

Abstract
Germline mutations in the tumor‐suppressor gene PTEN predispose to subsets of Cowden syndrome (CS), Bannayan–Riley–Ruvalcaba syndrome, and autism. Evidence‐based classification of PTEN variants as either deleterious or benign is urgently needed for accurate molecular diagnosis and gene‐informed genetic counseling. We studied 34 different germline PTEN intronic variants from 61 CS patients, characterized their PTEN mRNA processing, and analyzed PTEN expression and downstream readouts of P‐AKT and P‐ERK1/2. While we found that many mutations near splice junctions result in exon skipping, we also identified the presence of cryptic splicing that resulted in premature termination or a shift in isoform usage. PTEN protein expression is significantly lower in the group with splicing changes while P‐AKT, but not P‐ERK1/2, is significantly increased. Our observations of these PTEN intronic variants should contribute to the determination of pathogenicity of PTEN intronic variants and aid in genetic counseling.
Keywords: alternative splicing, BRRS, Cowden syndrome, cryptic splice sites, exon skipping, mutation, PTEN, transcription isoform
Cowden syndrome (CS) is an uncommon, but difficult to recognize, disorder that results in hamartomas and an increased risk of breast, thyroid, endometrial, and renal cancers (Ngeow & Eng, 2015; Tan et al., 2012). The majority of CS patients have macrocephaly (Mester, Tilot, Rybicki, Frazier, & Eng, 2011), and there is a subset of patients with autism spectrum disorder and macrocephaly with germline PTEN (MIM# 601728) mutations (Butler et al., 2005; Tilot, Frazier, & Eng, 2015). The CS susceptibility locus was initially mapped to 10q23.3 (Nelen et al., 1996) and 9‐exon PTEN was subsequently identified as the predisposition locus (Li & Sun, 1997; Liaw et al., 1997; Lynch et al., 1997). PTEN is a tumor suppressor comprising 403 amino acids and has dual‐specificity protein and lipid phosphatase activity (Myers et al., 1997; Stambolic et al., 1998). Its lipid phosphatase activity is regarded as essential to its tumor‐suppressor function by dephosphorylating phosphatidylinositol (3,4,5)‐triphosphate [PtdIns(3,4,5)P3 or PIP3], which is the key regulator in the PI3K/AKT signaling pathway. The protein phosphatase activity is important for its autodephosphorylation function and cell mobility (Zhang, Piccini, Myers, Van Aelst, & Tonks, 2012). The introduction of Pten mutations that impair Pten function or diminish protein levels in the mouse can lead to the development of tumor or neurological disorders (Barrott et al., 2016; Tilot et al., 2016).
In our cohort of over 6,000 CS and CS‐like patients and family members, we have identified germline PTEN variants near splice junctions that could possibly alter mRNA splicing but few have been fully characterized to date (Tan et al., 2011). With the exception of nine intronic variants previously described by our laboratory, little progress has been made in evaluating the role these variants play in the pathogenesis of CS/CSL (Agrawal, Pilarski, & Eng, 2005). Here, we performed a detailed study on the RNA processing and protein expression of 34 germline intronic variants of PTEN from 61 patients and family members (Supp. Table S1). Cleveland Clinic Institutional Review Board approval (Protocol #8458) and informed consent from all research participants were obtained for this study. The 34 intronic variants examined here span all eight introns of PTEN and may be helpful for predicting the molecular consequences of other nearby variants. NC_000010.11 is used as PTEN reference sequence for all PTEN intronic variants described here. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. We have submitted all novel variants reported in the paper to ClinVar. The URL is https://www.ncbi.nlm.nih.gov/clinvar/?term = PTEN%5Bgene%5D. The accession numbers are: SCV000579224‐SCV000579256.
mRNA was extracted from lymphoblast‐derived cell lines (LBL) from 61 patients and family members. In order to detect unstable mRNA transcripts that could be lost due to nonsense‐mediated decay, half of the cells from each subject were treated with puromycin at 100 μg/ml for 6 hr prior to harvest. cDNA was amplified using PTEN‐specific primer sets (Supp. Table S2). PTEN cDNA was sequenced for all 61 patients and family members. Splice aberrations were detected in 27 samples (Table 1). We then performed 3′RACE on six samples with intron 8 splice mutations. All six samples were found to have aberrant splicing (Table 1). This brings the total number of samples with detectable splicing defects to 33, with 29 samples still having no identified splicing aberration.
Table 1.
PTEN * intronic variation and resulting RNA alteration with predicted protein coding change
| Intronic variant | Intron | RNA change | Predicted protein coding change | Number of patients |
|---|---|---|---|---|
| c.165‐1G>C | 2 | Exon 3 skipping | p.(Arg55Ser)+p.(Phe56_Leu70del) | 1 |
| c.209+5G>A | 3 | Exon 3 skipping | p.(Arg55Ser)+p.(Phe56_Leu70del) | 2 |
| c.209+4_209+7delAGTA | 3 | Exon 3 skipping | p.(Arg55Ser)+p.(Phe56_Leu70del) | 1 |
| c.210‐4_210‐1delTTAG | 3 | Exon 4 skipping | p. (Ala72Thrfs*5) | 1 |
| c.210‐1G>A | 3 | Exon 4 skipping | p. (Ala72Thrfs*5) | 4 |
| c.253+1G>T | 4 | Exon 4 skipping | p. (Ala72Thrfs*5) | 2 |
| c.253+1G>A | 4 | Exon 4 skipping | p. (Ala72Thrfs*5) | 1 |
| c.253+5G>T | 4 | Exon 4 skipping | p. (Ala72Thrfs*5) | 1 |
| c.253+5G>A | 4 | Exon 4 skipping | p. (Ala72Thrfs*5) | 1 |
| c.492+1G>T | 5 | Exon 5 skipping | p.(Val85Glyfs*14) | 1 |
| c.493‐2A>G | 5 | Exon 6 skipping | p.(Gly165Ilefs*9) | 1 |
| c.634+1G>C | 6 | Exon 6 skipping | p.(Gly165Ilefs*9) | 1 |
| c.634+2T>C | 6 | Exon 6 skipping | p.(Gly165Ilefs*9) | 3 |
| c.634+4A>T | 6 | Exon 6 skipping | p.(Gly165Ilefs*9) | 1 |
| c.634+5G>C | 6 | Exon 6 skipping | p.(Gly165Ilefs*9) | 1 |
| c.165‐2A>G | 2 | Add 1 base at the beginning of exon 3 (r.164_165insG) | p. (Phe56Valfs*7) | 2 |
| c. 635‐1G>C | 6 | Use cryptic splice site at r.642_643, (7%) causing mutation r.635_642delAUCCUCAG | p.(Asn212Ilefs*28) (7%) | 1 |
| Or use cryptic splice site at r.688_689 (2%) causing mutation r.635_688del54 | p.(Asn212_Gly230delinsArg) (2%) | |||
| c.802‐2A>G | 7 | r.802‐844del43 | p.(Lys269Glnfs*8) | 1 |
| c.802‐2A>T | 7 | r.802‐844del43 | p.(Lys269Glnfs*8) | 1 |
| c.1026+1G>C | 8 | Retention of intron 8: r.1026+1_1026+190 | PTEN isoform (add two amino acid after exon 8) | 2 |
| c. 1026+1G>A | 8 | Retention of intron 8: r.1026+1_1026+190 | PTEN isoform (add two amino acid after exon 8) | 3 |
| c.1027‐2A>C | 8 | Transcript premature termination in exon 8 at r. 962 | p.(Thr321_403del) | 1 |
| c.79+7A>G | 1 | No change | No change | 2 |
| c.80‐3C>G | 1 | No change | No change | 3 |
| c.210‐7_210‐3delCTTTT | 3 | No change | No change | 11 |
| c.210‐8delT | 3 | No change | No change | 1 |
| c.210‐39A>G | 3 | No change | No change | 2 |
| c.254‐38dupT | 3 | No change | No change | 2 |
| c.254‐51A>T, c.254‐72A>T | 4 | No change | No change | 1 |
| c.493‐52A>G | 5 | No change | No change | 1 |
| c.493‐31A>G | 5 | No change | No change | 1 |
| c.802‐51_802‐14del38 | 7 | No change | No change | 6 |
Nucleotide numbering uses +1 as the A of ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1
*The PTEN reference sequence is NC_000010.11.
Among the splice mutation group, one prominent pattern, as expected, is exon skipping (Table 1; Supp. Fig. S1). A total of 15 intronic variants resulted in exon skipping. The intron 2, 3′ splice‐acceptor mutation c.165‐1G>C causes exon 3 skipping in the PTEN mRNA transcript, and alters the PTEN protein coding sequence dramatically, changing Arg55 to serine and deleting 15 amino acids from Phe56 to Leu70. Intron 3, 5′donor site mutations c.209+5G>A and c.209+4_209+7delAGTA also cause exon 3 skipping in the PTEN mRNA transcript. Six splice‐site mutations around exon 4 cause exon 4 skipping, resulting in a frameshift in PTEN protein coding at alanine 72 and introducing a stop codon four amino acids downstream. These mutations include intron 3, 3′splice‐acceptor mutations c.210‐4 _210‐1delTTAG and c.210‐1G>A and intron 4, 5′splice donor mutations c.253+1G>T, c.253+1G>A, c.253+5G>T, and c.253+5G>A. Intron 5, 5′splice donor mutation c.492+1G>T causes exon 5 skipping and results in a frameshift in the PTEN protein at valine 85 and terminates after 13 amino acids. Five mutations around exon 6 (c.493‐2A>G, c.634+1G>C, c.634+2T>C, c.634+4A>T, and c.634+5G>C) cause exon 6 skipping and result in a frameshift at glycine 165 that terminates after eight codons. It is important to note that intronic mutations at the +5 position often result in the skipping of the preceding exon. For example, the mutations c.209+5G>A in intron 3, c.253+5G>T and c.253+5G>A in intron 4, and intron 6 mutation c.634+5G>C, all result in the upstream exon being skipped. In addition, the intron 3 mutation c.209+4_209+7delAGTA also results in upstream exon 3 skipping. Overall, for the PTEN gene, exons 3–6 are often skipped even in the case of a one‐nucleotide change at the position of +5. This is in agreement with Buratti's finding that +5 position point mutation frequencies are significantly higher and prone to aberrant splicing after inspecting 166 human disease genes (Buratti et al., 2007).
In addition to exon skipping, there were four other types of alternative splice changes. One type is the creation of new, cryptic splice sites adjacent to the canonical site. An example of this type is the intron 2 splice‐acceptor mutant c.165‐2A>G. Instead of exon 3 skipping, the mutant uses the −3 adenosine and the mutant −2 guanidine as a new perfect acceptor that results in the addition of the −1 guanidine to the 5′ end of exon 3 (r.164_165insG), causing the frameshift Phe56Valfs*7 (Table 1).
A second type of splice change makes use of cryptic splice sites within exons. This is seen in the intron 6 splice‐acceptor mutation c.635‐1G>C and intron 7 acceptor mutation c.802‐2A>G (Table 1). Instead of skipping the downstream exon 7 or exon 8, we see cryptic splice sites being utilized in exon 7 and exon 8, respectively (Table 1; Supp. Fig. S1). For the intron 6 mutant, we identified two cryptic splice sites in exon 7, c.642_643 and c.688_689. When c.642_643 is used, the mutant transcript has an 8‐nucleotide deletion, resulting in a predicted frameshift in protein coding (Asn212Ilefs*28). Alternatively, when c.688_689 is used, the mutant transcript has a 54‐nucleotide deletion that causes the loss of 18 amino acids from Asp212 to Gly230 and the insertion of a single arginine residue. Both mutant transcripts are apparently unstable and could not be detected by Sanger sequencing directly after RT‐PCR when the LBL cells were not treated with puromycin. They could only be found after the RT‐PCR products were cloned and subjected to whole‐cell PCR and sequencing. We found the relative frequency of use for the two cryptic sites to be 3:1 (c.642_643:c.688_689). For the intron 7 splice‐acceptor mutation c.802‐2A>G, the cell uses a cryptic splice site in exon 8, c.844_845, resulting in a mutant transcript with 43 nucleotides deleted from 802 to 844, leading to a predicted frameshift in PTEN protein coding (Lys269Glnfs*8). This mutant transcript is stable and can be directly detected by RT‐PCR and Sanger sequencing even when LBL cells were not treated with puromycin. This is the first report that shows evidence of germline splice acceptor mutations in PTEN that result in the use of novel cryptic splice sites in exons 7 and 8.
The third type of splice change results in the retention of intronic sequence that, once again, is predicted to disrupt the protein coding sequence. An example is the intron 8 splice donor site mutations c.1026+1G>A and c.1026+1G>C (Table 1). Both mutant transcripts extend directly from exon 8 into intron 8, and retain 190 nucleotides from intron 8. A poly(A) tail can be detected (Supp. Fig. S2), but exon 9 is not included in the mutant transcript. These could only be detected by 3′RACE. To our surprise, the mutant transcript appears in Ensemble as a protein coding isoform of PTEN. Again, this is the first time a splice‐site mutation in PTEN has been reported to cause an isoform switch. In order to test whether this isoform exists in normal cells, we designed two primer sets with forward primers in exon 7 and reverse primers in intron 8. We were able to detect this isoform in reverse‐transcribed RNA taken from normal blood cells, control LBL cells, and in HeLa cells that contain wild‐type PTEN. We conclude that this isoform of PTEN coexists with canonical PTEN under normal conditions (Supp. Fig. S2).
Finally, a fourth type of splicing defect occurs where a transcript is terminated mid‐exon and polyadenylated because the canonical downstream splice acceptor has been lost and an alternate poly(A) signal is available (Matera & Wang, 2014). The intron 8 mutation c.1027‐2A>C (Table 1; Supp. Fig. S2) is an example of this type and as with other intron 8 mutations, could only be captured using 3′RACE. The transcript of this intron 8 mutation was cleaved within exon 8 at r.962 and a poly(A) tail was added directly onto the termination site. The polyadenylation signal sequence AAUAAA typically lies 10–30 bases ahead of a cleavage site (Colgan & Manley, 1997). There are two variations of this sequence, GAUAAT and AAUAUC, just 10–30 bases ahead of the exon 8 cleavage site. In addition, the consensus sequence GUUUU is often found downstream of the cleavage site (Colgan & Manley, 1997). For the PTEN intron 8 mutant, there is a variation of this sequence, CUUUU, just 30 bases downstream of the cleavage site.
Though we have characterized the effects of many PTEN intronic mutations, 12 intronic variants remain uncharacterized because we can only detect canonical PTEN transcript (Table 1). Most of them are outside of the −5 to +5 window, so they may not directly affect the splicing process. But, there are two interesting variants that are within the −5 range, yet are without splicing changes. The absence of detectable aberrant transcripts is still clinically relevant for these samples and provides valuable information for genetic counseling. The variants are intron 1 c.80‐3C>G and intron 3 c.210‐7_210‐3delCTTTT. In the former, we have three family members enrolled that share c.80‐3C>G, but only one has a phenotype of macrocephaly, other thyroid lesions and mucosal lesions (Supp. Table S1). Importantly, this patient has an additional intron 6, c.634+4A>T mutation, which results in exon 6 skipping. This suggests that the genetic driver of the phenotype is the intron 6 c.634+4A>T mutation and not the c.80‐3C>G.
The intron 3, c.210‐7_210‐3delCTTTT variation presents another facet of this complex splicing story. Although it is within five nucleotides of the splice junction and a rather significant deletion, we see no evidence of altered splicing. Upon closer inspection, the reason for this may lie in the sequence itself. There is a doublet of CTTTT in this region. If one copy is deleted, the remaining copy may still be able to act as a functional splice acceptor.
Patients harboring intronic PTEN mutations with aberrant RNA processing show more severe phenotypes reflected through higher CC scores compared with patients harboring intronic PTEN variants showing no detectable changes in RNA (Supp. Table S1). Specifically, the average CC score for the patient group with the intronic mutations that result in aberrant splicing is 24, and the median age of onset is 45, whereas the average CC score for the patient group with the intronic variants where we do not detect the aberrant splicing is 9.7 (P = 0.0002), and the median age of onset is 50 (P = 0.04). Compared with known pathogenic exonic mutations, PTEN intronic mutations show similar CC score (Supp. Table S3).
To determine the effect of the splice changes on PTEN and downstream protein expression, we separated the intron variants into two groups, those with splice alterations and those without. We compared the expression of PTEN and downstream proteins as detected by Western blot from the whole‐cell lysates of lymphoblast‐derived cells from 35 patients and 20 normal healthy controls (Fig. 1). There were 22 samples in the splice change group and 13 samples in the no splice change group. PTEN protein expression was significantly decreased in samples with intronic mutations that result in splice changes compared with samples with intronic variants that do not have splice changes. PTEN protein ratio between splice change group and no splice change group is almost decreased by half. This strongly suggests that only one allele of PTEN is making stable protein, whereas the mis‐spliced mutant allele is not fully expressed or is unstable. Conversely, P‐AKT levels are significantly increased in the samples carrying splicing changes compared with samples that harbor intronic variants that have no detectable splicing changes. This matches the canonical PTEN function in antagonizing the PI3K/Akt pathway where loss of PTEN protein increases the P‐AKT level, representing upregulation of the AKT pathway, with downstream cellular consequences such as cell survival and proliferation. In contrast, we do not see any overall significant change in P‐ERK1/2 level between these two groups of intronic variants. Indeed, a previous study from our laboratory also observed similar downstream effects of PTEN mutation from patient‐derived lymphoblastoid cells (LBLs). P‐AKT was activated in the group with exonic PTEN pathologic mutations compared with PTEN wt sequence group, PTEN SNPs group, and PTEN VUSs group, yet pERK did not show any significant change (Tan et al., 2011). What we may be observing here with our splice mutations is the predominant effect of cytoplasmic PTEN dysfunction, where AKT signaling is the predominant downstream effector (vs. nuclear PTEN where ERK is the predominant downstream effector) (Chung & Eng, 2005; Chung et al., 2006).
Figure 1.
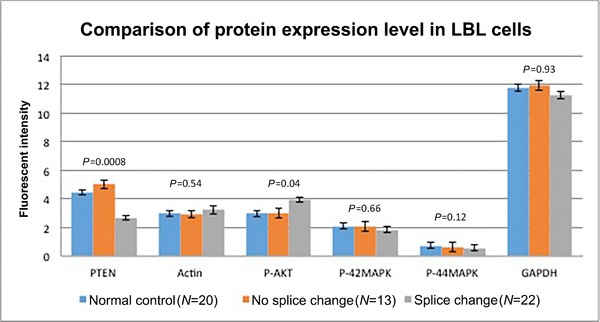
Protein expression of PTEN and downstream read‐out proteins by Western blot of protein lysates of lymphoblastoid cells from patients with germline PTEN intronic variants and controls. Blue bars, normal controls; orange bars, CS/CSL with intronic variants without detectable splice aberrations; and gray bars, CS/CSL with intronic variants resulting in splice aberrations. Actin and GAPDH are loading controls. The bar represents the mean, and error range, standard error of the mean. P values are derived from ANOVA test.
Although all the work was undertaken on LBLs, the aberrant splicing patterns we discussed here are generic, for the apparatus for splicing is not tissue specific and should be applicable to other tissues, for example, breast. We use LBLs as a model for germline mutations existing in our CS patients over the last 18 years. In the context of germline mutations, since all the tissues harbor the same mutations, we look for major changes in splicing concerning canonical PTEN transcript. Relatedly, we have tested several patients using blood leukocyte‐derived RNA (those available) instead of LBL‐derived RNA, and importantly noted a similar splicing pattern. For example, patient #20 has intron 3 mutation c.209+5G>A and when we used blood leukocyte‐derived RNA, we detected exon 3 skipping (Supp. Fig. S1). Patients #25 and #26 both have intron 4 mutation c.253+5G>T, and when we used their blood leukocyte‐derived RNA, we detected exon 4 skipping in both patients (Supp. Fig. S1).
Our data suggest that intronic variants that locate at five nucleotides upstream or downstream of an exon represent pathogenic splice‐site mutations. Since these variants are typically reported as VUS, we recommend scrutiny of such mutations, particularly in the diagnostic laboratory setting. For novel intronic variants not reported in our study, it is hence advisable to consider the −5 to +5 widow as a threshold for pathogenic splice‐site mutations. Additionally, any duplications or deletions that exist across the −5 and +5 window would be possibly considered as highly pathogenic splice‐site mutations.
In summary, we have analyzed 34 different germline PTEN intronic variants looking for evidence of aberrant splicing. We have found 22 mutations that alter splicing and have found no evidence of a splicing defect in the remaining 12. The spectrum of changes arising from these splice‐site perturbations includes exon skipping, the creation of new splice sites, the use of cryptic splice sites, the switch to rare PTEN isoforms and premature transcript termination, and polyadenylation within exon 8. Our functional data indicate that samples that harbor intronic mutations and have a splicing change have lower PTEN protein expression and higher P‐AKT expression compared with samples with intronic variants without splicing changes. We find it interesting that P‐ERK1/2 levels are unchanged in those with intronic variants. Our observations should help molecular diagnostics laboratories with such intronic variant calls and aid genetic counseling for patients and family members with these PTEN intronic variants.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the patients, their families, and their caregivers who have participated in this study. We thank Lamis Yehia for critical discussions and Dr. Rick Padgett for his insightful advice on RNA processing. We also thank the Genomic Medicine Biorepository of the Cleveland Clinic Genomic Medicine Institute, and our database and clinical research teams.
C.E. is the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinicand an American Cancer Society Clinical Research Professor.
Disclosure statement
The authors declare no conflict of interest.
Chen HJ, Romigh T, Sesock K, Eng C. Characterization of cryptic splicing in germline PTEN intronic variants in Cowden syndrome. Human Mutation. 2017;38:1372–1377. https://doi.org/10.1002/humu.23288
Funding InformationContract grant sponsors: National Cancer Institute(P01CA124570); Ambrose Monell Foundation; Zacconi Foundation.
Communicated by Claude Férec
REFERENCES
- Agrawal, S. , Pilarski, R. , & Eng, C. (2005). Different splicing defects lead to differential effects downstream of the lipid and protein phosphatase activities of PTEN. Human Molecular Genetics, 14(16), 2459–2468. [DOI] [PubMed] [Google Scholar]
- Barrott, J. J. , Kafchinski, L. A. , Jin, H. F. , Potter, J. W. , Kannan, S. D. , Kennedy, R. , … Jones, K. B. (2016). Modeling synovial sarcoma metastasis in the mouse: PI3 '‐lipid signaling and inflammation. Journal of Experimental Medicine, 213(13), 2989–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti, E. , Chivers, M. , Kralovicova, J. , Romano, M. , Baralle, M. , Krainer, A. R. , & Vorechovsky, I. (2007). Aberrant 5' splice sites in human disease genes: Mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Research, 35(13), 4250–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, M. G. , Dasouki, M. J. , Zhou, X. P. , Talebizadeh, Z. , Brown, M. , Takahashi, T. N. , … Eng, C. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. Journal of Medical Genetics, 42(4), 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, J. H. , & Eng, C. (2005). Nuclear‐cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Research, 65(18), 8096–8100. [DOI] [PubMed] [Google Scholar]
- Chung, J. H. , Ostrowski, M. C. , Romigh, T. , Minaguchi, T. , Waite, K. A. , & Eng, C. (2006). The ERK1/2 pathway modulates nuclear PTEN‐mediated cell cycle arrest by cyclin D1 transcriptional regulation. Human Molecular Genetics, 15(17), 2553–2559. [DOI] [PubMed] [Google Scholar]
- Colgan, D. F. , & Manley, J. L. (1997). Mechanism and regulation of mRNA polyadenylation. Genes & Development, 11(21), 2755–2766. [DOI] [PubMed] [Google Scholar]
- Li, D. M. , & Sun, H. (1997). TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Research, 57(11), 2124–2129. [PubMed] [Google Scholar]
- Liaw, D. , Marsh, D. J. , Li, J. , Dahia, P. L. , Wang, S. I. , Zheng, Z. , … Parsons, R. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics, 16(1), 64–67. [DOI] [PubMed] [Google Scholar]
- Lynch, E. D. , Ostermeyer, E. A. , Lee, M. K. , Arena, J. F. , Ji, H. L. , Dann, J. , … King, M. C. (1997). Inherited mutations in PTEN that are associated with breast cancer, Cowden disease, and juvenile polyposis. American Journal of Human Genetics, 61(6), 1254–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera, A. G. , & Wang, Z. (2014). A day in the life of the spliceosome. Nature Reviews Molecular Cell Biology, 15(2), 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester, J. L. , Tilot, A. K. , Rybicki, L. A. , Frazier, T. W. , & Eng, C. (2011). Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock‐in murine model. European Journal of Human Genetics, 19(7), 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, M. P. , Stolarov, J. P. , Eng, C. , Li, J. , Wang, S. I. , Wigler, M. H. , … Tonks, N. K. (1997). P‐TEN, the tumor suppressor from human chromosome 10q23, is a dual‐specificity phosphatase. Proceedings of the National Academy of Science of the United States of America, 94(17), 9052–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelen, M. R. , Padberg, G. W. , Peeters, E. A. J. , Lin, A. Y. , vandenHelm, B. , Frants, R. R. , … Eng, C. (1996). Localization of the gene for Cowden disease to chromosome 10q22‐23. Nature Genetics, 13(1), 114–116. [DOI] [PubMed] [Google Scholar]
- Ngeow, J. , & Eng, C. (2015). PTEN hamartoma tumor syndrome: Clinical risk assessment and management protocol. Methods, 77–78, 11–19. [DOI] [PubMed] [Google Scholar]
- Stambolic, V. , Suzuki, A. , de la Pompa, J. L. , Brothers, G. M. , Mirtsos, C. , Sasaki, T. , … Mak, T. W. (1998). Negative regulation of PKB/Akt‐dependent cell survival by the tumor suppressor PTEN. Cell, 95(1), 29–39. [DOI] [PubMed] [Google Scholar]
- Tan, M. H. , Mester, J. L. , Ngeow, J. , Rybicki, L. A. , Orloff, M. S. , & Eng, C. (2012). Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research, 18(2), 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, M. H. , Mester, J. , Peterson, C. , Yang, Y. , Chen, J. L. , Rybicki, L. A. , … Eng, C. (2011). A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. American Journal of Human Genetics, 88(1), 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilot, A. K. , Bebek, G. , Niazi, F. , Altemus, J. B. , Romigh, T. , Frazier, T. W. , & Eng, C. (2016). Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Molecular Psychiatry, 21(1), 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilot, A. K. , Frazier, T. W., 2nd , & Eng, C. (2015). Balancing proliferation and connectivity in PTEN‐associated autism spectrum disorder. Neurotherapeutics, 12(3), 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. C. , Piccini, A. , Myers, M. P. , Van Aelst, L. , & Tonks, N. K. (2012). Functional analysis of the protein phosphatase activity of PTEN. Biochemical Journal, 444(3), 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material