

Abstract
Pathogenic variants in genes encoding components of the BAF chromatin remodeling complex have been associated with intellectual disability syndromes. We identified heterozygous, novel variants in ACTL6A, a gene encoding a component of the BAF complex, in three subjects with varying degrees of intellectual disability. Two subjects have missense variants affecting highly conserved amino acid residues within the actin-like domain. Missense mutations in the homologous region in yeast actin were previously reported to be dominant lethal and were associated with impaired binding of the human ACTL6A to β-actin and BRG1. A third subject has a splicing variant that creates an in-frame deletion. Our findings suggest that the variants identified in our subjects may have a deleterious effect on the function of the protein by disturbing the integrity of the BAF complex. Thus, ACTL6A gene mutation analysis should be considered in patients with intellectual disability, learning disabilities or developmental language disorder.
Keywords: ACTL6A, BAF complex, speech delay, intellectual disability
Heterozygous pathogenic variants in genes encoding components of the BRG1-associated factor (BAF) complex, are causally related to syndromic and non-syndromic conditions with intellectual disability (Kosho, et al., 2014; López and Wood, 2015; Santen, et al., 2012b). Coffin-Siris syndrome (CSS [MIM# 135900, 614607, 614608, 614609, 616938]), characterized by developmental delay, coarse facial features, and hypoplastic fifth phalanges (Coffin and Siris, 1970), is associated with pathogenic variants in genes encoding ARID1B (MIM# 614556), ARID1A (MIM# 603024), SMARCB1 (MIM# 601607), SMARCA4 (MIM# 603254), SMARCE1 (MIM# 603111), and SOX11 (MIM# 600898) (Santen, et al., 2012a; Tsurusaki, et al., 2014; Tsurusaki, et al., 2012). Mutations in SMARCA2 (MIM# 600014) are thought to result in a phenotype better described as Nicolaides-Baraitser syndrome (Nicolaides and Baraitser, 1993), with overlapping clinical features (Mari, et al., 2015; Van Houdt, et al., 2012; Wolff, et al., 2012). Haploinsufficiency and loss-of-function variants in ARID1B are also found in 1% of individuals with intellectual disability without distinguishing physical features (Hoyer, et al., 2012). More recently, pathogenic variants in other chromatin remodeling proteins, or factors that closely interact with the BAF complex were implicated in autism and intellectual disability, including ADNP (MIM# 611386)(Helsmoortel, et al., 2014), ARID2 (MIM# 609539)(Shang, et al., 2015), DYRK1A (MIM# 600855) (Courcet, et al., 2012; Ji, et al., 2015; van Bon, et al., 2016), BCL11A (MIM# 606557)(Dias, et al., 2016) and BAZ1A (MIM# 605680)(Zaghlool, et al., 2016). With the advent of next-generation sequencing new intellectual disability genes are being discovered, and many of these genes play a role in neurodevelopment via epigenetic regulation of gene expression (Casanova, et al., 2016; López and Wood, 2015; Son and Crabtree, 2014).
We present clinical and molecular data from three unrelated subjects with developmental disability primarily affecting language. Whole exome sequencing (WES) identified pathogenic variants in ACTL6A (MIM# 604958), which encodes a member of the BAF complex that has not been previously associated with intellectual disability syndromes. A summary of the clinical and molecular findings in all three subjects is provided in Figure 1 and in the Supp. Table S1.
Figure 1. Clinical photographs of subjects and summary of ACTL6A pathogenic variants.
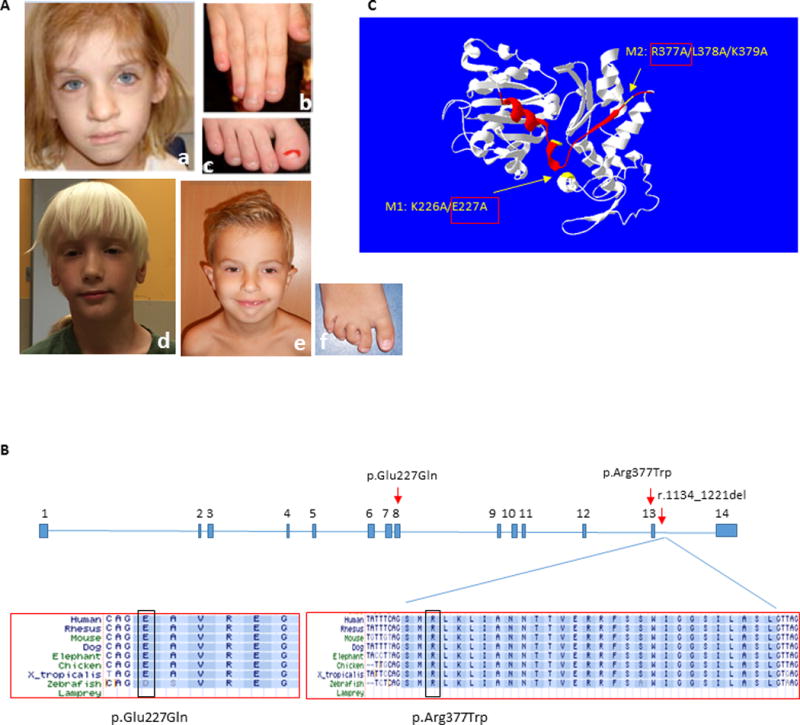
(A) Subject 1 at age 7 years, demonstrating coarse facial features with broad nasal tip (a), broad fingers and toes with dystrophic nails, and short distal phalanges (b, c); Subject 2 at age 6 years, demonstrating elongated face with large forehead, narrow eyelids, broad nasal tip and poorly developed philtrum ridge (d); and Subject 3 at age 6 years, showing prominent high forehead, low set everted ears, narrow eyelids, broad nasal tip, and small chin (e). Note digital anomalies, including overriding second toe, clinodactyly of 3rd–5th toes and sandal gap (f). The photographs of Subject 1 were reproduced with permission from the American Journal of Medical Genetics, Part A (Brautbar, et al., 2009).
(B) Annotation of the two amino acid residues in exons 8 and 13 affected in Subjects 1 and 2, and the splicing variant causing in-frame deletion of exon 13 in Subject 3. Conservation across species is shown for the three variants.
(C) ACTL6A protein structure model as predicted by I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER) (Zhang, 2008) and visualized using Swiss-Pdb viewer (http://spdbv.vital-it.ch/) (Guex and Peitsch, 1997), showing localization of the mutated amino acids in Subjects 1 and 2 (yellow arrows), and the deleted exon 13 in Subject 3 (red ribbon). Previous study (Nishimoto, et al., 2012) demonstrated that M1 and M2 mutants of human ACTL6A exhibit impaired binding capacity to beta actin (ACTB) and BRG1/SMARCA4, and thus disrupt ACTL6A recruitment to the BAF complex. This data supports a deleterious effect for p.Glu227Gln (E227Q/M1) and p.Arg377Trp (R377W/M2) on ACTL6A protein function.
Subject 1 is a 15-year-old female that was described in detail in a prior publication (Brautbar, et al., 2009), although she did not have an identified genetic etiology. Briefly, she presented with developmental delay that involved speech/language difficulties and inattention problems. She had to repeat first grade and continued to have significant learning difficulties in school. She had atrial septal defect and cleft mitral valve that required surgical repair. Her medical history was otherwise significant for torticollis, gastro-esophageal reflux, recurrent ear infections and urinary bladder irritability – all of which resolved during childhood. She also underwent a surgical repair of bilateral inguinal and umbilical hernias. She was evaluated for suspected autonomic dysfunction due to chronic fatigue, exercise intolerance and episodes of syncope. She was small for age at birth (weight and length at the 5th centile), but her current growth parameters are within the normal limits. She has dysmorphic features that include coarse facies, bushy eyebrows, prominent ears and broad nasal tip (Figure 1). She has hypoplastic nails on multiple digits, unusually broad thumbs and mildly persistent fetal fingertip pads. Neurological examination was non-focal. The patient’s chromosome analysis and chromosome microarray analysis were unremarkable. Clinically, her diagnosis was considered to be a potential mild CSS versus brachymorphism-onychodysplasia-dysphalangism syndrome (MIM# 113477) (Brautbar, et al., 2009). WES analysis revealed a de novo, heterozygous variant, NC_000003.11:g. 179304340C>T, in exon 13 of ACTL6A. This variant results in amino acid substitution at position 377 (p.Arg377Trp, Figure 1).
Subject 2 was identified via review of the Baylor Genetics WES database. This is a 6-year-old male with developmental delay, primarily affecting speech, and attention deficit hyperactivity disorder (ADHD) with behavioral problems (aggressive behavior, impulsiveness and tantrums, and sleep problems). Pregnancy was complicated by intrauterine growth restriction. He was born prematurely at 31 weeks gestation, and birth weight was 820 grams (2nd centile). He walked at 15 months. He said his first words at 12 months but started using phrases at 3 years of age and had articulation problems. He has poor fine motor skills and sensory integration deficits. Psychological evaluation at 5 years of age revealed borderline intelligence, with both receptive and expressive language skills below the mean for age. He has a history of hyperopia, recurrent respiratory infections, asthma and allergies. Early in life he had failure to thrive, gastro-esophageal reflux and constipation which resolved. He underwent bilateral inguinal hernia and umbilical hernia repair. The family history is significant for learning disabilities in both parents. His growth parameters on most recent exam were within the normal limits. The physical examination is notable for dysmorphic features, which include narrow face with prominent forehead, long eyelashes and poorly developed philtrum ridge. He has 5th finger clinodactyly, partial 3–4 finger syndactyly and 2–3 toe syndactyly. Neurological exam was normal. The patient’s chromosome analysis, SNP array and fragile X (MIM# 300624) testing were normal. He had negative testing for Russell-Silver syndrome (MIM# 180860). Transferrin isoelectric focusing screen for congenital disorders of glycosylation was normal. Newborn screen and other metabolic work up was normal, including plasma amino acids, urine organic acids, urine acylglycines, acylcarnitine analysis, and urine & plasma levels of creatine/guanidinoacetate. Brain MRI was normal. Clinical WES was done at Baylor Genetics. Research analysis of the data revealed a heterozygous variant, NC_000003.11:g.179294612G>C, in exon 8 of ACTL6A that results in amino acid substitution at position 227 (p.Glu227Gln, Figure 1). The mother tested negative for this variant. The father, who is not available for testing, has a history of ADHD and learning disabilities – thus this variant could be potentially de novo, or inherited from an affected parent.
Subject 3 was identified via GeneMatcher search (https://genematcher.org) (Sobreira, et al., 2015). This is a 6-year-old male with cognitive and language delay. He was born prematurely at 35 weeks gestation, and birth weight was 2635 grams (60th centile). He had prenatal diagnosis of bilateral hydronephrosis due to urethral valve obstruction for which he underwent a surgical repair after birth. He was also noted to have glandular hypospadias and laryngomalacia. Perinatal course was complicated by feeding difficulties, and he was hospitalized during the first 4 weeks of life. As for his development, he started walking at the age of 15 months and spoke his first words at 12 months. However, he required speech therapy for expressive and receptive language deficits. He had neurodevelopmental evaluation at 6 years of age and was found to have moderate cognitive impairment (IQ 56). In school, he is attending special education classes. He had extensive neuropediatric evaluation due to exercise intolerance (leg pain after minor exercise), which remained unexplained. Remarkably, he was noted to have a high pain threshold. The family history is significant for learning disabilities in the mother. His older brother and sister had some speech delay initially but do not have cognitive delays and attend regular education classes. His older sister also had epilepsy. Physical examination at the age of 6 years showed normal growth parameters, and dysmorphic facial features that include a prominent high forehead, low set everted ears, narrow eyelids with mild hypertelorism, small chin and a normal palate (Figure 1). He had flat feet with overriding 2nd toes and clinodactyly of the 3rd, 4th and 5th toes. He had joint hypermobility (Beighton score 6 out of 8), hypospadias with normally descended testes and multiple café-au-lait macules on his skin but without freckling in axillary and inguinal regions. He had a normal EEG. Several genetic tests were performed, including metabolic work up, SNP array, and DNA analysis of FMR1, NF1 and SPRED1, which all appeared normal. WES analysis revealed a de novo, heterozygous variant in ACTL6A. This variant, NC_000003.11 (NM_004301.3): c.1209+1G>C in intron 13, is predicted to affect splicing (Figure 1). To study the effect of the splicing variant on the ACTL6A transcript, total RNA was isolated from subject 3 and control lymphoblastoid cells, PCR-amplified, cloned and sequenced (Figure 2 A). Subject 3 cDNA sequencing results were aligned with control and reference ACTL6A mRNA sequence (UCSC genome browser, https://genome.ucsc.edu/). Sequencing revealed abnormal splicing that creates an in-frame deletion of exon 13. Western blot analysis detected decreased amount of the ACTL6A protein in subject 3 cell lysate, supporting a loss-of-function, or haploinsufficiency mechanism of this variant (Figure 2 B). Silencing of ACTL6A in HeLa cells resulted in decreased stability of the BAF complex (Nishimoto, et al., 2012). To evaluate the effect of ACTL6A deficiency on the BAF complex formation in subject 3 cells, we studied the interaction of ACTL6A and BRG1 by co-immunoprecipitation. The results demonstrate reduced binding of ACTL6A and BRG1 in subject 3 lymphoblasts, compared to control cells (Figure 2C).
Figure 2. RNA, protein and cell cycle studies in Subject 3 lymphoblasts encompassing a heterozygous ACTL6A splicing variant.
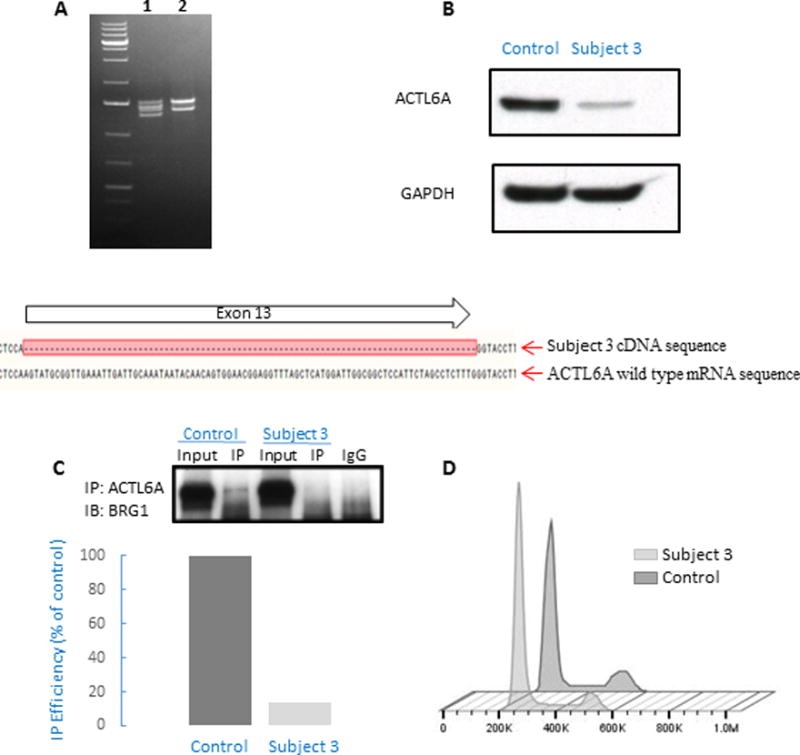
(A) RT-PCR amplification of the full-length cDNA from control sample (2) shows two transcripts due to alternative splicing at ACTL6A exon 1, while amplification of Subject 3 cDNA (1) shows four transcripts. PCR-cloning and sequencing, followed by alignment of the sequence results with ACTL6A mRNA sequence demonstrates that two of the four transcripts in Subject 3 correspond to expression of the two isoforms of wild-type allele, and two transcripts correspond to expression of the two isoforms of mutated allele containing an in-frame deletion of the full length of exon 13. (B) Western blot analysis of ACTL6A protein expression in Subject 3 and control cells, revealing reduced protein expression in Subject 3 cells that is suggestive of a haploinsufficiency mechanism.
(C) Immunoprecipitation (IP) with anti-ACTL6A antibody, showing decreased interaction with BRG1 in subject cells (Subject 3) compared to unrelated healthy control (Control). There was no binding to negative control (rabbit IgG). Immunoblot (IB) was quantified (using ImageJ program), showing 85% reduction in co-IP efficiency, presented here as % of control. The reduction of co-IP efficiency was repeated in two other independent experiments (showing 70% and 20% reduction, respectively). (D) Cell cycle analysis of cells from Subject 3 and control cells. As compared to control, a smaller fraction of cells from subject 3 resided in G2 phase (10.5 % vs. 16.8 % in control samples). This trend was observed in two independent experiments and is consistent with previous reports describing cell cycle perturbation in ACTL6A-depleted cells (Lee, et al., 2007).
Mutations in genes encoding components of the BAF complex, or interacting proteins, have been associated with CSS (Santen, et al., 2012a; Tsurusaki, et al.), and intellectual disability disorders (Hoyer, et al., 2012; Son and Crabtree, 2014). In the present report, we review clinical information and molecular data from three subjects with speech delay and varying degrees of developmental disability. We identified novel, heterozygous variants in ACTL6A, a gene encoding a component of the BAF complex that has not been previously associated with intellectual disability. The variants that were found in ACTL6A include two amino acid substitutions that involve highly conserved residues, and a splice variant (Figure 1). Two of the variants were confirmed to be de novo, and in one patient inheritance is unknown (due to unavailability of paternal testing). None of the variants has been previously reported in the Exome Aggregation Consortium database (ExAC, Cambridge, MA, http://exac.broadinstitute.org/) (Lek, et al., 2016) and the 1000 Genomes database (http://browser.1000genomes.org/) (Auton, et al., 2015). The variant found in subject 2 (p.Glu227Gln) was reported once in gnomAD database (GnomAD, http://gnomad.broadinstitute.org) (Lek, et al., 2016) with an allele frequency of 4.06e-6. Since Trio WES was not performed, we were unable to identify other de novo variants which may explain the phenotype seen in these subjects. We therefore cannot state beyond doubt that the ACTL6A variants are disease-causing vs. incidental findings in these patients. Importantly, WES analysis did not detect pathogenic variants in other genes that are known to be associated with intellectual disability. We reviewed DECIPHER (https://decipher.sanger.ac.uk) (Firth, et al., 2009) cases that involve ACTL6A. These include 13 gains and only 1 case with a deletion, all larger than 5 Mb and include 20 genes or more. Thus, data is lacking to directly support the contribution of ACTL6A solely in the pathogenicity of these copy number variants.
ACTL6A (actin-like 6a, also known as BAF53a/INO80K/Arp4) is a scaffold component of the BAF complex. It appears essential in maintaining the undifferentiated status of embryonic stem cells (Lu, et al., 2015) and epidermal progenitor cells (Bao, et al., 2013), and is implicated in neuronal and hematopoietic development (Krasteva, et al., 2012; Staahl, et al., 2013). Homozygous deletion of Actl6a in mice results in early embryonic lethality (Krasteva, et al., 2012). Conditional knock out of the gene within the hematopoietic lineage caused bone marrow failure and early death at about 3 weeks post deletion (Krasteva, et al., 2012). Knockdown of ACTL6A by siRNA in cells affected histone methylation and induced transcription of several cell cycle regulators (including MDM2, p21 and cyclin D1), resulting in cell cycle arrest (Lee, et al., 2007). We preformed cell cycle study in subject 3 and control lymphoblastoid cell lines. We observed a trend of cells from subject 3 to be retained in G1/S phase, with a smaller fraction of cells residing in G2 phase compared to control (9–10% of subject 3 cells in G2 phase, compared to 15–16.8% of control cells in G2 phase in two independent experiments, Figure 2 D). This finding is consistent with previous observation in ACTL6A-depleted cells (Lee, et al., 2007), and supports a mechanism of loss of function in our patient.
During epidermal differentiation, ACTL6A is significantly down-regulated, and conditional deletion is associated with loss of functional progenitor cells and tissue hypoplasia (Bao, et al., 2013). In neural development, ACTL6A substitution with ACTL6B (MIM# 612458) dictates a switch from neural progenitor npBAF complex to neural specific nBAF complex, to allow proper mitotic exit and neuronal differentiation (Staahl, et al., 2013). Altogether, these findings suggest that during development ACTL6A serves as an important regulator of progenitor/stem cell function.
ACTL6A was found to be highly homologous to actin; however, unlike actin it does not have ATPase activity and does not form actin-like filaments (Fenn, et al., 2011; Zhao, et al., 1998). Actin and actin-related proteins are involved in epigenetic regulation of transcription and chromatin dynamics (Chen and Shen, 2007; Olave, et al., 2002; Shen, et al., 2003). De novo heterozygous pathogenic variants in actin genes ACTB (MIM# 102630) and ACTG1 (MIM# 102560) are associated with Baraitser-Winter syndrome (MIM# 243310) (Baraitser and Winter, 1988), characterized by microcephaly, ocular and brain malformations, and intellectual disability (Di Donato, et al., 2014; Johnston, et al., 2013; Rivière, et al., 2012). ACTL6A directly interacts with beta actin (ACTB) and SMARCA4/BRG1 while enhancing chromatin binding and ATPase activity of the BAF complex (Nishimoto, et al., 2012; Zhao, et al., 1998).
WES identified three heterozygous variants in ACTL6A, two of which were confirmed to be de novo in our patients. The two missense variants: p.Glu227Gln and p.Arg377Trp, involve highly conserved amino acid residues and are predicted to be damaging by Polyphen2 and SIFT. In addition to these findings, analysis of the variants via Mutation Taster program, PhyloP score and the Cadd score, supports a deleterious effect of the identified variants (variant scores are detailed in Supp. Table S2). These two amino acids reside in a region with structural similarity to actin, where induced missense mutations were found to be dominant lethal in yeast (Wertman, et al., 1992). Interestingly, a previously published study in cells transiently transfected with human ACTL6A showed that introducing mutations into these same two residues found in our subjects impaired binding of the ACTL6A protein to ACTB, to SMARCA4/BRG1 (Nishimoto, et al., 2012), and to the TIP60 (MIM# 601409) chromatin remodeling complex (Lu, et al., 2015; Nishimoto, et al., 2012). A third subject was found to have a de novo splicing variant in intron 13. RNA and protein studies using a lymphoblastoid cell line derived from this subject showed abnormal splicing that creates an in-frame deletion of exon 13 (Figure 2A), resulting in reduced amount of ACTL6A protein (Figure 2B). Since all pathogenic variants are heterozygous and are predicted to result in loss of function, we hypothesize that haploinsufficiency is the mechanism underlying this disorder. The high haploinsufficiency score (HI index=3.57) reported in DECIPHER database (Firth, et al., 2009; Huang, et al., 2010) for ACTL6A supports this prediction.
Clinically, our subjects share a number of phenotypic features (Supp. Table S1). They all presented with developmental delay and variable degree of learning disabilities. Subjects 1 and 2 on the milder end of the spectrum, had inattention and learning problems with neurocognitive assessment at the low-normal range. Consistent with this observation, the variant that was identified in subject 2 was reported once in gnomAD database, which includes sequencing results from individuals that are not affected by severe pediatric disorders. On the other hand, subject 3 had a splicing variant causing a deletion of the full length of exon 13 that likely leads to loss of function of the mutated allele as demonstrated by western blot (Figure 2B), and presented with more severe cognitive impairment of moderate degree, compared to the other two subjects with mild intellectual disability and missense variants. The probability of loss-of-function intolerance (pLI) score reported in ExAC database is 0.99, predicting that ACTL6A is highly intolerant of loss-of-function variation. Potentially, variants that have less severe functional effects may give rise to the relatively milder neurocognitive phenotype, and may lead to under-ascertainment. In addition to delayed development, speech delay and learning disabilities, a number of organ systems were affected in some of the subjects, with cardiac defects in 1/3 subjects, genitourinary anomalies in 2/3 subjects, hernias in 2/3 subjects and finger and toe abnormalities in 3/3 subjects. Our subjects were born small for age or had failure to thrive early in life, but current growth parameters are within the normal limits. To further delineate the clinical spectrum associated with pathogenic variants of ACTL6A we established the website: www.humandiseasegenes.com/ACTL6A to collect detailed clinical information of additional individuals that will be identified over time.
In conclusion, we report here heterozygous pathogenic variants in ACTL6A in three subjects with varying degrees of intellectual disability. Subjects exhibit common physical features including genitourinary and skeletal defects. ACTL6A is a BAF-related gene that plays an essential role in neurodevelopment. Two of three variants localize to exon 13, shown here and in previous functional studies to affect protein-protein interaction at the BAF complex. We suggest that sequencing of ACTL6A should be considered in the diagnostic work-up of developmental delay and learning disabilities.
Supplementary Material
Acknowledgments
We thank the patients and their families for participation in our study. We thank Brian Dawson for his help with preparation of figures for this manuscript.
This work was supported by a Genzyme-ACMG Foundation Medical Genetics Training Award in Medical Biochemical Genetics (to R.M.), and Michael Geisman- Osteogenesis Imperfecta Foundation (OIF) Fellowship Award (to R.M.), by the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, NIH U54HG003273 (R.A.G.), NIH U54HG006542 (R.A.G.), NIH T32 GM07526 (M.J.), NIH K08DK106453 (L.C.B.), and NICHD P01 HD070394 (B.L), and by grants from the Netherlands Organization for Health Research and Development (917-86-319 to B.B.A.d.V., 912-12-109 to B.B.A.d.V). This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom.
Footnotes
Conflicts of Interest
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical testing done at Baylor Genetics Laboratories.
References
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, Consortium GP A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Tang J, Lopez-Pajares V, Tao S, Qu K, Crabtree GR, Khavari PA. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell. 2013;12:193–203. doi: 10.1016/j.stem.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraitser M, Winter RM. Iris coloboma, ptosis, hypertelorism, and mental retardation: a new syndrome. J Med Genet. 1988;25:41–3. doi: 10.1136/jmg.25.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautbar A, Ragsdale J, Shinawi M. Is this the Coffin-Siris syndrome or the BOD syndrome? Am J Med Genet A. 2009;149A:559–62. doi: 10.1002/ajmg.a.32671. [DOI] [PubMed] [Google Scholar]
- Casanova EL, Sharp JL, Chakraborty H, Sumi NS, Casanova MF. Genes with high penetrance for syndromic and non-syndromic autism typically function within the nucleus and regulate gene expression. Mol Autism. 2016;7:18. doi: 10.1186/s13229-016-0082-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Shen X. Nuclear actin and actin-related proteins in chromatin dynamics. Curr Opin Cell Biol. 2007;19:326–30. doi: 10.1016/j.ceb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Coffin GS, Siris E. Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child. 1970;119:433–9. doi: 10.1001/archpedi.1970.02100050435009. [DOI] [PubMed] [Google Scholar]
- Courcet JB, Faivre L, Malzac P, Masurel-Paulet A, Lopez E, Callier P, Lambert L, Lemesle M, Thevenon J, Gigot N, Duplomb L, Ragon C, et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J Med Genet. 2012;49:731–6. doi: 10.1136/jmedgenet-2012-101251. [DOI] [PubMed] [Google Scholar]
- Di Donato N, Rump A, Koenig R, Der Kaloustian VM, Halal F, Sonntag K, Krause C, Hackmann K, Hahn G, Schrock E, Verloes A. Severe forms of Baraitser-Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. Eur J Hum Genet. 2014;22:179–83. doi: 10.1038/ejhg.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias C, Estruch SB, Graham SA, McRae J, Sawiak SJ, Hurst JA, Joss SK, Holder SE, Morton JE, Turner C, Thevenon J, Mellul K, et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am J Hum Genet. 2016 doi: 10.1016/j.ajhg.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn S, Breitsprecher D, Gerhold CB, Witte G, Faix J, Hopfner KP. Structural biochemistry of nuclear actin-related proteins 4 and 8 reveals their interaction with actin. EMBO J. 2011;30:2153–66. doi: 10.1038/emboj.2011.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84:524–33. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, Schuurs-Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE, Yntema HG, Bakshi M, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet. 2014;46:380–4. doi: 10.1038/ng.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, Zweier M, Göhring I, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90:565–72. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Lee H, Argiropoulos B, Dorrani N, Mann J, Martinez-Agosto JA, Gomez-Ospina N, Gallant N, Bernstein JA, Hudgins L, Slattery L, Isidor B, et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur J Hum Genet. 2015;23:1473–81. doi: 10.1038/ejhg.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Wen KK, Keppler-Noreuil K, McKane M, Maiers JL, Greiner A, Sapp JC, Demali KA, Rubenstein PA, Biesecker LG, Center NIS. Functional analysis of a de novo ACTB mutation in a patient with atypical Baraitser-Winter syndrome. Hum Mutat. 2013;34:1242–9. doi: 10.1002/humu.22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosho T, Miyake N, Carey JC. Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet. 2014;166C:241–51. doi: 10.1002/ajmg.c.31415. [DOI] [PubMed] [Google Scholar]
- Krasteva V, Buscarlet M, Diaz-Tellez A, Bernard MA, Crabtree GR, Lessard JA. The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood. 2012;120:4720–32. doi: 10.1182/blood-2012-04-427047. [DOI] [PubMed] [Google Scholar]
- Lee K, Kang MJ, Kwon SJ, Kwon YK, Kim KW, Lim JH, Kwon H. Expansion of chromosome territories with chromatin decompaction in BAF53-depleted interphase cells. Mol Biol Cell. 2007;18:4013–23. doi: 10.1091/mbc.E07-05-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López AJ, Wood MA. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci. 2015;9:100. doi: 10.3389/fnbeh.2015.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Fang L, Ouyang B, Zhang X, Zhan S, Feng X, Bai Y, Han X, Kim H, He Q, Wan M, Shi FT, et al. Actl6a protects embryonic stem cells from differentiating into primitive endoderm. Stem Cells. 2015;33:1782–93. doi: 10.1002/stem.2000. [DOI] [PubMed] [Google Scholar]
- Mari F, Marozza A, Mencarelli MA, Lo Rizzo C, Fallerini C, Dosa L, Di Marco C, Carignani G, Baldassarri M, Cianci P, Vivarelli R, Vascotto M, et al. Coffin-Siris and Nicolaides-Baraitser syndromes are a common well recognizable cause of intellectual disability. Brain Dev. 2015;37:527–36. doi: 10.1016/j.braindev.2014.08.009. [DOI] [PubMed] [Google Scholar]
- Nicolaides P, Baraitser M. An unusual syndrome with mental retardation and sparse hair. Clin Dysmorphol. 1993;2:232–6. [PubMed] [Google Scholar]
- Nishimoto N, Watanabe M, Watanabe S, Sugimoto N, Yugawa T, Ikura T, Koiwai O, Kiyono T, Fujita M. Heterocomplex formation by Arp4 and β-actin is involved in the integrity of the Brg1 chromatin remodeling complex. J Cell Sci. 2012;125:3870–82. doi: 10.1242/jcs.104349. [DOI] [PubMed] [Google Scholar]
- Olave IA, Reck-Peterson SL, Crabtree GR. Nuclear actin and actin-related proteins in chromatin remodeling. Annu Rev Biochem. 2002;71:755–81. doi: 10.1146/annurev.biochem.71.110601.135507. [DOI] [PubMed] [Google Scholar]
- Rivière JB, van Bon BW, Hoischen A, Kholmanskikh SS, O’Roak BJ, Gilissen C, Gijsen S, Sullivan CT, Christian SL, Abdul-Rahman OA, Atkin JF, Chassaing N, et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat Genet. 2012;44:440–4. S1–2. doi: 10.1038/ng.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012a;44:379–80. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- Santen GW, Kriek M, van Attikum H. SWI/SNF complex in disorder: SWItching from malignancies to intellectual disability. Epigenetics. 2012b;7:1219–24. doi: 10.4161/epi.22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Cho MT, Retterer K, Folk L, Humberson J, Rohena L, Sidhu A, Saliganan S, Iglesias A, Vitazka P, Juusola J, O’Donnell-Luria AH, et al. Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics. 2015;16:307–14. doi: 10.1007/s10048-015-0454-0. [DOI] [PubMed] [Google Scholar]
- Shen X, Ranallo R, Choi E, Wu C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol Cell. 2003;12:147–55. doi: 10.1016/s1097-2765(03)00264-8. [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son EY, Crabtree GR. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am J Med Genet C Semin Med Genet. 2014;166C:333–49. doi: 10.1002/ajmg.c.31416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staahl BT, Tang J, Wu W, Sun A, Gitler AD, Yoo AS, Crabtree GR. Kinetic analysis of npBAF to nBAF switching reveals exchange of SS18 with CREST and integration with neural developmental pathways. J Neurosci. 2013;33:10348–61. doi: 10.1523/JNEUROSCI.1258-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Koshimizu E, Ohashi H, Phadke S, Kou I, Shiina M, Suzuki T, Okamoto N, Imamura S, Yamashita M, Watanabe S, Yoshiura K, et al. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat Commun. 2014;5:4011. doi: 10.1038/ncomms5011. [DOI] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–8. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- van Bon BW, Coe BP, Bernier R, Green C, Gerdts J, Witherspoon K, Kleefstra T, Willemsen MH, Kumar R, Bosco P, Fichera M, Li D, et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry. 2016;21:126–32. doi: 10.1038/mp.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet. 2012;44:445–9, S1. doi: 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- Wertman KF, Drubin DG, Botstein D. Systematic mutational analysis of the yeast ACT1 gene. Genetics. 1992;132:337–50. doi: 10.1093/genetics/132.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff D, Endele S, Azzarello-Burri S, Hoyer J, Zweier M, Schanze I, Schmitt B, Rauch A, Reis A, Zweier C. In-Frame Deletion and Missense Mutations of the C-Terminal Helicase Domain of SMARCA2 in Three Patients with Nicolaides-Baraitser Syndrome. Mol Syndromol. 2012;2:237–244. doi: 10.1159/000337323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghlool A, Halvardson J, Zhao JJ, Etemadikhah M, Kalushkova A, Konska K, Jernberg-Wiklund H, Thuresson AC, Feuk L. A Role for the Chromatin-Remodeling Factor BAZ1A in Neurodevelopment. Hum Mutat. 2016 doi: 10.1002/humu.23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, Crabtree GR. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 1998;95:625–36. doi: 10.1016/s0092-8674(00)81633-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.