

Abstract
Background
Histone deacetylase (HDAC) inhibitors are promising therapeutics for various forms of cardiac disease. The purpose of this study was to assess cardiac HDAC catalytic activity and expression in children with single ventricle heart disease of right ventricular morphology (SV), as well as in a rodent model of right ventricular hypertrophy (RVH).
Methods
Homogenates of RV explants from non-failing controls and SV children were assayed for HDAC catalytic activity and HDAC isoform expression. Postnatal 1-day old rat pups were placed in hypoxic conditions and echocardiographic analysis, gene expression, HDAC catalytic activity and isoform expression studies of the RV were performed.
Results
Class I, IIa, and IIb HDAC catalytic activity and protein expression were elevated in hearts of SV children. Hypoxic neonatal rats demonstrated RVH, abnormal gene expression and elevated class I and class IIb HDAC catalytic activity and protein expression in the RV compared to control.
Conclusions
These data suggest that myocardial HDAC adaptations occur in the SV heart and could represent a novel therapeutic target. While further characterization of the hypoxic neonatal rat is needed, this animal model may be suitable for pre-clinical investigations of pediatric RV disease and could serve as a useful model for future mechanistic studies.
INTRODUCTION
Right ventricular hypertrophy (RVH) and pulmonary hypertension (PH) are common sequelae in pediatric patients with various forms of congenital heart disease. On the severe end of the spectrum are children with single ventricle heart disease (SV), especially those with a single right ventricle (RV), such as hypoplastic left heart syndrome. SV is a rare but important form of hypoxic congenital heart disease that is fatal without intervention(1). Current treatment strategies for infants born with SV include a 3-stage surgical palliation, or primary heart transplantation. Primary heart transplant is significantly limited by donor availability. Although there have been advancements in perioperative and surgical approaches for SV, in the largest prospective study to date, 32% of those managed with surgical palliation died or were transplanted prior to 1 year of age(2). While there are many causes of poor outcome in SV patients, systemic right ventricular dysfunction, for which there are no proven therapies, is a risk factor for death and listing for transplant.(3–6)
Histone deacetylases (HDACs) are epigenetic enzymes that function canonically through removal of acetyl groups from lysine residues within nucleosomal histone tails. The 18 mammalian HDACs are categorized into 4 classes: class I (HDACs 1, 2, 3, 8), class II (class IIa: HDACs 4, 5, 7, 9; class IIb: HDACs 6, 10), class III (SIRT1-7), and class IV (HDAC11). Class III HDACs are also known as sirtuins and use NAD+ as a cofactor, while HDACs 1-11 employ Zn2+ as a cofactor for catalytic activity. Class IIa HDACs are thought to be protective in the setting of heart failure due to their ability to bind to and inhibit the transcriptional activity of myocyte enhancer factor 2 (MEF2)(7). Elevated catalytic activity of sirtuins appears to be beneficial in failing hearts(8), whereas elevated catalytic activity of class I and IIb HDACs is thought to be maladaptive(9, 10).
Two HDAC inhibitors, Vorinostat (suberoylanilide hydroxamic acid, Zolinza™) and Romidepsin (Istodax™), are FDA-approved to treat cutaneous T-cell lymphoma. These and other HDAC inhibitors are currently in clinical trials, investigating efficacy in a variety of oncologic and non-oncologic diseases. HDAC inhibition has beneficial effects in pre-clinical models of adult heart failure, reducing cardiac hypertrophy(11) and fibrosis(12), and suppressing the fetal gene program associated with adverse cardiac remodeling(13). These findings suggest that HDACs could be therapeutically targeted for the treatment of adult human heart failure(14). However, nothing is known about the roles of HDACs in pediatric heart failure.
The purpose of this study was to examine the catalytic activity and protein expression of HDACs in pediatric SV patients with a single RV, and characterize a neonatal animal model with potential to elucidate molecular mechanisms controlling pathological remodeling of the pediatric RV. We hypothesized that pediatric SV patients would exhibit elevated HDAC catalytic activity, and that hypoxia would result in elevated HDAC catalytic activity coincident with adverse remodeling in neonatal rat RVs.
METHODS
This study used heart tissue samples from pediatric patients (<18 years of age) that donated their hearts to the University of Colorado Institutional Review Board-approved pediatric heart tissue bank (informed consent is obtained for all patients). Patients included males and females of all races and ethnic background undergoing heart transplantation at Children’s Hospital Colorado. Nonfailing (NF) control hearts were obtained from pediatric donors (<18 years of age) with structurally normal hearts and normal heart function whose hearts could not be placed for technical reasons, usually size or blood type mismatch. SV tissue was obtained from explanted hearts of patients with single ventricle physiology and a morphologic single RV. Patients with a single LV or indeterminate morphology of the single ventricle were excluded. All heart tissue was rapidly flash frozen in the operating room immediately after removal from the subject. A detailed description of patients included in this study is outlined in Table 1.
Table 1.
Patient Demographics. NF- non-failing control. SV- single ventricle patients. M- male. F- female. Inotropes included dopamine and norepinephrine. PDE- phosphodiesterase. ACEI- Angiotensin converting enzyme inhibitor. BB- beta-blocker (the non-selective beta-blocker labetalol was used in NF1). PGE-prostaglandin E1, used to maintain ductus arteriosus patency. PDA- patent ductus arteriosus.
| Study ID | Age (years) | Sex | HDAC Activity | HDAC Westerns | Inotrope | Digoxin | ACEI | BB | Diuretic | Palliation History/Indication for transplant |
|---|---|---|---|---|---|---|---|---|---|---|
| NF1 | 13 | M | X | X | Y | N | N | Y | N | NA |
| NF2 | 8 | F | X | X | N | N | N | N | N | NA |
| NF3 | 7 | M | X | X | N | N | N | N | N | NA |
| NF4 | 1.4 | M | X | X | N | N | N | N | N | NA |
| NF5 | 12 | M | X | X | N | N | N | N | N | NA |
| NF6 | 14 | M | X | X | NA | NA | NA | NA | NA | NA |
| SV1 | 0.1 | F | X | X | N | N | N | N | Y | No palliation, on PGE/Primary transplant, RVH dilation |
| SV2 | 0.1 | M | X | X | N | N | N | N | Y | Aortic valvuloplasty and atrial septal stent on PGE/Not a candidate for additional surgical palliation, ventilator dependent |
| SV3 | 0.2 | M | X | X | N | N | Y | N | Y | PDA stent/Primary transplant, RV dilation |
| SV4 | 0.2 | M | X | X | N | N | N | N | Y | PDA stent/Primary transplant, RVH |
| SV5 | 0.2 | M | X | NA | NA | NA | NA | NA | No palliation, on PGE/Primary transplant, RVH | |
| SV6 | 0.1 | M | X | X | N | Y | Y | N | Y | No palliation, on PGE/Primary transplant, RVH and RV dilation |
Histone deacetylase catalytic activity assays
Measurement of HDAC catalytic activity was performed as previously described(15). Reagents were purchased from indicated vendors. HDAC inhibitor, Trichostatin A (TSA; Sigma) diluted in dimethylsulfoxide (DMSO). Synthetic HDAC substrates: class I HDAC substrate (custom synthesis by Genscript), class IIa HDAC substrate (I-1985; Bachem), class I/IIb HDAC substrate (I-1875; Bachem). Trypsin, Triton X-100, and DMSO were obtained from Sigma. 7-Amino-4-methylcoumarin (AMC; Alfa Aesar).
Frozen explants from human pediatric RV were prepared in PBS (pH 7.4) containing 0.5% Triton X-100, 300mM NaCl and HALT™ protease/phosphatase inhibitor cocktail (ThermoFisher) using a Bullet Blender homogenizer (Next Advance). Protein concentrations were determined by BCA Protein Assay Kit (ThermoFisher). Incubation of tissue lysates with class selective HDAC substrates results in a cleavable AMC product. AMC fluorescence was measured using a BioTeK Synergy 2 plate reader, with excitation and emission filters of 360 nm and 460 nm. Background fluorescence from buffer blanks were subtracted from raw signals, and data were normalized as needed using appropriate controls.
Immunoblotting
Immunoblotting was performed as previously described(16). Briefly, pediatric human and neonatal rat RV homogenates were prepared and concentrations quantified as above for HDAC catalytic activity assay. Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes (BioRad) and probed with antibodies for HDAC1 (Cell Signaling Technology, 5356), HDAC2 (Cell Signaling Technology, 5113), HDAC3 (Cell Signaling Technology, 3949), HDAC4 (Cell Signaling Technology, 5392), HDAC5 (Cell Signaling Technology, 2082), HDAC6 (Santa Cruz Biotechnology, 11420), HDAC7 (Cell Signaling Technology, 2882), calnexin (Santa Cruz Biotechnology, 11397).
Animal model
The pediatric neonatal rat RV hypertrophy model was adapted from a previous study(17). Timed-pregnant E17 Sprague Dawley rats were obtained from Charles River Laboratories. Rat mothers birthed their litters at Denver altitude and equilibrated for one day. Post-natal day 1 normoxic animals were placed in a hypobaric chamber simulating sea level altitude (21% oxygen), while post-natal day 1 hypoxic animals were placed in a hypobaric chamber simulating 18,000 feet (~10% oxygen) for seven days. Neonatal rats were anesthetized with isoflurane and sacrificed by thoracotomy. Hearts were flushed with saline through the aorta, atria were removed, and right ventricles were carefully separated from left ventricle and septum prior to flash freezing in liquid nitrogen.
Cardiac Imaging
Echocardiographic analysis was performed on the neonatal rats using a Vevo2100 system equipped with a MS400 18–38 mHz transducer (VisualSonics), as previously described(9).
Real-time polymerase chain reaction
Real-time polymerase chain (RT-PCR) reactions were performed as previously described(18). Total RNA was extracted using a mirVana kit (Ambion, Austin, TX), RNA was reverse transcribed into complementary DNA using I-script (Bio-Rad, Hercules, CA), and Power SYBR Green PCR Master Mix was (Applied Biosystems/Life Technologies, Carlsbad, CA) were used in the RT-PCR reactions. Reactions were performed using the ABI 7300 system. Primer sequences are indicated below:
ANF: Forward 5′- gcgaaggtcaagctgctt; Reverse 5′- ctgggctccaatcctgtcaat
BNP: Forward 5′- ggtgctgccccagatgatt; Reverse 5′- ggtgctgccccagatgatt
Serca2a: Forward 5′- ggccagatcgcgctaca; Reverse 5′- gggccaattagagagcaggttt
Data analysis
GraphPad Prism software was used to generate graphs and analyze data. Student’s T-test with Welch’s correction or ANOVA with Bonferroni’s post-test (P<0.05) was used to determine statistical differences between groups.
RESULTS
Patient characteristics
The age range of the NF controls was 1.4–14 years, with 17% female. The SV group was composed of infants less than 3 months of age, with 17% female. The indication for transplantation for the SV group is outlined in Table 1, and encompasses those undergoing primary transplant (listed for transplant shortly after birth prior to surgical palliation) and includes one patient that failed an attempt at interventional palliation.
Cardiac HDAC catalytic activity is increased in pediatric single-ventricle patients
Eleven zinc-dependent HDACs are grouped into four classes (Figure 1a). HDAC catalytic activity has yet to be quantified in the human heart. To investigate whether pediatric SV patients have altered HDAC catalytic activity, RV homogenates from pediatric NF controls and pediatric SV patients were incubated with HDAC class-specific fluorescent substrates. A significant increase in class I, IIa and IIb HDAC catalytic activity was observed in RVs of SV patients relative to NF control RVs (Figure 1b). Class IIb HDAC catalytic activity was most dramatically increased (~3.5 fold), followed by a 2-fold increase in class I HDAC catalytic activity, and a modest, but statistically significant increase in class IIa HDAC catalytic activity. These observations are consistent with reported increases in HDAC catalytic activity in hearts of adult rats with RVH due to PH(15).
Figure 1.
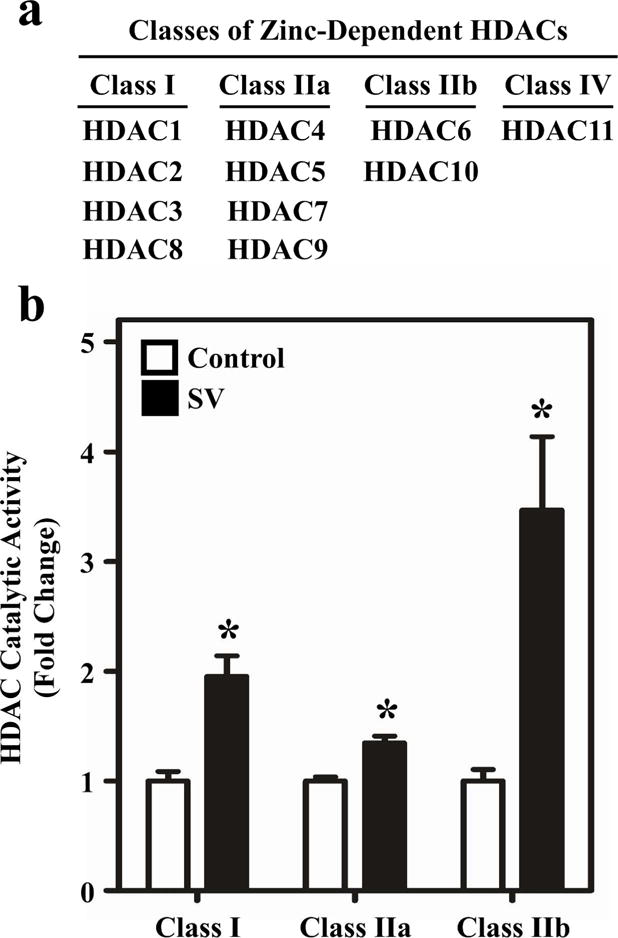
HDAC catalytic activity is elevated in the RV of children with SV. (a) Classifications of zinc-dependent HDACs. (b) HDAC Catalytic Activity: RV tissue from non-failing pediatric and SV hearts was homogenized in a mild lysis buffer and incubated with HDAC class I, IIa, and IIb-specific substrates. All classes of HDACs displayed elevated catalytic activity relative to control. Results are displayed as the mean with standard error; N = 6 per group, *P<0.05 vs controls.
Protein expression of distinct HDAC isoforms is elevated in hearts of pediatric single-ventricle patients
Since all classes of HDACs measured in the enzymatic assay exhibited elevated catalytic activity in RVs of SV patients, we investigated whether protein expression of HDACs belonging to class I, IIa and IIb was concomitantly augmented. Protein expression of HDAC1-7 was measured by immunoblotting (Figure 2a), and quantified by normalization to a house-keeping protein, calnexin (Figure 2b). Expression of class I HDACs -1, -2, and -3 was elevated and reached statistical significance in SV patient RVs compared to NF controls. Of the class IIa HDACs, only HDAC5 expression was significantly elevated in SV patient RVs. Expression of class IIb HDAC6 was not increased in SV patient RVs.
Figure 2.
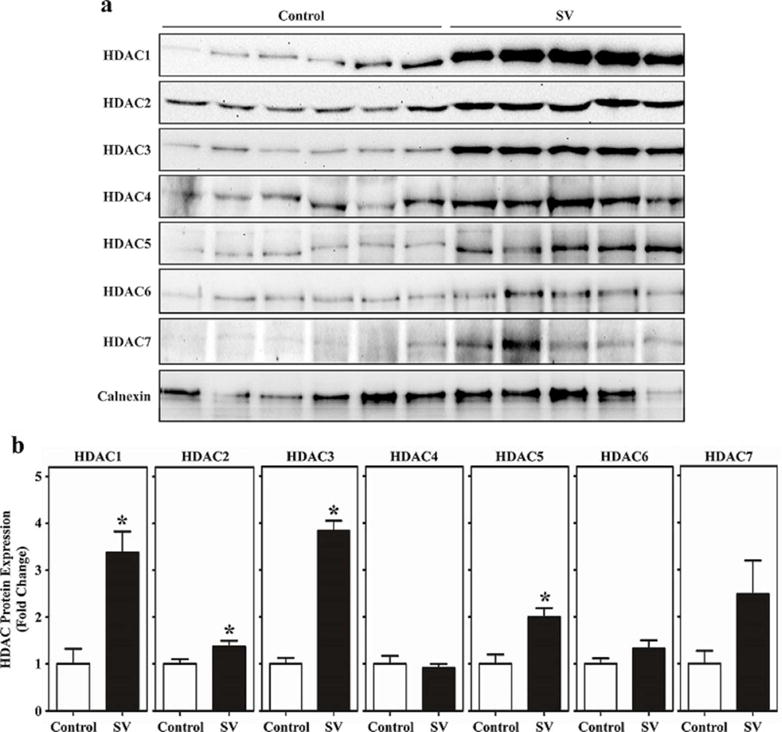
HDAC protein expression is elevated in pediatric RVs from SV patients. (a) Immunoblot analysis was performed with the same homogenates used for the HDAC catalytic activity assays employing antibodies specific for class I HDACs (HDAC1-3), class IIa HDACs (HDAC4, 5, 7) and class IIb HDAC6. (b) Densitometry was used to quantify immunoblot signals; *P<0.05 vs. controls.
Activation of cardiac HDACs in a hypoxic neonatal rat model
RVH and failure is a common phenotype in pediatric patients with PH and various congenital heart lesions, including SV. In an effort to re-create elements of pediatric RVH, neonatal rats were placed in a hypobaric chamber simulating 18,000 feet in elevation and 10% O2 for seven days. Control animals were simultaneously housed at sea level altitude (21% O2) for seven days (Figure 3a). The use of neonatal rats, as opposed to mice, facilitated echocardiographic assessment of RV hypertrophy and pulmonary artery blood flow.
Figure 3.
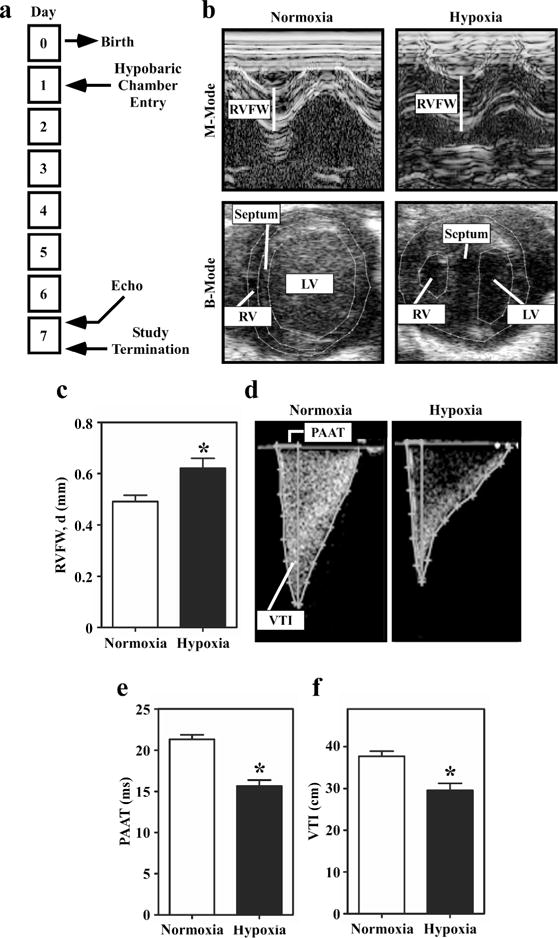
Echocardiographic analyses of right ventricular hypertrophy and pulmonary hypertension in hypoxic neonatal rats. (a) Study design. (b) Representative M-mode and B-mode images of hearts from neonatal rats exposed to hypobaric hypoxia or normoxic conditions for 7 days. (c) RV Free Wall Thickness, Diastole: Quantitative assessment of RV free wall (RVFW) thickness demonstrates increased thickness in the hypoxic rats; *P<0.05 vs. controls. (d) Representative Doppler images of pulmonary blood flow in normoxic and hypoxic neonatal rats. (e) Pulmonary artery acceleration time (PAAT) is significantly decreased in the hypoxic neonatal rats compared to control. (f) Pulmonary valve velocity-time integral (VTI) is also significantly decreased in hypoxic neonatal rats compared to control. Results are displayed as the mean with standard error. N = 11 per condition; P<0.05 vs. controls.
In response to seven days of hypobaric hypoxia, thickness of the free wall of the neonatal rat RV was significantly increased in both systole and diastole compared to sea level controls, as quantified by M-mode echocardiography (Figure 3b and 3c). Assessment of heart chamber remodeling by B-mode parasternal short axis evaluation demonstrated septal wall flattening and chamber dilation in the RVs of hypoxic neonatal rats (Figure 3b, lower images). Septal wall flattening occurred in 100% of the hypoxic animals.
Pulmonary artery blood flow was assessed using color Doppler and pulse-wave Doppler. Hypoxic neonatal rats exhibited a significant decrease in pulmonary artery acceleration time (PAAT) compared to sea level controls. Pulmonary valve velocity-time integral (VTI) and pulmonary valve peak velocity (PVPV) were also significantly decreased in hypoxic rats (Figure 3d – f and data not shown); PAAT, PVPV and PV VTI are well-accepted measurements of PH in humans(19, 20). These findings suggest that PH exists in neonatal rats exposed to 7 days of hypobaric hypoxia.
Morphometric analysis performed at the time of necropsy confirmed significant RV hypertrophy in neonatal rats exposed to hypobaric hypoxia. After 7 days of hypobaric hypoxia, the ratio of RV-to-left ventricle (LV) + septum (S) was 1.5-fold higher in hypoxic rats compared to sea level control animals (Figure 4a).
Figure 4.
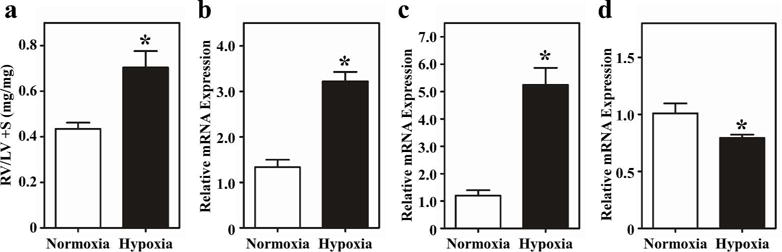
Pathological RV remodeling in neonatal rats exposed to hypoxia. (a) RV hypertrophy was assessed by comparing weights of RV vs. LV + septum (S). The RV/LV+septum ratio was higher in the hypoxic rats. (b) Atrial natriuretic peptide, (c) B-type natriuretic peptide (BNP), and (d) sarcoplasmic reticulum calcium ATPase 2a (Serca2a) mRNA expression was determined by quantitative PCR. ANP and BNP expression was increased, while Serca2a expression was decreased in the hypoxic animals. Results are presented as the mean with standard error. N = 3 (normoxia), N = 6 (hypoxia); *P<0.05 vs. control.
Myocardial failure in humans is characterized by activation of a “fetal” gene program, as embryonically-expressed genes are reactivated(7). This gene program is indicative of ventricular remodeling and is typified by increases in expression of B-type natriuretic peptide (BNP) and atrial natriuretic peptide (ANP), and a coordinate decrease in sarcoplasmic reticulum calcium-ATPase 2a (SERCA2a) expression. Quantitative PCR revealed that ANP and BNP mRNA levels were elevated 3-fold and 5-fold, respectively, while SERCA2a expression was significantly attenuated in the hypoxic neonatal rat RVs compared to sea level control RVs (Figure 4b – d).
HDAC catalytic activity was subsequently quantified in homogenates of RVs from sea level and hypoxic neonatal rats. Significant increases in class I HDAC and class IIb HDAC catalytic activity were observed in hypertrophic RVs, which was consistent with pediatric SVs. However, unlike pediatric SVs, there was no difference in class IIa HDAC catalytic activity in hypoxic RVs (Figure 5a). Increases in class I and class IIb HDAC activity correlated with the abundance of HDAC1, HDAC2, HDAC3 and HDAC6 in RVs of hypoxic neonatal rats (Figure 5b).
Figure 5.
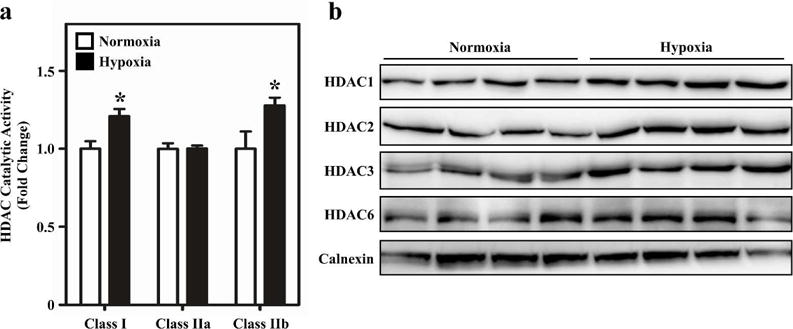
Activation of HDACs during pathological RV remodeling in neonatal rats. (a) HDAC catalytic activity was quantified in RV homogenates from neonatal rats exposed to hypobaric hypoxia (N = 6) or normoxic (N = 3) conditions for 7 days. Class I and IIb activity was increased in the RV of the hypoxic rats. Results are displayed as the mean with standard error; *P<0.05 vs. normoxic controls. (b) Immunoblot analysis was performed with the same rat RV homogenates used for the HDAC catalytic activity assays employing antibodies specific for class I HDACs (HDAC1-3) and class IIb HDAC6. HDAC 1, 2, 3 and 6 protein expression was increased in the RV of the hypoxic rats.
Discussion
Pharmacotherapy to prevent or treat myocardial failure in patients with single-ventricle physiology does not exist, underscoring the need for research to define the molecular mechanisms of this complex congenital heart lesion(4). Treatment of adult heart failure (HF) has dramatically advanced over the past several decades, yet many of the drugs used to treat adult HF have not been proven to be beneficial in treating pediatric HF(21). Part of the challenge lies in the fact that pediatric HF is a much more heterogeneous disease than in adults, with a low incidence of ischemic disease. Pediatric HF ranges from idiopathic dilated cardiomyopathy (IDC), which is the leading indication for heart transplant in children, to single ventricle heart disease, the leading indication for transplant in infants(22). There are inherent differences in myocardial adaptation in pediatric versus adult HF, including altered gene, protein, adrenergic receptor and microRNA expression, as well as protein phosphorylation and enzyme activity(18, 23–25). In addition, children are a vulnerable population, limiting the ability to perform the invasive studies that have been so useful in advancing the care of adults with HF. This study was designed to characterize the adaptations occurring in myocardial HDACs in the pediatric SV population, and to develop and perform initial characterization of an animal model that could serve to facilitate future mechanistic studies and drug development relevant to RV remodeling in the young.
We found that the catalytic activity of class I, IIa and IIb HDACs is elevated in RVs of children with SV disease compared to NF controls (Figure 1b). For class I and IIa HDACs, enhanced catalytic activity correlated with increased levels of HDAC isoform protein expression (class I HDACs -1, -2 and -3; class IIa HDAC5) (Figure 2). In contrast, activation of class IIb HDAC activity occurred independently of altered expression of the predominant isoform within this class, HDAC6. In this regard, multiple post-translational mechanisms for regulation of HDAC6 activity have described in non-cardiac cells. For example, phosphorylation of HDAC6 by glycogen synthase kinase-3β was shown to activate HDAC6 catalytic activity in neurons(26). Elucidation of the mechanisms controlling HDAC catalytic activity in SV disease awaits further investigation.
The molecular mechanisms by which HDACs contribute to adult pressure overload-induced LV hypertrophy, myocardial interstitial fibrosis, cardiac inflammation, pulmonary hypertension and RV remodeling are only beginning to be understood(27–29). HDAC inhibition is effective in improving HF symptoms in animal models of spontaneously hypertensive rats(30), mouse models of myocardial infarction/ischemia-reperfusion(31) and an angiotensin II-infusion model of cardiac fibrosis(12). Consistent with our hypothesis, the class I HDAC inhibitor apicidin attenuated hypoxia-induced RVH and pulmonary vascular remodeling in neonatal mice(32). A recent proteomic study using 19 different HDAC inhibitors in three different human cell lines demonstrated the complexity of individual small molecule effects on the acetylome, with differential effects on the magnitude of acetylation signal and specific acetylation targets between compounds(33). Due to these findings, selection of the class of HDAC inhibitor and specific small molecule for therapeutic development requires careful consideration.
In combination with our previously described abnormalities in the beta-AR system(18), elevated HDAC catalytic activity and expression in the RVs of children with SV suggests that SV hearts are undergoing molecular remodeling. Consistent with these findings, clinically the single RV does not tolerate chronic pressure overload indefinitely, and patients remain at risk for the development of ventricular failure.
While prenatal models of SV exist(34–36), a postnatal animal model is not feasible due to complexities of maintaining the SV circulation after birth. Therefore, we developed a hypoxic neonatal rat model that reasonably recapitulates some of the echocardiographic and molecular signatures of pediatric SV. Children born with SV are hypoxic until the 3rd stage of surgical palliation (Fontan) is completed at 2–5 years of age and systemic afterload on the RV is present throughout their lifespan, with hypertrophy essentially a universal finding. Hypoxia in neonatal rats results in PH and RVH by echocardiographic and morphometric assessment, altered myocardial gene expression, with partial recapitulation of the fetal gene program, elevated HDAC class I and IIb activity, and elevated HDAC protein expression. From both a physiologic and molecular perspective, the hypoxic neonatal rat model has similarities to the human SV condition that support its use for future mechanistic studies of RV remodeling, as well as for therapeutic development. While HDAC IIa activity was not increased in the rat RV as it was in the pediatric SV, whether this difference is clinically significant is unknown. Interestingly, HDAC 4 is associated with the cardiac sarcomere and may play a role in regulating cardiac contractility(37), warranting future study. The use of echocardiography in this model not only provides translational utility, but also allows for longitudinal investigation. RVH and failure are not uncommon in the pediatric cardiovascular disease population, and this model has potential to affect a myriad of populations beyond SV. PH, intrinsic lung disease, various forms of biventricular congenital heart disease (e.g. tetralogy of Fallot, Ebstein’s anomaly) also result in pediatric RV remodeling and failure.
There are important limitations to this study. The tissue bank-based aspect of this study is inherently cross-sectional, and proof of mechanistic associations based on our results is not possible. Due to the rarity of this form of congenital heart disease, and thus the limited tissue available for study, it is not possible for us to determine the influence of age, prior surgical procedures, medications, clinical status of the patient or the temporal relationship of gene expression changes on our findings. We recognize that we have not developed a rodent model of SV. However, many of the findings in this study show consistent changes in phenotype and molecular signatures of hypoxic neonatal rats and SV patients versus their respective controls. This model could be used as a platform for the study of mechanisms of pressure-overloaded RV failure in the young and determination of the influence of aging and development vs disease on myocardial adaptations.
In conclusion, though HDACs are ubiquitously expressed and are essential enzymes, HDAC inhibitors are surprisingly well tolerated(38–40). There is an evolving body of literature suggesting that HDAC-inhibition has value for the treatment of adult cardiovascular diseases(14). The results of this study demonstrate that HDACs are altered in the ventricle of young children with single ventricle heart disease. Given the challenges of studying a rare disease in a vulnerable population, the hypoxic neonatal rat represents a reasonable platform for extending pre-clinical studies to explore mechanisms of disease and investigate novel therapeutics, such as HDAC inhibitors, in preventing or limiting pathologic RV remodeling.
Acknowledgments
Statement of Financial Support
This work was supported by NIH R21 HL113846 to S.D.M., T.A.M and C.S.L. S.D.M. was also supported by NIH (R01 HL126928), the Addison Scott Memorial Fund, the Boedecker Foundation, the Nair Family and the Millisor Chair in Pediatric Heart Disease. T.A.M. was also supported by NIH (HL116848, HL127240 and AG043822) and the American Heart Association (Grant-in-Aid, 14510001). W.W.B was supported by the University of Colorado-Denver Pharmacology Program NIH T32 Training Grant (GM007635).
Footnotes
Disclosure Statement
There are no disclosures or conflicts of interest to report.
Category of Study
This is a translational study.
References
- 1.Correa-Villasenor A, Ferencz C, Loffredo C, Magee C. Paternal exposures and cardiovascular malformations. The Baltimore-Washington Infant Study Group. J Expo Anal Environ Epidemiol. 1993;3(Suppl 1):173–85. [PubMed] [Google Scholar]
- 2.Ohye RG, Sleeper LA, Mahony L, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362(21):1980–92. doi: 10.1056/NEJMoa0912461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulkarni A, Neugebauer R, Lo Y, et al. Outcomes and risk factors for listing for heart transplantation after the Norwood procedure: An analysis of the Single Ventricle Reconstruction Trial. J Heart Lung Transplant. 2016;35(3):306–11. doi: 10.1016/j.healun.2015.10.033. [DOI] [PubMed] [Google Scholar]
- 4.Kirk R, Dipchand AI, Rosenthal DN, et al. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary. [Corrected] J Heart Lung Transplant. 2014;33(9):888–909. doi: 10.1016/j.healun.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Frommelt PC, Gerstenberger E, Cnota JF, et al. Impact of initial shunt type on cardiac size and function in children with single right ventricle anomalies before the Fontan procedure: the single ventricle reconstruction extension trial. J Am Coll Cardiol. 2014;64(19):2026–35. doi: 10.1016/j.jacc.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newburger JW, Sleeper LA, Frommelt PC, et al. Transplantation-free survival and interventions at 3 years in the single ventricle reconstruction trial. Circulation. 2014;129(20):2013–20. doi: 10.1161/CIRCULATIONAHA.113.006191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A. 2000;97(8):4070–5. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanno M, Kuno A, Horio Y, Miura T. Emerging beneficial roles of sirtuins in heart failure. Basic Res Cardiol. 2012;107(4):273. doi: 10.1007/s00395-012-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demos-Davies KM, Ferguson BS, Cavasin MA, et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol. 2014;307(2):H252–8. doi: 10.1152/ajpheart.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mani SK, Kern CB, Kimbrough D, et al. Inhibition of class I histone deacetylase activity represses matrix metalloproteinase-2 and -9 expression and preserves LV function postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2015;308(11):H1391–401. doi: 10.1152/ajpheart.00390.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao DJ, Wang ZV, Battiprolu PK, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–8. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams SM, Golden-Mason L, Ferguson BS, et al. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol. 2014;67:112–25. doi: 10.1016/j.yjmcc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antos CL, McKinsey TA, Dreitz M, et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278(31):28930–7. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- 14.McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–19. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- 15.Lemon DD, Horn TR, Cavasin MA, et al. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol. 2011;51(1):41–50. doi: 10.1016/j.yjmcc.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blakeslee WW, Wysoczynski CL, Fritz KS, Nyborg JK, Churchill ME, McKinsey TA. Class I HDAC inhibition stimulates cardiac protein SUMOylation through a post-translational mechanism. Cell Signal. 2014;26(12):2912–20. doi: 10.1016/j.cellsig.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavasin MA, Demos-Davies K, Horn TR, et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012;110(5):739–48. doi: 10.1161/CIRCRESAHA.111.258426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyamoto SD, Stauffer BL, Polk J, et al. Gene expression and beta-adrenergic signaling are altered in hypoplastic left heart syndrome. J Heart Lung Transplant. 2014;33(8):785–93. doi: 10.1016/j.healun.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cevik A, Kula S, Olgunturk R, et al. Assessment of pulmonary arterial hypertension and vascular resistance by measurements of the pulmonary arterial flow velocity curve in the absence of a measurable tricuspid regurgitant velocity in childhood congenital heart disease. Pediatr Cardiol. 2013;34(3):646–55. doi: 10.1007/s00246-012-0520-4. [DOI] [PubMed] [Google Scholar]
- 20.Vlahos AP, Feinstein JA, Schiller NB, Silverman NH. Extension of Doppler-derived echocardiographic measures of pulmonary vascular resistance to patients with moderate or severe pulmonary vascular disease. J Am Soc Echocardiogr. 2008;21(6):711–4. doi: 10.1016/j.echo.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Rossano JW, Shaddy RE. Update on pharmacological heart failure therapies in children: do adult medications work in children and if not, why not? Circulation. 2014;129(5):607–12. doi: 10.1161/CIRCULATIONAHA.113.003615. [DOI] [PubMed] [Google Scholar]
- 22.Rossano JW, Dipchand AI, Edwards LB, et al. The Registry of the International Society for Heart and Lung Transplantation: Nineteenth Pediatric Heart Transplantation Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J Heart Lung Transplant. 2016;35(10):1185–1195. doi: 10.1016/j.healun.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 23.Miyamoto SD, Karimpour-Fard A, Peterson V, et al. Circulating microRNA as a biomarker for recovery in pediatric dilated cardiomyopathy. J Heart Lung Transplant. 2015;34(5):724–33. doi: 10.1016/j.healun.2015.01.979. [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto SD, Stauffer BL, Nakano S, et al. Beta-adrenergic adaptation in paediatric idiopathic dilated cardiomyopathy. Eur Heart J. 2014;35(1):33–41. doi: 10.1093/eurheartj/ehs229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano SJ, Miyamoto SD, Movsesian M, Nelson P, Stauffer BL, Sucharov CC. Age-related differences in phosphodiesterase activity and effects of chronic phosphodiesterase inhibition in idiopathic dilated cardiomyopathy. Circ Heart Fail. 2015;8(1):57–63. doi: 10.1161/CIRCHEARTFAILURE.114.001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5(5):e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavasin MA, Stenmark KR, McKinsey TA. Emerging roles for histone deacetylases in pulmonary hypertension and right ventricular remodeling (2013 Grover Conference series) Pulm Circ. 2015;5(1):63–72. doi: 10.1086/679700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gillette TG, Hill JA. Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res. 2015;116(7):1245–53. doi: 10.1161/CIRCRESAHA.116.303630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuetze KB, McKinsey TA, Long CS. Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J Mol Cell Cardiol. 2014;70:100–7. doi: 10.1016/j.yjmcc.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardinale JP, Sriramula S, Pariaut R, et al. HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension. 2010;56(3):437–44. doi: 10.1161/HYPERTENSIONAHA.110.154567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granger A, Abdullah I, Huebner F, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22(10):3549–60. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Q, Sun M, Ramchandran R, Raj JU. IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: Role of epigenetic regulation. Vascul Pharmacol. 2015;73:20–31. doi: 10.1016/j.vph.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholz C, Weinert BT, Wagner SA, et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol. 2015;33(4):415–23. doi: 10.1038/nbt.3130. [DOI] [PubMed] [Google Scholar]
- 34.deAlmeida A, Sedmera D. Fibroblast Growth Factor-2 regulates proliferation of cardiac myocytes in normal and hypoplastic left ventricles in the developing chick. Cardiol Young. 2009;19(2):159–69. doi: 10.1017/S1047951109003552. [DOI] [PubMed] [Google Scholar]
- 35.Kowalski WJ, Teslovich NC, Menon PG, Tinney JP, Keller BB, Pekkan K. Left atrial ligation alters intracardiac flow patterns and the biomechanical landscape in the chick embryo. Dev Dyn. 2014;243(5):652–62. doi: 10.1002/dvdy.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedmera D, Hu N, Weiss KM, Keller BB, Denslow S, Thompson RP. Cellular changes in experimental left heart hypoplasia. Anat Rec. 2002;267(2):137–45. doi: 10.1002/ar.10098. [DOI] [PubMed] [Google Scholar]
- 37.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283(15):10135–46. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klimek VM, Fircanis S, Maslak P, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14(3):826–32. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 39.Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19(9):1049–66. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Connor OA, Heaney ML, Schwartz L, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24(1):166–73. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]