cells per 6-cm dish to form ESC colonies. After 7 days, colonies were stained using the VECTOR Blue Alkaline Phosphatase Substrate Kit (Vector Laboratories) and counted.
Chromatin Immunoprecipitation Assays
ChIP assays were carried out as previously described with some modifications (Li et al., 2010). Detailed procedures are provided in Supplemental Experimental Procedures. The primers for ChIP-qPCR are listed in Table S2.
Somatic Cell Reprogramming
Somatic cell reprogramming was induced as previously described (Sun et al., 2016). Detailed procedures are provided in Supplemental Experimental Procedures.
Microarray Analysis
Microarray was carried out with the Affymetrix GeneChip Mouse Genome 430 2.0 Arrays. Detailed procedures are provided in Supplemental Experimental Procedures.
Statistical Analysis
Data are presented as the mean ± SD and analyzed by the unpaired Student's t test. p Values of ≤0.05 were considered statistically significant.
Author Contributions
F.X. and Y.J. designed the study, interpreted the data, and wrote the manuscript; F.X., B.L., J.H., S.L., H.Z., M.S., and J.G. performed the experiments and analyzed the data.
Acknowledgments
We thank Makoto Tachibana from the University of Tokushima for generously providing the pCAG-Jmjd1c-FLAG-IP plasmid and the B6.Cg-Jmjd1c (RBRC06532) mice; Jinsong Li from the Institute of Biochemistry and Cell Biology, CAS, for providing TgOG2 Oct4-GFP transgenic mice; Guoliang Xu from the Institute of Biochemistry and Cell Biology, CAS, for providing the retroviral plasmids of miR-200s/pMXs; Austin Smith from the University of Cambridge for providing CGR8 ESC lines; Xiaohua Shen from Tsinghua University for providing J1 ESC lines; and Huan Zhang from the Institute of Microbiology, CAS, for data analysis and technical support. This work was supported by grants from the Ministry of Science and Technology of China (2016YFA0100100 and 2013CB966801), the National Natural Science Foundation of China (91419309), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA01010102 and XDB19020100).
Published: August 17, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.07.013.
Accession Numbers
The microarray data from this publication have been submitted to the GEO database with an accession number GEO: GSE89709.
Supplemental Information
References
- Ang Y.S., Tsai S.Y., Lee D.F., Monk J., Su J., Ratnakumar K., Ding J., Ge Y., Darr H., Chang B. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland M.J., Nazor K.L., Loring J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014;115:311–324. doi: 10.1161/CIRCRESAHA.115.301517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro U., Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Cell Stem Cell Editorial Team 10 questions: clinical outlook for iPSCs. Cell Stem Cell. 2016;18:170–173. doi: 10.1016/j.stem.2016.01.023. [DOI] [PubMed] [Google Scholar]
- Chambers J.C., Zhang W., Sehmi J., Li X., Wass M.N., Van der Harst P., Holm H., Sanna S., Kavousi M., Baumeister S.E. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011;43:1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Liu H., Liu J., Qi J., Wei B., Yang J., Liang H., Chen Y., Chen J., Wu Y. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- Chen M., Zhu N., Liu X., Laurent B., Tang Z., Eng R., Shi Y., Armstrong S.A., Roeder R.G. JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors. Genes Dev. 2015;29:2123–2139. doi: 10.1101/gad.267278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry A.B., Daley G.Q. Reprogramming cellular identity for regenerative medicine. Cell. 2012;148:1110–1122. doi: 10.1016/j.cell.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Ruggiero D., Sorice R., Song C., Nutile T., Vernon Smith A., Concas M.P., Traglia M., Barbieri C., Ndiaye N.C. Six novel loci associated with circulating VEGF levels identified by a meta-analysis of genome-wide association studies. PLoS Genet. 2016;12:e1005874. doi: 10.1371/journal.pgen.1005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Y., Ding J., Theunissen T.W., Faiola F., Hore T.A., Shliaha P.V., Fidalgo M., Saunders A., Lawrence M., Dietmann S. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–374. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P.P., Shao Z., Beyaz S., Apostolou E., Pinello L., De Los Angeles A., O'Brien K., Atsma J.M., Fujiwara Y., Nguyen M. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell. 2014;53:32–48. doi: 10.1016/j.molcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Los Angeles A., Ferrari F., Xi R., Fujiwara Y., Benvenisty N., Deng H., Hochedlinger K., Jaenisch R., Lee S., Leitch H.G. Hallmarks of pluripotency. Nature. 2015;525:469–478. doi: 10.1038/nature15515. [DOI] [PubMed] [Google Scholar]
- Ding J., Xu H., Faiola F., Ma'ayan A., Wang J. Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res. 2012;22:155–167. doi: 10.1038/cr.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Huang X., Shao N., Zhou H., Lee D.F., Faiola F., Fidalgo M., Guallar D., Saunders A., Shliaha P.V. Tex10 coordinates epigenetic control of super-enhancer activity in pluripotency and reprogramming. Cell Stem Cell. 2015;16:653–668. doi: 10.1016/j.stem.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M.J., Kaufman M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Feng X., Wang Z., Fillmore R., Xi Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014;344:166–173. doi: 10.1016/j.canlet.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas G., Vlachos I.S., Zagganas K., Vergoulis T., Paraskevopoulou M.D., Kanellos I., Tsanakas P., Dellis D., Fevgas A., Dalamagas T. DIANA-miRGen v3.0: accurate characterization of microRNA promoters and their regulators. Nucleic Acids Res. 2016;44:D190–D195. doi: 10.1093/nar/gkv1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve T.S., Judson R.L., Blelloch R. microRNA control of mouse and human pluripotent stem cell behavior. Annu. Rev. Cell Dev. Biol. 2013;29:213–239. doi: 10.1146/annurev-cellbio-101512-122343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B., Lee M.G. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013;3:39. doi: 10.1186/2045-3701-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J.A., Surani M.A. Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell. 2014;15:416–430. doi: 10.1016/j.stem.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Hazzalin C.A., Mahadevan L.C. MAPK-regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell Biol. 2002;3:30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- He J., Shen L., Wan M., Taranova O., Wu H., Zhang Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013;15:373–384. doi: 10.1038/ncb2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Zhang L., Mao S.Q., Li Z., Chen J., Zhang R.R., Wu H.P., Gao J., Guo F., Liu W. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell. 2014;14:512–522. doi: 10.1016/j.stem.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Huang G., Ye S., Zhou X., Liu D., Ying Q.L. Molecular basis of embryonic stem cell self-renewal: from signaling pathways to pluripotency network. Cell. Mol. Life Sci. 2015;72:1741–1757. doi: 10.1007/s00018-015-1833-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T., Tachibana M., Magoori K., Kudo H., Tanaka T., Okamura M., Naito M., Kodama T., Shinkai Y., Sakai J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells. 2009;14:991–1001. doi: 10.1111/j.1365-2443.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- Ivanova N., Dobrin R., Lu R., Kotenko I., Levorse J., DeCoste C., Schafer X., Lun Y., Lemischka I.R. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- Kim S.M., Kim J.Y., Choe N.W., Cho I.H., Kim J.R., Kim D.W., Seol J.E., Lee S.E., Kook H., Nam K.I. Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res. 2010;38:6389–6403. doi: 10.1093/nar/gkq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kunath T., Saba-El-Leil M.K., Almousailleakh M., Wray J., Meloche S., Smith A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- Kuroki S., Akiyoshi M., Tokura M., Miyachi H., Nakai Y., Kimura H., Shinkai Y., Tachibana M. JMJD1C, a JmjC domain-containing protein, is required for long-term maintenance of male germ cells in mice. Biol. Reprod. 2013;89 doi: 10.1095/biolreprod.113.108597. [DOI] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent B., Ruitu L., Murn J., Hempel K., Ferrao R., Xiang Y., Liu S., Garcia B.A., Wu H., Wu F. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell. 2015;57:957–970. doi: 10.1016/j.molcel.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.F., Su J., Sevilla A., Gingold J., Schaniel C., Lemischka I.R. Combining competition assays with genetic complementation strategies to dissect mouse embryonic stem cell self-renewal and pluripotency. Nat. Protoc. 2012;7:729–748. doi: 10.1038/nprot.2012.018. [DOI] [PubMed] [Google Scholar]
- Lessard J.A., Crabtree G.R. Chromatin regulatory mechanisms in pluripotency. Annu. Rev. Cell Dev. Biol. 2010;26:503–532. doi: 10.1146/annurev-cellbio-051809-102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li L., Sun L., Gao F., Jiang J., Yang Y., Li C., Gu J., Wei Z., Yang A., Lu R. Stk40 links the pluripotency factor Oct4 to the Erk/MAPK pathway and controls extraembryonic endoderm differentiation. Proc. Natl. Acad. Sci. USA. 2010;107:1402–1407. doi: 10.1073/pnas.0905657107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zhu L., Yang A., Lin J., Tang F., Jin S., Wei Z., Li J., Jin Y. Calcineurin-NFAT signaling critically regulates early lineage specification in mouse embryonic stem cells and embryos. Cell Stem Cell. 2011;8:46–58. doi: 10.1016/j.stem.2010.11.027. [DOI] [PubMed] [Google Scholar]
- Liang G., Zhang Y. Embryonic stem cell and induced pluripotent stem cell: an epigenetic perspective. Cell Res. 2013;23:49–69. doi: 10.1038/cr.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Han Q., Peng T., Peng M., Wei B., Li D., Wang X., Yu S., Yang J., Cao S. The oncogene c-Jun impedes somatic cell reprogramming. Nat. Cell Biol. 2015;17:856–867. doi: 10.1038/ncb3193. [DOI] [PubMed] [Google Scholar]
- Liu N., Zhang Z., Wu H., Jiang Y., Meng L., Xiong J., Zhao Z., Zhou X., Li J., Li H. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 2015;29:379–393. doi: 10.1101/gad.254425.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y.H., Zhang W., Chen X., George J., Ng H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane L.A., Murphy P.R. MicroRNA: biogenesis, function and role in cancer. Curr. Genomics. 2010;11:537–561. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E., Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 2006;7:540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N., Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- Nakajima R., Okano H., Noce T. JMJD1C exhibits multiple functions in epigenetic regulation during spermatogenesis. PLoS One. 2016;11:e0163466. doi: 10.1371/journal.pone.0163466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng H.H., Surani M.A. The transcriptional and signalling networks of pluripotency. Nat. Cell Biol. 2011;13:490–496. doi: 10.1038/ncb0511-490. [DOI] [PubMed] [Google Scholar]
- Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- Pedersen M.T., Agger K., Laugesen A., Johansen J.V., Cloos P.A., Christensen J., Helin K. The demethylase JMJD2C localizes to H3K4me3-positive transcription start sites and is dispensable for embryonic development. Mol. Cell. Biol. 2014;34:1031–1045. doi: 10.1128/MCB.00864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen M.T., Kooistra S.M., Radzisheuskaya A., Laugesen A., Johansen J.V., Hayward D.G., Nilsson J., Agger K., Helin K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016;35:1550–1564. doi: 10.15252/embj.201593317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J., Shi G., Jia Y., Li J., Wu M., Li J., Dong S., Wong J. The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res. 2010;20:908–918. doi: 10.1038/cr.2010.81. [DOI] [PubMed] [Google Scholar]
- Rauch N., Rukhlenko O.S., Kolch W., Kholodenko B.N. MAPK kinase signalling dynamics regulate cell fate decisions and drug resistance. Curr. Opin. Struct. Biol. 2016;41:151–158. doi: 10.1016/j.sbi.2016.07.019. [DOI] [PubMed] [Google Scholar]
- Saez M.A., Fernandez-Rodriguez J., Moutinho C., Sanchez-Mut J.V., Gomez A., Vidal E., Petazzi P., Szczesna K., Lopez-Serra P., Lucariello M. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet. Med. 2016;18:378–385. doi: 10.1038/gim.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikhibrahim Z., Wernert N. ETS transcription factors and prostate cancer: the role of the family prototype ETS-1 (review) Int. J. Oncol. 2012;40:1748–1754. doi: 10.3892/ijo.2012.1380. [DOI] [PubMed] [Google Scholar]
- Shakya A., Callister C., Goren A., Yosef N., Garg N., Khoddami V., Nix D., Regev A., Tantin D. Pluripotency transcription factor oct4 mediates stepwise nucleosome demethylation and depletion. Mol. Cell. Biol. 2015;35:1014–1025. doi: 10.1128/MCB.01105-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroczynska P., Cruickshank V.A., Bukowski J.P., Miyagi S., Bagger F.O., Walfridsson J., Schuster M.B., Porse B., Helin K. shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood. 2014;123:1870–1882. doi: 10.1182/blood-2013-08-522094. [DOI] [PubMed] [Google Scholar]
- Stupack D.G., Cheresh D.A. Get a ligand, get a life: integrins, signaling and cell survival. J. Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- Sun M., Liao B., Tao Y., Chen H., Xiao F., Gu J., Gao S., Jin Y. Calcineurin-NFAT signaling controls somatic cell reprogramming in a stage-dependent manner. J. Cell. Physiol. 2016;231:1151–1162. doi: 10.1002/jcp.25212. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tan T.K., Zheng G., Hsu T.T., Wang Y., Lee V.W., Tian X., Wang Y., Cao Q., Wang Y., Harris D.C. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am. J. Pathol. 2010;176:1256–1270. doi: 10.2353/ajpath.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K., Okada Y., Kallin E.M., Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg D.L., Snoek T., Mullin N.P., Yates A., Bezstarosti K., Demmers J., Chambers I., Poot R.A. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Guo X., Hong W., Liu Q., Wei T., Lu C., Gao L., Ye D., Zhou Y., Chen J. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl. Acad. Sci. USA. 2013;110:2858–2863. doi: 10.1073/pnas.1212769110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.H., Yamaguchi S., Burstein M., Terashima K., Ng H.K., Nakamura H., He Z.X., Suzuki T., Nishikawa R., Natsume A. Association of rare germline variants of Jmjd1c with intracranial germ cell tumors. Neuro Oncol. 2014;16:38. [Google Scholar]
- Wang L.H., Yamaguchi S., Burstein M.D., Terashima K., Chang K., Ng H.K., Nakamura H., He Z.X., Doddapaneni H., Lewis L. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. 2014;511:241–245. doi: 10.1038/nature13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B., Wu H., Shinkai Y., Irizarry R.A., Feinberg A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Rodriguez-Aznar E., Yabuta N., Owen R.J., Mingot J.M., Nojima H., Nieto M.A., Longmore G.D. Lats2 kinase potentiates Snail1 activity by promoting nuclear retention upon phosphorylation. EMBO J. 2012;31:29–43. doi: 10.1038/emboj.2011.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.S., He C. Fate by RNA methylation: m6A steers stem cell pluripotency. Genome Biol. 2015;16:43. doi: 10.1186/s13059-015-0609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Chen M., Eng R., DeJong J., Sinha A.U., Rahnamay N.F., Koche R., Al-Shahrour F., Minehart J.C., Chen C.W. MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J. Clin. Invest. 2016;126:997–1011. doi: 10.1172/JCI82978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.