

Summary
Many intracellular pathogens exploit host secretory trafficking to support their intracellular cycle, but knowledge of these pathogenic processes is limited. The bacterium Brucella abortus uses a Type IV secretion system (VirB T4SS) to generate a replication-permissive Brucella-containing vacuole (rBCV) derived from the host endoplasmic reticulum (ER), a process that requires host early secretory trafficking. Here we show that the VirB T4SS effector BspB contributes to rBCV biogenesis and Brucella replication by interacting with the Conserved Oligomeric Golgi (COG) tethering complex, a major coordinator of Golgi vesicular trafficking, thus remodeling Golgi membrane traffic and redirecting Golgi-derived vesicles to the BCV. Altogether, these findings demonstrate that Brucella modulates COG-dependent trafficking via delivery of a T4SS effector to promote rBCV biogenesis and intracellular proliferation, providing mechanistic insight into how bacterial exploitation of host secretory functions promotes pathogenesis.
Keywords: Type IV secretion, Brucella, COG complex, secretory pathway, Golgi, pathogenesis, macrophage, Rab GTPases
eTOC blurb
How bacterial pathogens exploit host secretory functions to achieve their intracellular cycle remains poorly understood. Miller et al. show here that Brucella abortus delivers into macrophages a Type IV-secretion effector that remodels Golgi-associated membrane traffic to promote biogenesis of the Brucella replicative vacuole and bacterial proliferation.
Introduction
Many bacterial pathogens use secretion systems to deliver effector proteins into target host cells, which modulate specific cellular and immune functions to the pathogen’s advantage. Intracellular bacteria use these pathogenic attributes to remodel cellular compartments into niches dedicated to their survival, proliferation or persistence, making the characterization of effectors essential to understanding the molecular basis of their pathogenesis. Bacteria of the genus Brucella are the causative agents of brucellosis, a world-widespread chronic debilitating disease that affects both animals and humans (Atluri et al., 2011). Essential to Brucella pathogenesis is its ability to invade, survive and proliferate within phagocytes, such as macrophages and dendritic cells (Archambaud et al., 2010; Copin et al., 2012). Once phagocytosed, Brucella resides within a membrane-bound vacuole, the Brucella-containing vacuole (BCV), which undergoes remodeling from a compartment with endosomal/lysosomal features (eBCV) into an organelle derived from the host endoplasmic reticulum (ER) that supports bacterial replication (rBCV)(Celli et al., 2003; Comerci et al., 2001; Pizarro-Cerdá et al., 1998; Starr et al., 2008). rBCV biogenesis occurs through sustained interactions and fusion with the ER (Celli et al., 2003; Pizarro-Cerdá et al., 1998), and invokes ER-associated functions, including the Unfolded Protein Response (UPR) IRE1α signaling pathway (Qin et al., 2008; J. A. Smith et al., 2013; Taguchi et al., 2015), the autophagy-related protein Atg9 (Taguchi et al., 2015) and Sar1/COPII-dependent early secretory trafficking (Celli et al., 2005; Taguchi et al., 2015). Whether downstream secretory trafficking is important for rBCV biogenesis is unclear. While inhibition of Arf1-dependent traffic does not alter eBCV interactions with the ER (Celli et al., 2003) or cause any major defect in bacterial replication in HeLa epithelial cells (Celli et al., 2005), the small GTPase Rab2, which controls ER-Golgi trafficking (Tisdale et al., 1992), is important for rBCV biogenesis and bacterial replication in HeLa cells (Fugier et al., 2009). This argues for a possible role of additional compartments of the secretory pathway in the bacterium’s intracellular cycle.
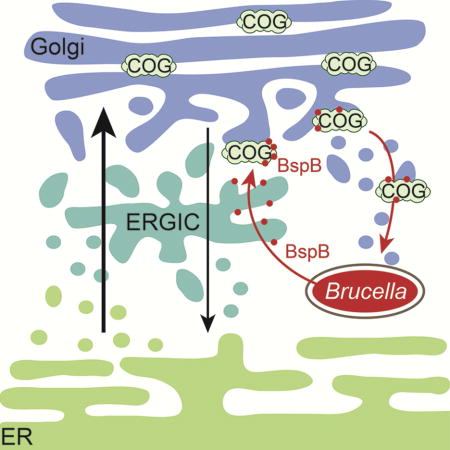
rBCV biogenesis requires a functional VirB Type IV secretion system (T4SS), which mediates Brucella intracellular growth and supports chronic infection in a murine model of brucellosis (Celli et al., 2003; Comerci et al., 2001; Hartigh et al., 2008; O’Callaghan et al., 1999; Paixão et al., 2009; Sieira et al., 2000), suggesting that remodeling of eBCVs into rBCVs is mediated by delivery of bacterial effectors. Although several VirB T4SS effectors have been identified (de Barsy et al., 2011; De Jong et al., 2008; Döhmer et al., 2014; Ines Marchesini et al., 2011; Myeni et al., 2013), only a few have been associated with rBCV biogenesis and bacterial replication, or with potential modulation of secretory trafficking. SepA is required for early BCV trafficking (Döhmer et al., 2014) and RicA binds GDP-bound Rab2 and negatively modulates eBCV remodeling into rBCV (de Barsy et al., 2011), leaving the question of its role unanswered. We recently identified a series of VirB T4SS effectors (BspA, BspB, BspF) that target secretory compartments and inhibit constitutive host protein secretion when ectopically expressed in HeLa cells (Myeni et al., 2013). Among these, BspB prominently interferes with constitutive secretion during infection (Myeni et al., 2013). Multiple deletions of bspA, bspB and bspF in B. abortus caused a replication defect in macrophages (Myeni et al., 2013), arguing for a coordinated role of these effectors in promoting bacterial intracellular growth via the modulation of host secretory functions. Here we have characterized the role of BspB in the bacterium’s intracellular cycle and dissected its mode of action on secretory compartments. We show that this effector is required for optimal rBCV biogenesis and bacterial replication via its targeting to the Golgi apparatus, interaction with the Conserved Oligomeric Golgi (COG) tethering complex, a major coordinator of intra-Golgi vesicular trafficking (Willett et al., 2013), remodeling of ER-Golgi secretory traffic and redirecting of Golgi-derived vesicles to the BCVs. These findings therefore establish the molecular mode of action of a Brucella VirB T4SS effector in promoting rBCV biogenesis via modulation of secretory trafficking, and assign a role of Golgi secretory functions in this bacterium’s intracellular cycle.
Results
Golgi-associated vesicular trafficking is required for rBCV biogenesis and Brucella replication
Brucella impairs host ER-to-Golgi secretory trafficking during infection of HeLa cells, via delivery of specific T4SS effectors (Myeni et al., 2013). These bacterial-driven trafficking alterations suggest that Brucella modulates ER-to-Golgi trafficking, yet whether they relate to rBCV biogenesis is unclear. We therefore examined the effect of the reversible Arf1 inhibitor, Brefeldin A (BFA), which disrupts ER-to-Golgi traffic (Ward et al., 2001), on rBCV biogenesis and bacterial replication. BMMs could be treated with BFA for up to 6 h without showing cytotoxic effects, which defined the time window of our analyses. In an assay measuring the kinetics of eBCV to rBCV conversion via acquisition (eBCV stage), then exclusion (rBCV stage), of the late endosomal/lysosomal marker LAMP1 on BCV membranes (Starr et al., 2008; 2012), BFA treatment during the eBCV to rBCV conversion stage (6–12 h post-infection (pi)), but not during the eBCV stage (0–6 h pi), nor once rBCVs were formed (12–18 h pi) (Celli et al., 2005; 2003; Starr et al., 2008), significantly blocked LAMP1 exclusion (Fig. 1A), indicating that BFA-sensitive compartments are important for rBCV biogenesis. Consistently, BFA treatment from 6–12 h pi, but not 0–6 h pi nor 12–18h pi, significantly decreased intracellular bacterial loads at 24 h pi (mean of 6.4, 11.0 and 8.1 bacteria/cell, respectively, compared to 11.2 in untreated BMMs; P<0.0001), as measured by fluorescence microscopy (Fig. 1B). We corroborated these findings via siRNA nucleofection of BMMs, where Arf1 depletion (Fig. 1C; 74.3±0.1% knockdown) delayed rBCV biogenesis and decreased bacterial replication (Fig. 1D–E), compared to a non-targeting control (siNT), as did depletion of the ER-associated small GTPase Sar1 (Fig. 1C; 84.0±0.1% knockdown), which is required for rBCV biogenesis and bacterial replication (Celli et al., 2005; Qin et al., 2008). Hence, Golgi-associated secretory trafficking plays a significant role in the intracellular cycle of Brucella.
Figure 1. Golgi-associated trafficking and the Brucella T4SS effector BspB are required for rBCV biogenesis and bacterial replication.
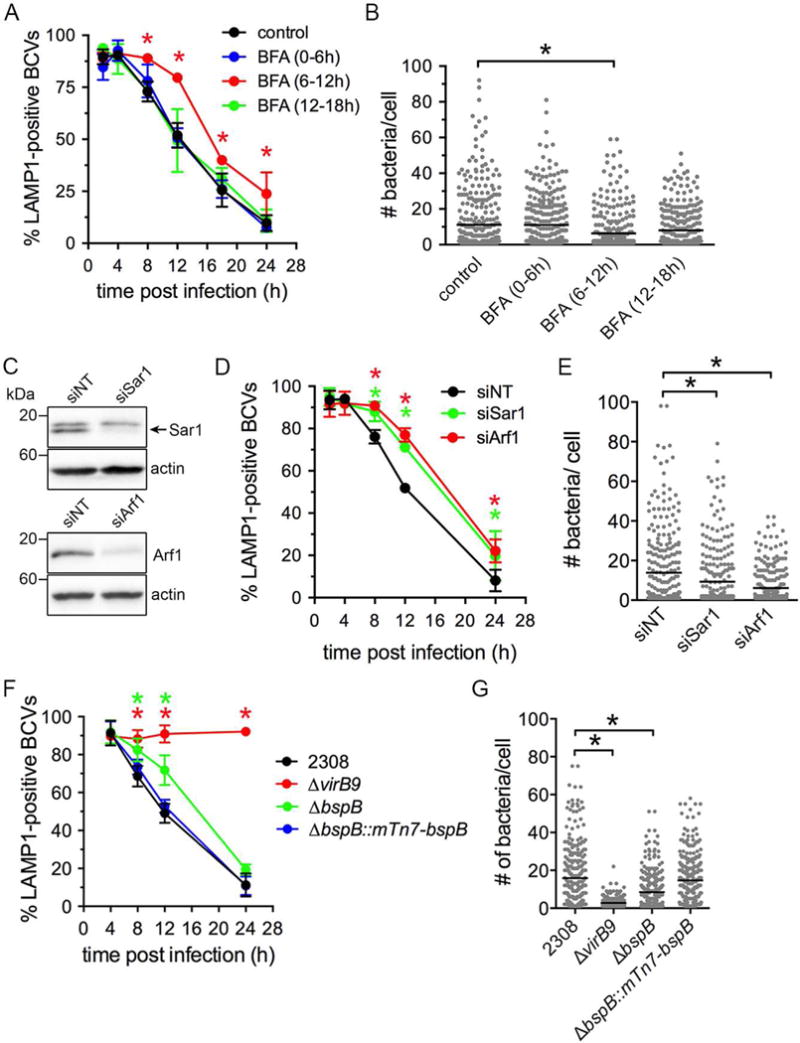
(A) BCV remodeling in BMMs treated with BFA (5 μg/ml) for 6 h intervals at 0, 6, and 12 h post infection (pi). Data are means ± SD of 3 independent experiments.
(B) Brucella replication in either control or BFA-treated BMMs, measured as number of bacteria per cell at 24 h pi. Each dot represents one cell (n>300 from 3 independent experiments) and lines indicate mean.
(C) Western blot analysis of Sar1 and Arf1 levels in BMMs upon their depletion via siRNA nucleofection, compared to non-targeting siRNA (siNT) treatment. β-actin was used as loading control.
(D) BCV remodeling in BMMs treated with either non-target (siNT), Sar1, or Arf1 siRNAs. Data are means ± SD of 3 independent experiments.
(E) Brucella replication in either control NT, Sar1, or Arf1 siRNA treated BMMs at 24 h pi. Lines indicate the means from 3 independent experiments.
(F) BMMs were infected with either wild type (2308), VirB-deficient (ΔvirB9), ΔbspB, or complemented ΔbspB (ΔbspB::mTn7-bspB) bacteria, and BCV remodeling measured via fluorescence microscopy. Data are means ± SD of 3 independent experiments.
(G) Bacterial replication in BMMs at 24 h pi. Data are means (indicated by lines) from 3 independent experiments.
Asterisks in all panels indicate statistically significant differences compared to control as determined by a two-way ANOVA with Dunnett’s multiple comparisons test (panels A, D, F) or by a Kruskal-Wallis test with Dunn’s multiple comparisons test (panels B, E, G) (P <0.05).
See also Figure S1.
BspB is required for rBCV biogenesis and optimal bacterial replication
Given that Brucella impairs ER-to-Golgi secretory traffic in a VirB T4SS-dependent manner (Myeni et al., 2013), we next considered a role of the T4SS effector BspB in rBCV biogenesis, as it prominently inhibits constitutive secretion during this stage (Myeni et al., 2013). Compared to wild type bacteria, and a VirB-deficient strain (ΔvirB9) that remained within LAMP1-positive eBCVs (Celli et al., 2005; 2003; Comerci et al., 2001), in-frame deletion of bspB significant delayed LAMP1 exclusion from eBCVs at both 8 h (P=0.0004) and 12 h (P<0.0001) pi (Fig. 1F), a phenotypic defect restored by genetic complementation with a single chromosomal copy of bspB (Fig. 1F). Consistently, the number of ΔbspB bacteria/cell at 24 h pi was intermediate to that of wild type and ΔvirB9 mutant bacteria, which showed strong and failed replication patterns, respectively (Fig. 1G). This phenotype was restored to wild type levels upon genetic complementation (Fig. 1G) and did not result from differential uptake, as all strains were similarly phagocytosed by BMMs (Fig. S1). Hence, the secretion-interfering effector BspB contributes to rBCV biogenesis and optimal bacterial replication, similar to Golgi-associated trafficking.
BspB impairs ER-Golgi secretory trafficking
Having established the contribution of BspB to the Brucella intracellular cycle, we next examined its specific role in Brucella-induced alteration of secretory trafficking. Compared to uninfected cells, traffic of the rapamycin-inducible secretory reporter ss-eGFP-FKBPF36M in HeLa-M (C1) cells (Gordon et al., 2010) infected with wild type bacteria for 24 h showed i) a slight delay in exit from the ER, ii) significant retention in the ER-Golgi intermediate compartment (ERGIC) and Golgi compartments and iii) a strong delay in traffic through the Trans-Golgi network (TGN) and into secretory vesicles (SV) (Fig. 2A–B), consistent with our previous observations (Myeni et al., 2013). By contrast, infection with the ΔbspB mutant failed to elicit any of these effects and chromosomal complementation restored the trafficking defects (Fig. 2B). Moreover, ectopic expression of HA-tagged BspB in HeLa-M (C1) cells reproduced the secretory trafficking defects caused by bacterially translocated BspB (Fig. 2B–C), altogether demonstrating that BspB is necessary and sufficient for Brucella-induced alterations of ER-to-Golgi secretory trafficking.
Figure 2. BspB impairs ER-to-Golgi secretory trafficking.
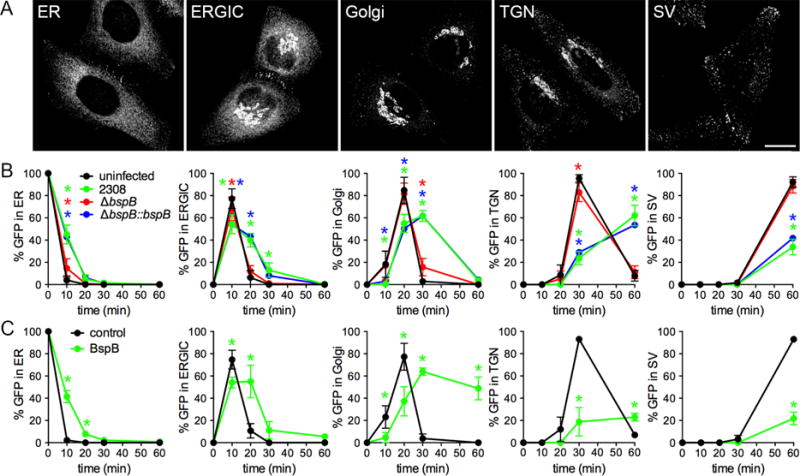
(A) Representative micrographs of ss-eGFP-FKBPF36M trafficking through secretory compartments [ER, ERGIC, Golgi, TGN, and post TGN secretory vesicles (SV)] in uninfected HeLa-M-(C1) cells post-rapamycin addition. Scale bar, 10 μm.
(B) Quantification of ss-eGFP-FKBPF36M trafficking in uninfected or Brucella-infected cells post-rapamycin addition over time. HeLa-M-(C1) cells were either uninfected or infected with B. abortus wild type (2308), ΔbspB, or complemented (ΔbspBⵆbspB) bacteria for 24 h. Rapamycin was added to initiate secretory traffic of ss-eGFP-FKBPF36M (t) and 0 its colocalization with Calnexin (ER), ERGIC-53 (ERGIC), GM130 (Golgi), p230 (TGN), or secretory vesicles was scored over a 60 min time course. Data are means ± SD from 3 independent experiments. Asterisks indicate statistical significant differences compared to uninfected cells as determined by a two-way ANOVA with Tukey’s multiple comparisons test (P<0.05).
(C) Quantification of ss-eGFP-FKBPF36M trafficking in HeLa-M-(C1) cells transfected for 24 h with pCMV-HA (control) or pCMV-HA-BspB (BspB) prior to rapamycin addition. Data are means ± SD from 3 independent experiments. Asterisks indicate statistical significant differences compared to control cells as determined by a two-way ANOVA with Bonferroni’s multiple comparisons test (P<0.05).
BspB targets the Golgi apparatus and interacts with the Conserved Oligomeric Golgi (COG) complex
To understand BspB’s mode of action on secretory traffic, we next sought to identify its host molecular target(s). Using ectopic expression of BspB, which recapitulates the effect of Brucella-delivered BspB (Fig. 2), we found that HA-BspB specifically co-immunoprecipitated with a number of 80–120 kDa proteins, which were identified as the COG1, COG2, COG3, COG4, COG5 and COG 7 subunits of the Conserved Oligomeric Golgi (COG) complex (Fig. 3A). The COG complex is a 8-subunit multimeric tethering complex (MTC) belonging to the CATCHR (complexes associated with tethering containing helical rods) family (Willett et al., 2013). Composed of two lobes (lobe A, COG1-4; lobe B COG5-8), it associates with Golgi membranes and serves as an interaction hub for vesicular trafficking proteins such as small GTPases, SNAREs (Soluble NSF Attachment protein Receptors), tethers, coat proteins and molecular motors, and orchestrates vesicle docking and fusion within the Golgi apparatus, contributing to intra-Golgi retrograde trafficking and transport of Golgi glycosylation enzymes (Lees et al., 2010; Willett et al., 2013). Co-immunoprecipitation of HA-BspB with both lobes of the COG complex was confirmed using a series of COG subunit-specific antibodies (Fig. 3B). Consistent with the Golgi-associated role of the COG complex, ectopically expressed HA-BspB in HeLa cells not only localized to the ER, as we previously reported (Myeni et al., 2013), but also to the ERGIC and Golgi apparatus (Fig. 3C and 4A), accumulating in COG8-positive structures (Fig. 3C). HA-BspB expressed in BMMs predominantly accumulated in the ERGIC and Golgi apparatus (Fig. S2A). Hence, BspB interacts with the COG complex and associates with the ERGIC and Golgi apparatus.
Figure 3. The Conserved Oligomeric Golgi (COG) complex co-immunoprecipitates with BspB and is required for rBCV biogenesis and bacterial replication.
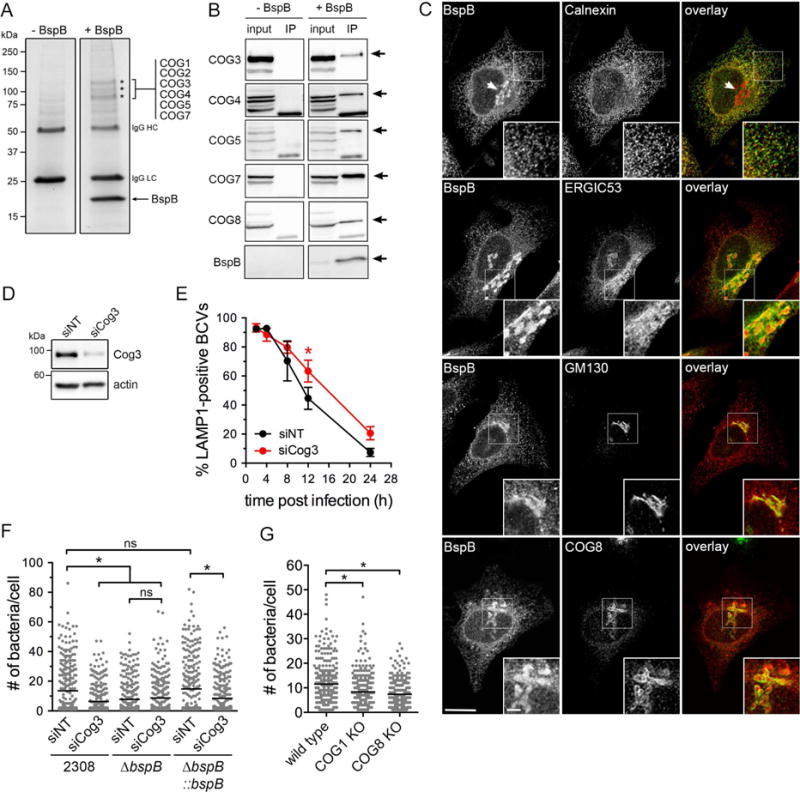
(A) Representative co-immunoprecipitation of HA-BspB with COG subunits. HA-BspB expressed in HeLa cells was immunoprecipitated using anti-HA-conjugated magnetic beads and co-immunoprecipitated proteins (asterisks) absent from control (−BspB) were identified by SDS-PAGE followed by micro-capillary LC/MS/MS.
(B) Representative Western blot analyses of co-immunoprecipitates from control (−BpsB) and HA-BspB-expressing (+BspB) HeLa cells probed with antibodies against COG3, 4, 5, 7 and 8 subunits. Arrows indicate the corresponding COG subunit band. Input was 4% of clarified cell lysates.
(C) Representative confocal micrographs of HeLa cells transfected with a plasmid expressing HA-tagged BspB (red) and immunostained for calnexin, ERGIC53, GM130 or COG8 (green). Scale bar, 10 μm.
(D) Representative Western blot analysis of Cog3 depletion in BMMs upon siRNA nucleofection, compared to non-targeting siRNA (siNT) treatment. β-actin was used as loading control.
(E) BCV remodeling in BMMs treated with either siNT or siCog3 siRNAs for 72 h prior to infection with DsRedm-expressing B. abortus wild type bacteria (2308::mTn7K-DsRedm). Data are means ± SD from 3 independent experiments. Asterisks indicate statistically significant differences compared to siNT control as determined by a two-way ANOVA with Bonferroni’s multiple comparisons test (P<0.05).
(F) Brucella replication in BMMs treated with either siNT or siCog3 siRNAs for 72 h prior to infection with DsRedm-expressing B. abortus wild type (2308), ΔbspB or complemented ΔbspB strains for 24 h. Lines indicate the means from 3 independent experiments.
(G) Replication of DsRedm-expressing Brucella in either wild type, COG1, or COG8 KO HEK293T cells at 12 h pi. Lines indicate the means from 3 independent experiments. Asterisks in (F) and (G) indicate statistically significant differences compared to control determined by a Kruskal-Wallis test with Dunn’s multiple comparisons test (P<0.05). ns, not significant.
See also Figure S2.
Figure 4. BspB Golgi targeting and interaction with the COG complex are required for bacterial replication.
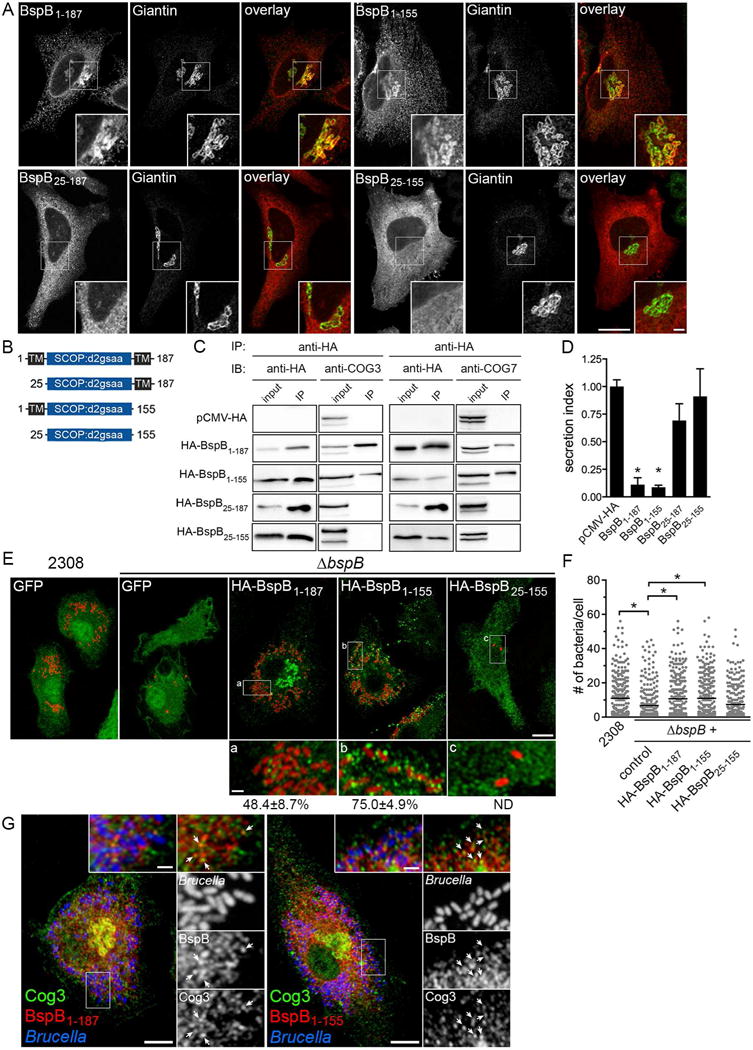
(A) Representative confocal micrographs of HeLa cells expressing either HA-BspB1–187 (full-length), HA-BspB1–155, HA-BspB25–187 or HA-BspB25–155 (red) and immunostained for Giantin (green). Scale bars, 10 μm (main panels) or 2 μm (insets).
(B) Schematic depicting predicted domains of BspB and corresponding truncations. Numbers refer to amino acid residues.
(C) Representative Western blot analysis of anti-HA co-immunoprecipitation of either HA-BspB1–187, HA-BspB1–155, HA-BspB25–187 or HA-BspB25–155 with COG3 (left panels) or COG7 (right panels) in HeLa cells. Transfection with empty vector (pCMV-HA) was used as negative control. Input was 4% of clarified cell lysates.
(D) SEAP secretion in HeLa cells expressing either HA-BspB1–187, HA-BspB1–155, HA-BspB25–187 or HA-BspB25–155 for 24 h, compared to cells transfected with empty vector pCMV-HA. Data are means ± SD from 3 independent experiments. Asterisks indicate statistical significance compared to pCMV-HA as determined by one-way ANOVA with Tukey’s multiple comparisons test (P< 0.05).
(E) Representative confocal micrographs showing expression and localization of GFP, HA-BspB or HA-BspB truncations in Brucella-infected BMMs. BMMs were transduced to express GFP, HA-BspB1–187, HA-BspB1–155 or HA-BspB25–155 for 24 h, then infected for 24 h with either DsRedm-expressing B. abortus wild type (2308) or ΔbspB strains, and immunostained with anti-HA antibodies. Insets show recruitment of BspB-positive vesicles to BCVs. Scale bars, 10 μm (main panels) and 1 μm (insets).
(F) Bacterial replication in BMMs expressing GFP (control), HA-BspB1–187, HA-BspB1–155 or HA-BspB25–155, and infected with DsRedm-expressing B. abortus wild type (2308) or ΔbspB strains. Lines indicate means from 3 independent experiments. Asterisks indicate statistical significance determined by a Kruskal-Wallis test with Dunn’s multiple comparisons test (P<0.05).
(G) Representative confocal micrographs of BMMs transduced to either express HA-BspB1–187 or HA-BspB1–155 for 24 h, infected with DsRedm-expressing B. abortus ΔbspB bacteria (pseudocolored in blue) for 24 h and immunostained with anti-HA and anti-Cog3 antibodies. Insets show that BspB-positive vesicles recruited to BCVs are also Cog3-positive (arrowheads). Scale bars, 10 μm (main panels) and 1 μm (insets).
See also Figure S3.
COG functions are required for BspB-dependent rBCV biogenesis and bacterial replication
To establish whether COG functions play a role in the Brucella intracellular cycle, we examined the effect of COG knockdown on rBCV biogenesis and bacterial replication in BMMs. siRNA-mediated depletion of Cog3 (77.3±0.5% depletion; Fig. 3D), which inactivates COG functions (Pokrovskaya et al., 2011; Zolov, 2005), or Cog 4 (80.6±0.1% depletion; Fig. S2B) significantly delayed rBCV biogenesis (P=0.007 and P=0.006, respectively; Fig. 3E and Fig. S2C), similar to the effect of bspB deletion (Fig. 1F). Depletion of either Cog3 or Cog4 also caused a significant decrease in bacterial replication at 24 h pi (P<0.0001 and P=0.0114, respectively; Fig. 3F and S2D), which was independently confirmed by deletion of either COG1 or COG8 in HEK293T cells (Bailey Blackburn et al., 2016) (P<0.0001; Fig. 3G). Bacterial replication at 12 h pi in HEK293T cells was comparable to that seen in BMMs at 24 h pi, as rBCV biogenesis was dramatically accelerated (Fig. S2E). Depletion of either Cog3 (Fig. 3F) or Cog4 (Fig. S2D) did not impact the replication defect of the ΔbspB mutant, as expected if BspB and the COG complex are functionally linked, yet it did impair replication of the complemented strain (Fig. 3F and S2D). Altogether, these data demonstrate that COG functions are required for BspB-dependent rBCV biogenesis and bacterial replication.
BspB Golgi targeting and interaction with the COG complex are required for inhibition of secretion
To further establish the functional relevance of BspB-COG interactions, we next examined the molecular determinants of BspB targeting to the Golgi apparatus and interaction with the COG complex. BspB is predicted to contain a central domain (SCOP:d2gsaa) structurally related to pyridoxal phosphate-dependent (PLP) transferases (Myeni et al., 2013), flanked by N- and C-terminal putative transmembrane domains (TM; amino acid residues 5–23 and 161–178; Fig. 4B). Microscopy analysis showed that both BspB1–187 (full-length) and BspB1–155 localized to the Golgi apparatus (65.3±1.5 and 46.0±4.4% of expressing cells, respectively), indicating that the C-terminal TM domain of BspB contributes to, but is not essential for, Golgi targeting (Fig. 4A). By contrast, neither BspB25–187 nor BspB25–155 localized to the Golgi apparatus, instead decorating either undefined perinuclear structures (BspB25–187) or appearing cytosolic (BspB25–155) (Fig. 4A), indicating that N-terminal TM domain is required for Golgi targeting. Biochemical fractionation of HeLa cells expressing BspB truncations showed enrichment of both BspB1–187 and BspB1–155 in the membrane fraction, consistent with their Golgi targeting, while BspB25–187 was enriched in the insoluble/cytoskeletal fraction and BspB25–155 in the cytosolic fraction (Fig. S3A–B). Furthermore, only Golgi-targeted BspB (BspB1–187 and BspB1–155) co-immunoprecipitated with the COG complex (Fig. 4C). Altogether, this demonstrates that the BspB N-terminal TM domain determines its association with membranes, Golgi targeting and interaction with the COG complex. However, Golgi targeting of BspB does not require a functional COG complex since full-length BspB still accumulated in the Golgi apparatus in either COG1 or COG8 KO HEK293T cells (Fig. S3C).
Given BspB’s effect on secretory trafficking (Fig. 2), we next examined whether BspB truncations affect the secretory reporter SEAP in HeLa cells (Myeni et al., 2013). While expression of either full-length BspB1–187 or BspB1–155 similarly inhibited SEAP secretion, neither BspB25–187 nor BspB25–155 significantly decreased it (Fig. 4D), indicating that only truncations that target the Golgi apparatus and interact with the COG complex inhibit constitutive secretion. Collectively, these findings establish a functional link between BspB intracellular targeting, its interaction with the COG complex and inhibitory effect on the secretory pathway.
BspB-COG interaction is required for BspB-dependent rBCV biogenesis and bacterial replication
To establish a link between BspB-COG interaction (Fig. 3), BspB’s effect on secretory trafficking (Fig. 2 and 4) and role in rBCV biogenesis and bacterial replication (Fig. 1), we next determined whether bacterial expression of the BspB truncations complements replication of the ΔbspB mutant in BMMs. Unfortunately, BspB truncations were not produced to comparable levels in B. abortus, which prevented us from conclusively testing their VirB T4SS-mediated translocation and complementation abilities. We tested instead whether production of BspB truncations in BMMs via retroviral transduction could complement in trans the replication defect of the ΔbspB mutant. Compared to expression of GFP, which did not restore normal replication of the ΔbspB mutant, BMMs expressing full length BspB1–187 supported replication of the ΔbspB mutant to levels seen with wild type bacteria (Fig. 4E and F). Hence, ectopically expressed BspB complements in trans the replication defect of the ΔbspB mutant. Expression of BspB1–155 complemented replication of the ΔbspB mutant to levels similar to full length BspB1–187, while that of BspB25–155 failed to do so (Fig. 4E and F), consistent with their respective abilities to target the Golgi apparatus, interact with the COG complex, and inhibit constitutive secretion (Fig. 4A–D). Interestingly, replicating bacteria in BspB1–187-expressing BMMs were surrounded by BspB-positive structures (48.4±8.7% of BCVs), a process enhanced in BspB1–155 expressing BMMs (75.0±4.9% of BCVs; Fig. 4E), suggesting that Brucella recruits vesicles generated from Golgi-targeted BspB to the BCV. Moreover, BspB-positive vesicles recruited to BCVs were also Cog3-positive (Fig. 4G), indicating BspB-associated recruitment of vesicular carriers containing the COG complex to rBCVs. BspB25–187 was not detectable upon retroviral transduction of BMMs, so it was not tested, and BspB25–155 did not associate with BCVs (Fig. 4E). Altogether, this data demonstrates that BspB modulation of host secretory trafficking via its interaction with the COG complex mediates its role in rBCV biogenesis and bacterial replication, and causes recruitment of COG-containing vesicles to rBCVs.
Interference with Rab1- and Rab2-dependent trafficking restores BspB-dependent bacterial replication
Since BspB alters secretory transport within the ERGIC and Golgi apparatus (Fig. 2), we next tested whether its role in bacterial replication is linked to its negative effect on vesicular transport. Both ER-Golgi anterograde and retrograde pathways are controlled by the small GTPases Rab1 and Rab2 (Tisdale, 1999; Tisdale et al., 1992; Tisdale and Balch, 1996), which interact with the COG complex(Fukuda et al., 2008; Miller et al., 2013). We therefore tested whether altering transport controlled by these GTPases influenced BspB-dependent replication of B. abortus in BMMs. Depletion of Rab1a (86.8±0.08% knockdown; Fig. 5A), but not that of Rab2a (88.4±0.04% knockdown; Fig. 5A), impaired anterograde transport of the Golgi protein Giantin upon BFA removal (Fig. S4A), demonstrating inhibition of anterograde traffic. Individual depletion of Rab1a and Rab2a impaired retrograde transport of Giantin upon BFA addition (Fig. S4B), indicating that both GTPases influence this transport pathway. Under these experimental conditions, depletion of Rab1a decreased replication of wild type, but not of ΔbspB bacteria (Fig. 5B), indicating that Rab1-dependent traffic is important for Brucella replication and possibly linked to BspB function. The combined depletions of Cog3 and Rab1a (81.5±0.1% and 83.4±0.1% knockdown, respectively; Fig. 5A) did not have an additive effect on the replication defect of wild type bacteria (Fig. 5C), suggesting they act on the same membrane trafficking process. Depletion of Rab2a altered growth of wild type bacteria, as previously reported(Fugier et al., 2009), and surprisingly restored replication of ΔbspB bacteria to wild type levels (Fig. 5B and E). Hence, the contribution of BspB to bacterial replication is phenocopied by inhibition of Rab2-dependent traffic. Furthermore, restored replication of BspB-deficient bacteria by Rab2a depletion was abolished in Cog3-depleted BMMs (Fig. 5C), demonstrating its dependency on COG functions. Conversely, using a gain-of-function approach, overexpression in BMMs of either GFP-Rab1a or its constitutively active allele (GFP-Rab1aQ70L) restored replication of ΔbspB bacteria to wild type levels, but did not significantly alter growth of wild type bacteria (Fig. 5D–F), compared to replication in control GFP-expressing BMMs. By contrast, overexpression of GFP-Rab2a or its constitutively active allele (GFP-Rab2aQ65L) did not alter growth of any of the strains (Fig. 5D and F). GFP-Rab1a expression in Cog3-depleted (77.0±0.02% knockdown) BMMs still restored growth of the ΔbspB mutant (Fig. 5D), suggesting a dominant effect of Rab1a function over COG deficiency. Considering that Rab2a depletion specifically impairs retrograde transport, whereas Rab1a depletion affects both anterograde and retrograde transport (Fig. S4), these findings collectively indicate that suppressing Rab2a-dependent retrograde traffic restores normal replication of BspB-deficient bacteria, demonstrating that BspB’s function in bacterial replication invokes remodeling of COG-dependent trafficking events within the ERGIC and Golgi compartments.
Figure 5. Interference with Rab1- and Rab2-dependent trafficking restores BspB-dependent replication.
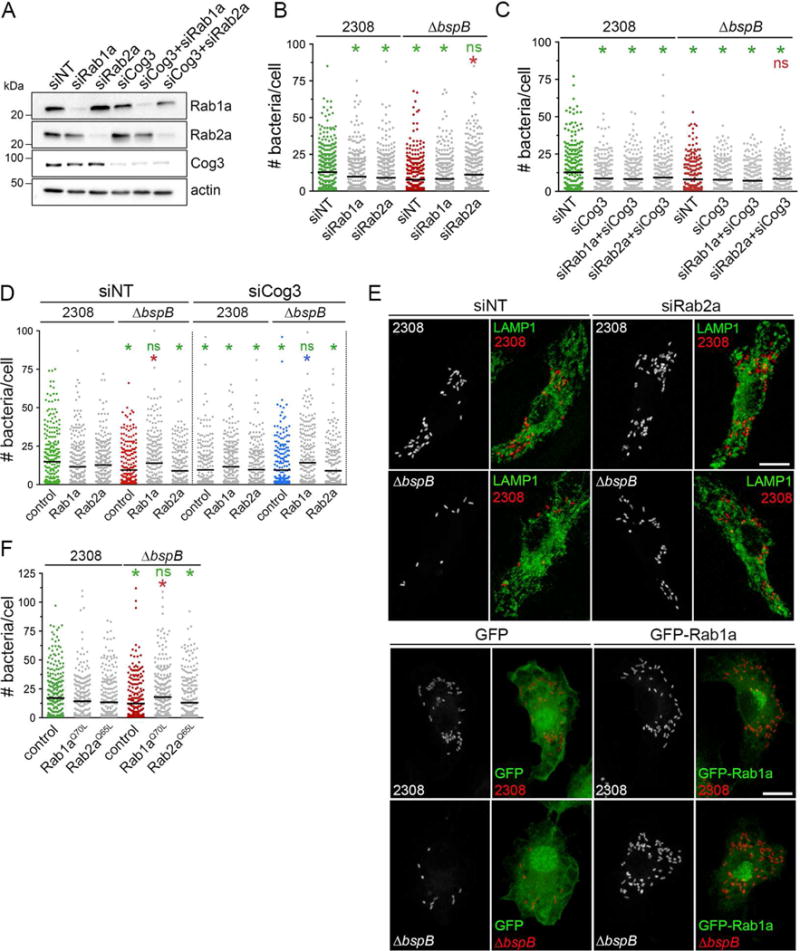
(A) Representative Western blots analysis of Rab1a, Rab2a and Cog3 depletions in BMMs treated with individual or combinations of siRNAs for 72 h. β-actin was used as loading control.
(B) Brucella replication in BMMs treated with either NT, Rab1a or Rab2a siRNAs for 72 h prior to infection with DsRedm-expressing B. abortus wild type (2308) or ΔbspB strains for 24 h.
(C) Brucella replication in BMMs treated with NT, Cog3, Rab1a or Rab2a siRNAs, or their combinations, for 72 h prior to infection with DsRedm-expressing B. abortus wild type (2308) or ΔbspB strains for 24 h.
(D) Brucella replication in BMMs treated with either NT or Cog3 siRNAs for 72 h, and transduced to express GFP (control), GFP-Rab1a or GFP-Rab2a for 24 h prior to infection with either DsRedm-expressing B. abortus wild type (2308) or ΔbspB strains for 24 h.
(E) Representative confocal micrographs of siRNA-treated BMMs (upper panels) or BMMs expressing GFP or GFP-Rab1a via retroviral transduction (lower panels), that were infected with either B. abortus wild type (2308) or ΔbspB bacteria for 24 h. siRNA-treated BMMs were immunostained for LAMP1 to identify bacteria in rBCVs. Single channel panels show bacteria. Scale bars, 10 μm.
(F) Brucella replication in BMMs transduced to express GFP (control), GFP-Rab1aQ70L or GFP-Rab2aQ65L for 24 h prior to infection with either DsRedm-expressing B. abortus wild type (2308) or ΔbspB strains for 24 h.
In panels B, C, D and F, lines indicate the means from 3 independent experiments. Color-coded asterisks indicate statistically significant differences compared to control datasets, as determined by a Kruskal-Wallis test with Dunn’s multiple comparisons test (P<0.05). ns, not significant.
See also Figure S4.
BspB redirects COG-dependent vesicular traffic to the BCV
Given the potential remodeling of vesicular traffic between the ERGIC and Golgi compartment by BspB, we next tested whether this effector causes morphological changes to these compartments. While infection of BMMs with wild type B. abortus did not cause any overt changes in cis/medial Golgi structures at 24 h pi (Fig. S5A), the ERGIC marker GFP-p58 was redistributed to the cis/medial Golgi at both 12 and 24 h pi (Fig. 6A–B). The ΔbspB strain did not, however, cause GFP-p58 redistribution at 12 h pi and only induced a reduced redistribution at 24 h pi, compared to uninfected cells (Fig. 6B). The complemented strain (ΔbspBⵆmTn7-bspB) behaved like the wild type strain at both time points (Fig. 6B), indicating a BspB-dependent process. Moreover, ectopic expression of HA-BspB via retroviral transduction in BMMs also caused GFP-p58 redistribution to the Golgi apparatus (Fig. S5B), indicating that BspB is necessary and sufficient for this alteration.
Figure 6. BspB redirects COG-dependent vesicular traffic to the BCV.
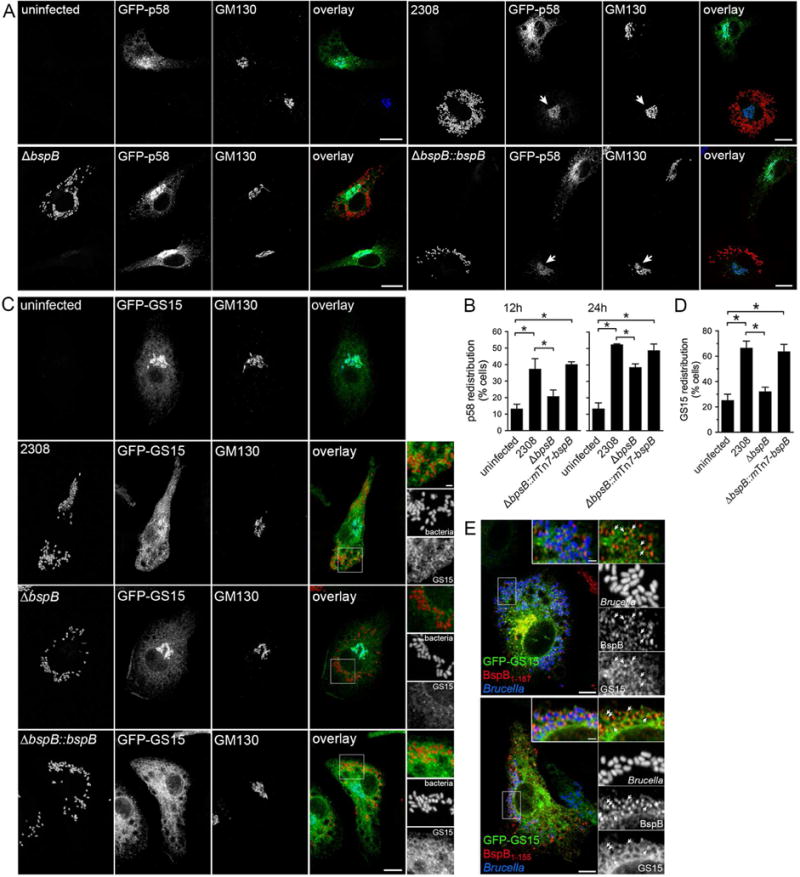
(A) Representative confocal micrographs of BMMs expressing GFP-p58 via retroviral transduction and either left uninfected or infected with either B. abortus wild type (2308), ΔbspB or complemented (ΔbspB::bspB) bacteria for 24 h, and immunostained for GM130. Arrows indicate redistribution of GFP-p58 to the Golgi apparatus. Scale bars, 10 μm.
(B) Quantification of GFP-p58 redistribution in uninfected BMMs or those infected with B. abortus wild type (2308), ΔbspB or complemented (ΔbspBⵆmTn7-bspB) bacteria for either 12 h or 24 h. Data are means ± SD from 3 independent experiments. Asterisks indicate statistically significant differences compared to uninfected BMMs as determined by one-way ANOVA with Tukey’s multiple comparisons test (P < 0.05).
(C) Representative confocal micrographs of BMMs expressing GFP-GS15 via retroviral transduction and either left uninfected or infected with either B. abortus wild type (2308), ΔbspB or complemented (ΔbspBⵆbspB) bacteria for 24 h, then immunostained for GM130. Insets illustrate GS15 redistribution to BCVs containing wild type or complemented bacteria only. Scale bars, 10 μm (main panels) and 2 μm (insets).
(D) Quantification of GFP-GS15 redistribution in uninfected BMMs or those infected with B. abortus wild type (2308), ΔbspB or complemented (ΔbspBⵆmTn7-bspB) bacteria for 24 h. Data are means ± SD from 3 independent experiments. Asterisks indicate statistically significant differences compared to uninfected BMMs as determined by one-way ANOVA with Tukey’s multiple comparisons test (P < 0.05).
(E) Representative confocal micrographs of BMMs transduced to express GFP-GS15 and either HA-BspB1–187 or HA-BspB1–155 for 24 h, infected with DsRedm-expressing B. abortus ΔbspB bacteria (pseudocolored in blue) for 24 h and immunostained with anti-HA antibodies. Insets show that BspB-positive vesicles recruited to BCVs are also GS15-positive (arrowheads). Scale bars, 10 (main panels) and 1 μm (insets).
See also Figure S5
Since redistribution of ERGIC53/p58 to the cis-Golgi compartment has been associated with COG complex dysfunction (Steet and Kornfeld, 2006), we also examined whether Brucella infection alters the distribution of COG-interacting Golgi SNAREs, as a readout of COG-dependent traffic alterations. No changes in the cellular distribution of the Golgi t-SNARE Syntaxin5 (Stx5) were observed upon infection of BMMs expressing GFP-Stx5 with wild type or ΔbspB bacteria (Fig. S5D). By contrast, distribution of the Golgi v-SNARE GS15, which depends upon a functional COG complex (Oka et al., 2004), was dramatically altered by infection with wild type bacteria, from a Golgi localization to a diffuse pattern (Fig. 6C–D). This effect was BspB-dependent, as the ΔbspB strain failed to cause this redistribution but the complemented strain did (Fig. 5C–D), and ectopic expression of HA-BspB in BMMs partially recapitulated redistribution of GS15 (Fig. S5C). Importantly, BspB-induced redistribution of GS15 was associated with GS15 recruitment to BCVs (Fig. 6C), indicating acquisition of COG-dependent, Golgi-derived vesicular carriers. Consistently, BspB-positive vesicles recruited to ΔbspB BCVs upon in trans complementation also contained GS15 (Fig. 6E), further indicating their Golgi-derived origin. Altogether, these findings demonstrate that BspB delivery during infection impairs COG-dependent transport to recruit Golgi-derived vesicular carriers to BCVs.
Discussion
Understanding the mode of action of bacterial effectors has been key to deciphering the pathogenic molecular mechanisms of many intracellular bacteria. The importance of the host secretory compartment in the biogenesis of Brucella’s rBCV has long been known (Celli et al., 2003; Pizarro-Cerdá et al., 1998), yet our understanding of the underlying molecular mechanisms has been limited by our lack of knowledge of the VirB T4SS effectors mediating these processes. Here we uncover a T4SS effector-dependent molecular mechanism of Brucella modulation of Golgi-associated traffic that promotes rBCV biogenesis and bacterial replication. Based on the role of BFA-sensitive and Arf1-dependent secretory compartments in both rBCV biogenesis and bacterial growth, our finding that BspB targets the COG complex, a Golgi MTC, demonstrate that this bacterium modulates Golgi-associated secretory processes to generate a replicative organelle. This expands upon our previous understanding of the role of the ER and ERES (Celli et al., 2005; 2003; Pizarro-Cerdá et al., 1998) in these processes, arguing that Brucella modulates multiple secretory functions to achieve its intracellular cycle.
BspB interaction with the COG complex, which plays a role in Brucella’s optimal growth, implicates this complex in pathogenesis. COG functions have been previously linked to Chlamydia trachomatis intracellular growth (Pokrovskaya et al., 2012) and HIV replication (Liu et al., 2014), yet no microbial effector was known to directly target this MTC. Through its interactions with many vesicular trafficking and fusion machinery proteins (Willett et al., 2013), the COG complex controls multiple aspects of Golgi-associated membrane trafficking, making it an ideal target for a bacterial effector to coordinately modulate various secretory trafficking stages. How BspB acts on the COG complex remains to be established. Its targeting to the Golgi apparatus does not require a functional COG complex, suggesting that its interaction with this MTC has a functional, rather than a targeting, role. The N-terminal TM domain requirement for Golgi targeting suggests that this region of BspB serves as a trafficking signal upon ectopic expression. Whether it acts similarly upon T4SS delivery of BspB remains to be established, as expression of the BspB25–187 truncation could not be detected in Brucella, possibly because this domain is required for protein stability and T4SS delivery. Nonetheless, our in trans complementation assay established that BspB Golgi targeting is important for its role in rBCV biogenesis, suggesting that bacterially delivered BspB also traffics to the Golgi compartment.
Both bacterially delivered and ectopically expressed BspB altered the steady-state distribution of the ERGIC lectin p58/ERGIC53, invoking changes in anterograde and retrograde transport pathways. Consistently, depletion of either Rab1a or Rab2a, two GTPases that bind the COG complex (Fukuda et al., 2008; Miller et al., 2013) and regulate anterograde and retrograde traffic (Tisdale et al., 1992; Tisdale and Balch, 1996; Tisdale and Jackson, 1998), impaired replication of wild type bacteria, confirming the importance of Golgi-associated vesicular transport for Brucella replication. This finding is consistent with the previously reported role of Rab2a in Brucella replication in HeLa cells (Fugier et al., 2009). Overexpression of Rab1a or depletion of Rab2a rescued the replication defect of a ΔbspB mutant, arguing that suppressing retrograde transport within the ERGIC/Golgi, or possibly enhancing the anterograde pathway, phenocopies BspB’s effect on secretory trafficking and function in Brucella replication. Interestingly, BspB deficiency was still rescued by Rab1a overexpression in COG-deficient cells. This dominant effect of Rab1a over COG functions is consistent with the suppressive effect of the yeast Rab1 homolog Ypt1 on COG-dependent growth defects (Suvorova et al., 2002; VanRheenen et al., 1998; 1999). By contrast, rescue of BspB deficiency by Rab2a depletion required a functional COG complex, in agreement with the role of Rab2 in COG-dependent retrograde trafficking of COPI-coated vesicles (Tisdale, 1999; Tisdale and Jackson, 1998). Altogether, these findings suggest that BspB’s effect on secretory traffic may cause remodeling of specific COG-dependent anterograde and retrograde transport events within the ERGIC and Golgi that invoke Rab1a and Rab2a functions. Additionally, both bacterially delivered and ectopically expressed BspB induced redistribution of GS15-containing COG complex-dependent vesicular carriers, which were recruited to replicating rBCVs in BspB-positive structures. Together with the recruitment of COG-positive, BspB-containing vesicles to rBCVs, this supports a model in which BspB causes redirection of COG-dependent, Golgi-associated vesicular traffic to BCVs. In further agreement with this model, BspB induced redistribution of the v-SNARE GS15, a COG-sensitive “GEAR” protein (Oka et al., 2004), but not of the COG-insensitive t-SNARE Syntaxin5 (Oka et al., 2004). Since GS15 preferentially associates with Golgi-derived vesicles rather than Golgi stack membranes (Willett et al., 2016), our findings argue for the recruitment of a specific subset of COG-dependent carriers by BspB.
Based on these findings, we propose a model of BspB function in rBCV biogenesis and bacterial replication whereby T4SS-delivered BspB traffics to the ERGIC and Golgi apparatus, where it interacts with the COG complex and interferes with its functions. As a consequence, ERGIC-Golgi trafficking is remodeled to redirect a subset of Golgi-derived vesicles to BCVs during rBCV biogenesis and bacterial replication. Our findings therefore support a concept by which Brucella modulates ER-Golgi trafficking to foster interactions with, or accretion of, membranes from secretory compartments. The upregulation of COPII components during infection (Taguchi et al., 2015) may also increase secretory carrier formation from ERES, further contributing to secretory membrane acquisition by BCVs. Accretion of secretory vesicles is also supported by the detection of both Rab1 and Rab2 on purified rBCVs (Fugier et al., 2009), further arguing for the recruitment of membranes from different secretory compartments.
We show here that BspB remodels membrane traffic between the ERGIC and Golgi apparatus for rBCV biogenesis and replication purposes. We previously identified additional secretion-interfering effectors (Myeni et al., 2013), so Brucella delivers multiple proteins that likely target various secretory trafficking stages, including BspA, BspF (Myeni et al., 2013) and possibly RicA via its binding of GDP-bound Rab2 (de Barsy et al., 2011). BspB therefore appears to be one of multiple virulence factors that Brucella uses to reshape the host secretory pathway to promote its intracellular cycle. Future studies of these effectors need to identify the host functions they target and characterize their mode of action to comprehend the complex role of the secretory compartment in the Brucella intracellular cycle, and advance our understanding of the importance of this compartment as a target of microbial pathogens.
STAR Methods
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jean Celli (jcelli@vetmed.wsu.edu). Distribution of Brucella strains will follow regulations of the CDC/USDA Select Agent Program to possess and use Brucella spp., in agreement with Select Agent Federal regulations (42 C.F.R. part 73 and 9 C.F.R. part 121).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Bacterial strains and culture
Brucella abortus strains 2308, 2308ΔvirB9 and 2308ΔbspB have been described previously (Celli et al., 2005; Myeni et al., 2013) and were grown on tryptic soy agar (TSA, Difco™) for 3 days at 37°C and 5% CO2, and subsequently in tryptic soy broth (TSB, Difco™) at 37°C with shaking to an OD600~1.0 for infections. For fluorescence microscopy purposes, all strains were modified to express the fluorescent protein DsRedm via integration of the miniTn7K-dsRed at the attTn7 locus by electroporation of pUC18T-miniTn7K-dsRed (Smith et al., 2016) with the helper plasmid pUC18T-Tn7tnp, as described previously(Myeni et al., 2013). Electroporants were selected on TSA plates containing 30 μg/ml of kanamycin, and correct insertion of miniTn7K-dsRed was confirmed using PCR primers RC603 and RC604 (Table S1). For genetic complementation of B. abortus strain 2308ΔbspB via chromosomal insertion of a single copy of bspB, a 714 bp DNA fragment containing 150 bp upstream of BAB1_0711 fused to bspB was PCR amplified from pUC18T-miniTn7K-bspB(Myeni et al., 2013) using primers WSU0203 and WSU0204 (Table S1), and cloned into pUC18T-miniTn7K-dsRed using EcoRI and KpnI restriction sites to generate pUC18T-miniTn7K-dsRed-bspB, which was electroporated into B. abortus 2308ΔbspB as described above.
All experiments with B. abortus strains were performed in a Biosafety Level 3 facility in compliance with the CDC Division of Select Agents and Toxins regulations in accordance with standard operating procedures approved by Washington State University Institutional Biosafety Committee. Escherichia coli strains used for cloning (DH10B, DH5α: Invitrogen) were grown in Luria-Bertani (Difco™ LB Lennox, BD) broth at 37°C, supplemented with 50 μg/ml of kanamycin or 100 μg/ml of ampicillin (Thermo Fisher Scientific) when necessary.
Mammalian cells
Human embryonic kidney 293T cells (HEK293T/17; ATCC CRL-11268; RRID:CVCL_1926; sex of cells: female) and HEK293T COG1 and COG8 knockout cells (Bailey Blackburn et al., 2016) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM 4.5 g/l glucose and sodium pyruvate; Corning Cat#15013) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Life Technologies) and 4 mM L-glutamine at 37 °C and 10% CO2. HeLa cells (ATCC clone CCL-2; RRID:CVCL_0030; sex of cells: female) were cultured in Minimum Essential Medium (MEM; Corning Cat#15010) supplemented with 2 mM L-glutamine and 10% FBS and grown at 37°C in 5% CO2. HeLa-M clone 1 (C1) cells (Gordon et al., 2010) were grown in 4.5 g/liter glucose DMEM supplemented with 10% FBS, 2mM L-glutamine and 1.66 μg/ml puromycin (ThermoFisher Scientific) at 37°C and 10% CO2.
Murine bone marrow-derived macrophages (BMMs) were generated from bone marrow cells collected from 6–12 week-old female C57BL/6NHsd mice (Envigo), following procedures approved by the Institutional Animal Care and Use Committee (IACUC) at Washingon State University (Protocol #4586), in compliance with relevant regulatory standards. Bone marrow cells were differentiated into macrophages for 5 days at 37°C and 10% CO2 in 1g/L glucose Dulbecco’s Modified Eagle’s Medium (DMEM with L-glutamine, and sodium pyruvate, Corning Cat#10014) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 20% L-929 mouse fibroblasts-conditioned medium in non tissue culture-treated Petri dishes. After 5 days, loosely adherent BMMs were washed with PBS, harvested by incubation in chilled cation-free PBS supplemented with 1g/l D-glucose on ice for 10 min, resuspended in complete medium and replated in either 6- or 24-well tissue culture treated plates at a density of 1×106 or 5×104 cell/well, respectively. The number of mice needed for each experiment was determined based on an average yield of 2 × 107 BMMs/mouse. BMMs were further incubated at 37°C under 10% CO2 atmosphere for 48 h, replenishing with complete medium 24 h before infection.
METHOD DETAILS
Construction of retroviral vectors
To facilitate cloning, we first added restriction sites to pCLXSN, by excising the multiple cloning site of p3XFLAG-CMV™-7.1 (Sigma-Aldrich #E4026) using EcoRI and BamHI restriction sites, and cloning it into the respective restriction sites of pCLXSN to create pCLXSN-MCS2. The eGFP gene was amplified from pEGFP-N1 (Clontech) using primers WSU0247 and WSU0350 (Table S1) and cloned as an EcoRI-BamHI fragment into pCLXSN-MCS2 to generate pCLXSN-GFP. Rab1a cDNA from Canis lupus was amplified from pET11d-HA-Rab1a (a gift from Dr William Balch, The Scripps Research Institute) using primers WSU0298 and WSU0265 (Table S1) and cloned into pEGFP-C1 (Clontech) using SacI and BamHI restriction sites. The resulting eGFP-Rab1a fragment was then amplified using primers WSU0249 and WSU0265 (Table S1) and cloned as a BamHI fragment into pCLXSN-MCS2 to generate pCLXSN-GFP-Rab1a. The constitutively active Rab1aQ70L mutant was made using site-directed mutagenesis of pCLXSN-GFP-Rab1a (mutation of base pairs 209–210 AA>TG) through amplification with primers WSU0262 and WSU0263 (Table S1). Human Rab2a cDNA was amplified from pGEM-Rab2 and pGEM-Rab2aQ65L (kindly provided by Dr Craig Roy, Yale University) using primers WSU0305 and WSU0255 (Table S1) and cloned into pEGFP-C1 using XhoI and BamHI restriction sites. The resulting eGFP-Rab2a fragments were then amplified using primers WSU0247 and WSU0255 and cloned as an EcoRI-BamHI fragment into pCLXSN-MCS2 to generate pCLXSN-GFP-Rab2a and pCLXSN-GFP-Rab2aQ65L. Human GS15 cDNA was cloned directly from pEGFP-C1-GS15(Shestakova et al., 2007) into pCLXSN-GFP-C1 using EcoRI and BamHI restriction sites resulting in pCLXSN-GFP-GS15. GFP-Syntaxin5 cDNA from Rattus norvegicus (Kudlyk et al., 2013) was amplified using the primers WSU0354 and WSU0353 (Table S1) and subsequently cloned into pCLXSN-MCS2 using ClaI and BamHI restriction sites to generate pCLXSN-GFP-Stx5. All plasmids were confirmed by sequencing.
Construction of BspB truncations
The pCMV-HA-bspB plasmid was constructed previously(Myeni et al., 2013). BspB truncation constructs were generated via PCR amplification from pCMV-HA-bspB using either primers WSU0003 and WSU0006 (bspB1–155), WSU0004 and WSU0005 (bspB25–187), and WSU0004 and WSU0006 (bspB25–155) (Table S1). PCR products were subsequently cloned into pCMV-HA (Clontech) using EcoRI and XhoI restriction sites. The same HA-tagged bspB truncations were cloned into pCLXSN (Imgenex) using EcoRI and BamHI restrictions sites, after amplification from pCMV-HA-bspB using either primers WSU0301 and(bspB1–187), WSU0303 and WSU0302 (bspB1–155), WSU0304 and WSU0301 (bspB25–187), or WSU0304 and WSU0302 (bspB25–155) (Table S1). All plasmids were confirmed by sequencing.
siRNA treatment and retroviral transduction of BMMs
After 5 days of differentiation, BMMs were collected and 1×106 cells transfected with 2–3 μM ON-TARGETplus SMARTpool siRNAs (GE Dharmacon) directed against either mouse Arf1 (L-060268-01-0005), mouse Sar1a (L-042764-00-0005), mouse and human Cog3 (L-057180-01-0020; L-013499-02-00020), mouse Cog4 (L-051963-01-0005), mouse Rab1a (L-040850-01-0005), mouse and human Rab2a (L-040851-01-0005; L-010533-00-0005), or a non-targeting (siNT; J-001810) siRNA, in an Amaxa™ Nucleofector II using the Mouse Macrophage Nucleofector® Kit (Lonza). BMMs were immediately diluted in pre-warmed medium, plated either onto coverslips in a 24-well plate, or in a 6-well plate, and incubated for 72 h prior to infection. Protein depletions were evaluated by Western blot analysis using β-actin levels for normalization. Retroviral transductions of BMMs were performed using derivatives of pCLXSN and the ecotropic helper plasmid pCL-Eco (Retromax, Imgenex). pCLXSN-GFP-p58 was described previously (Celli et al., 2005; Kagan and Roy, 2002). Retroviral supernatants were generated as follows: HEK 293T cells were seeded in 10 cm tissue culture dishes at 2.5×106 in 20ml medium and transfected after 24 h with a mix of 800 μl DMEM, 8 μg pCL-Eco, 8 μg pCLXSN derivative, and 48 μl FuGene® 6 following the manufacturer’s protocol, and incubated for 48 h prior to collection. Retroviral supernatants filtered through a 0.45 μm filter were added to BMMs (2:5 ratio v/v), and retroviral transduction proceeded for 24 h before BMM infections were performed.
Infection of mammalian cells
For infections, bacterial cultures diluted in chilled tissue culture medium were added to chilled BMMs at a multiplicity of infection (MOI) of 10, or to HeLa-M (C1) or HEK 293T cells at a MOI of 1000 in the presence of 200 μg/ml recombinant human epidermal growth factor (EGF; EMD Millipore). Bacteria were centrifuged onto cells at 400 × g for 10 min at 4°C and incubated for 20 min at 37°C for BMM and 30 min at 37°C for HeLa-M (C1) or HEK293T cells. Infected cells were then washed five times with DMEM or MEM to remove extracellular bacteria, then treated with 100 μg/ml of gentamicin (Gibco) between 1 and 2 h pi to kill extracellular bacteria, after which gentamicin was omitted for the remainder of the experiment. Brefeldin A (MP Biomedicals) was added to BMM medium at 5 μg/ml for 6 h intervals when necessary. All infections were repeated independently at least 3 times and treatments within experiments were randomized in each repeat.
Antibodies
For immunofluorescence: rabbit polyclonal anti-BspB (1:500; generated by QED Bioscience, Inc. using purified recombinant GST-tagged BspB), rabbit polyclonal anti-Calnexin (1:1000; Enzo Life Sciences; RRID:AB_10616095), mouse monoclonal anti-GM130 (1:100; BD Biosciences; RRID:AB_398142), mouse monoclonal anti-p230 (1:100; BD Transduction Laboratories; RRID:AB_398808), mouse monoclonal anti-LAMP1 (1:1000; clone H4A3; RRID:AB_2296838) and rat monoclonal anti-LAMP1 (1:500; clone 1D4B; RRID:AB_2134500) (both obtained from the Developmental Studies Hybridoma Bank and developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242), mouse monoclonal anti-HA (1:1000; Biolegend®; RRID:AB_2565006), rabbit polyclonal anti-Giantin (1:500; Biolegend®; RRID:AB_2565451), mouse monoclonal anti-ERGIC-53 (1:200; Alexis® Biochemicals; RRID:AB_2051363). Rabbit affinity purified anti-hCOG8 antibodies were previously described(Pokrovskaya et al., 2011). For Western blotting: rabbit polyclonal anti-Arf1 (1:1,000; Proteintech; RRID:AB_2227609), anti-Sar1 (1:1,000; Abcam; RRID:AB_11128857), anti-Rab1 (1:500; Cell Signaling; RRID:AB_2665537), anti-Rab2a (1:1000; Proteintech; RRID:AB_2176874), anti-BspB (1:5,000), anti-Calnexin (1:20,000; Enzo Life Sciences), anti-Lamin A/C (1:5,000; Cell Signaling; RRID:AB_2136278), anti-β-actin (1:20,000; Bethyl Laboratories; RRID:AB_451020) antibodies; mouse monoclonal anti-HA, (1:10,000; clone 16B12, Biolegend®), anti-Hsp27 (1:10,000; clone G31, Cell Signaling; RRID:AB_331761), anti-β-actin (1:40,000; clone 8H10D10, Cell Signaling; RRID:AB_2242334) antibodies. COG antibodies were rabbit affinity purified anti-human COG3, COG4, COG8 (Pokrovskaya et al., 2011), and rabbit polyclonal anti-COG5 (Walter et al., 1998) and anti-COG7 (Oka et al., 2004) antibodies. Secondary antibodies used were anti-mouse TrueBlot Ultra IgG HRP (1:1,000; Rockland, 18-8817-33), HRP-conjugated horse anti-mouse IgG (1:10,000; Cell Signaling Technology, 7076S) and goat anti-rabbit IgG (1:10,000; Cell Signaling Technology, 7074S).
Immunofluorescence microscopy
Mammalian cells seeded onto 12 mm glass coverslips were processed for immunofluorescence staining as follows: coverslips were washed three times in 1 × PBS, then fixed in 3% paraformaldehyde (EMD) in 1 × PBS for 20 min at 37°C. Samples were then washed three times with 1 × PBS, and free aldehydes quenched in 50 mM ammonium chloride in PBS for 30 min at room temperature. Samples were blocked and permeabilized for 30 min in 0.1% saponin (w/v), 10% normal horse serum (v/v), 1 × PBS, then incubated for 20 min to 1 h with primary antibodies diluted in permeabilization buffer at room temperature. Samples were washed in 0.1% saponin/PBS, then 1 × PBS, and incubated for 30 min with either Alexa Fluor™ 488-conjugated donkey anti-mouse IgG, anti-rat IgG, Alexa Fluor™ 568-conjugated donkey anti-mouse IgG or anti-rabbit IgG antibodies (1:500; Invitrogen, Life Technologies) at room temperature. Coverslips were washed in PBS, then rinsed in distilled H2O, and mounted on glass slides in Mowiol (Calbiochem). Samples were viewed with a Leica DM4000 epifluorescence upright microscope for quantitative analysis or a Leica SP8 confocal laser-scanning microscope for image acquisition. Representative confocal micrographs of 1024 × 1024 pixels were acquired and assembled using Adobe Photoshop CS6.
Bacterial replication assay
To monitor intracellular replication of Brucella strains, BMMs infected with DsRedm-expressing bacteria were processed for immunofluorescence staining of LAMP1 as counterstain, coverslips were mounted blind on glass slides and analyzed by independent observers by epifluorescence microscopy. Numbers of intracellular bacteria were scored in all individual infected BMMs within a series of random fields (~ 100 BMMs analyzed/experiment), and each experiment was repeated independently at least 3 times (n ~ 300 BMMs from 3 different mice).
Secretory trafficking assay
The HeLa-M (C1) cell line (Gordon et al., 2010) produces the secretory reporter protein ss-eGFP-FKBPF36M as aggregates in the ER that become solubilized upon addition of rapamycin, thereby initiating its traffic through the ERGIC, Golgi apparatus, TGN and secretory vesicles. HeLa-M (C1) cells were either infected with B. abortus at a MOI of 1000 in the presence of 200 μg/ml EGF, or transfected with pCMV-HA control (Clontech) or pCMV-HA-bspB for 24 h to allow for either bacterial replication or production of HA-BspB. Transfections were conducted following the FuGENE® 6 manufacturer’s instructions (Roche). At 24 h pi or post-transfection, rapamycin (200 nM, LC Laboratories®) was added to trigger solubilization, and initiate trafficking of, ss-eGFP-FKBPF36M, which was followed over a 60 min time course. Cells were PFA fixed and analyzed via immunofluorescence microscopy. To identify where ss-eGFP-FKBPF36M localized over time, the ER, ERGIC, Golgi apparatus, and trans-Golgi network (TGN) were individually counterstained using anti-Calnexin, anti-ERGIC-53, anti-GM130 and anti-p230 antibodies, respectively.
SEAP secretion assay
Secreted embryonic alkaline phosphatase (SEAP) secretion assays were performed as previously described (Myeni et al., 2013) with the following modifications: HeLa cells were seeded in 24-well plates to 70% confluency and co-transfected using FuGENE® 6 with plasmids expressing various truncations of BspB (300 ng DNA) and the secreted embryonic alkaline phosphatase (200 ng DNA; pSEAP2-control vector, Clontech); 500 μl of supernatant were collected and cells were lysed in 500 μl of DMEM containing 0.2% Triton X-100. Samples were centrifuged at 100 × g for 5 min to remove cell debris and SEAP activity was measured in triplicate wells, using the SEAP Reporter Gene Assay Chemiluminescent kit (Roche Cat# 11779842001). Data are represented as SEAP secretion index, which is the ratio of extracellular SEAP activity to intracellular SEAP activity, normalized to that of cells transfected with empty pCMV-HA control vector. Data are from 3 independent repeats, each performed in triplicate.
Immunoprecipitation
HeLa cells seeded in 10 cm tissue culture dishes (1×106 cells/dish) were transfected at 70–80% confluency either with pCMV-HA (empty vector) or pCMV-HA-bspB following the FuGENE® 6 manufacturer’s instructions. After 16 to 24 h of transfection, cells were washed in cold PBS and lysed in 2 ml of lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM MgCl2, 0.5% Triton X-100 (v/v), and 1:500 HALT protease inhibitor (Thermo Scientific)) on ice for 30 min, and lysates were clarified at 12,000 × g for 5 min at 4°C. Anti-HA-conjugated Dynabeads (Novex, Life Technologies; 400 μl/6×106 cells) were rinsed in 1× PBS, 0.01% Tween-20 (v/v) (Calbiochem), incubated for 2 h at 4°C with the clarified lysate, then washed five times in lysis buffer. Bound proteins were eluted in 40 μl of 120 mM Tris HCl pH 6.8, 1% glycerol (v/v), 120 mM SDS and 0.4% bromophenol blue (w/v), then heated at 95°C for 5 min. Immunopr ecipitated proteins were separated by SDS-PAGE, stained with GelCode™ Blue Safe Protein Stain (Thermo Fisher Scientific) and protein bands of interest were excised for micro-capillary LC/MS/MS mass spectrometry identification (Taplin Mass Spectrometry Facility, Harvard Medical School). Immunoprecipitations were repeated independently at least 3 times.
Differential detergent fractionation
HeLa cells were seeded in 6-well dishes at 2.4 × 105 cells/well, transfected for 17 h and subjected to differential detergent fractionation as described previously(Knodler et al., 2011) with the following modifications. Cells were washed in cold 1× PBS and incubated in 100 μl saponin lysis buffer (50 mM Tris-HCl (pH 7.5), 0.1% (w/v) saponin with protease inhibitors (Calbiochem Complete Mini EDTA-free)) on ice for 5 min. Cells were scraped and collected, and wells washed with an additional 100 μl saponin lysis buffer. Samples were centrifuged at 3,000 × g for 5 min at 4°C. Saponin-soluble proteins were precipitated with 10% (w/v) trichloroacetic acid, washed in acetone and solubilized in 120 μl of 1.5× SDS-PAGE sample buffer. Saponin-insoluble proteins were solubilized in 100 μl Triton X-100 lysis buffer (50 mM Tris-Cl pH 7.5, 0.5% (v/v) Triton X-100 with protease inhibitors) and incubated on ice for 15 min, followed by centrifugation at 5,000 × g for 10 min at 4°C. Supernatants containing Triton X-100 soluble proteins were collected and 20 μl 6× SDS-PAGE sample buffer added. Finally, the Triton X-100 insoluble pellet was solubilized in 1.5× SDS-PAGE sample buffer. Saponin-, Triton X-100- and SDS-soluble fractions were analyzed by Western blotting. Fractionations were repeated independently at least 3 times.
Western blotting
Bacterial and mammalian cell lysates were generated using 2× SDS-PAGE sample buffer (0.12 M Tris (pH 6.8), 10% (v/v) glycerol, 3.4% (w/v) SDS, 0.2 M dithiothreitol [DTT], 0.004% (w/v) bromophenol blue). Samples were boiled at 95°C for 10 min and loaded at equal volumes or according to loading controls. Proteins were resolved on SDS-PAGE and transferred onto 0.45 μm or 0.2 μm nitrocellulose membranes (Amersham Hybond-ECL, GE Healthcare). Membranes were blocked in TBST (0.14 M NaCl, 0.02 M Tris (pH 7.6), 0.1% (w/v) Tween-20), 5% (w/v) non fat dry skim milk powder for 2 h at room temperature, and probed with primary antibodies overnight at 4°C, then with HRP-conjugated secondary antibodies, all diluted in TBST-milk. Western blots were developed using the Super Signal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and imaged using a BioRad Chemi-Doc gel imaging system, and representative figures were assembled using Adobe Photoshop CS6.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 6 software. The assumptions of data distributions and variances were based on data plotting, but not on statistical tests. All results are presented as the means ± standard deviations (SD) of results from at least three independent experiments, except for bacterial replication assays, where means are presented. Statistical significance of comparisons between treatment groups was determined using either an unpaired, two-tailed Student’s t test, or for group analysis, using one-way or two-way analysis of variance (ANOVA) followed by either Bonferroni’s, Dunnett’s, or Tukey’s multiple-comparison test. For bacterial replication data analysis, a non-parametric Kruskal-Wallis test with Dunn’s multicomparison test was performed, as measures did not follow a normal distribution. A P value < 0.05 was considered significant. The specific statistical tests used are indicated in the corresponding figure legends.
Supplementary Material
Highlights.
The Brucella Type IV effector BspB promotes rBCV biogenesis and bacterial replication.
BspB targets the COG complex to redirect vesicular traffic to the rBCV.
COG-dependent membrane traffic is required for optimal bacterial replication.
Acknowledgments
We would like to thank Dr Ross Tomaino (Taplin Mass Spectrometry Facility, Harvard Medical School) for mass spectrometry, Dr Daniel Ungar for the gift of antibodies, Dr Andrew Peden for HeLa-M (C1) cells, Drs William Balch and Craig Roy for plasmids, Tetyana Kudlyk for technical assistance, and Dr Eric Lofgren for assistance with statistical analysis. This work was supported by NIH grants AI112649 and AI129992 to Jean Celli, GM083144 to Vladimir Lupashin, by Washington State University funds from the Paul G. Allen School for Global Animal Health and USDA NIFA to Jean Celli, and by USDA NIFA postdoctoral fellowship 2016-67012-25179 to Cheryl Miller.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
Conceptualization: C.N.M., V.L. and J.C; Methodology: C.N.M., E.P.S., L.A.K., V.L., J.C.; Investigation: C.N.M., E.P.S., L.A.K., J.A.C., J.C.; Writing – Original Draft: C.N.M., E.P.S., L.A.K. and J.C.; Writing – Review and Editing: C.N.M., E.P.S., J.A.C., L.A.K., V.L., J.B.B., J.C.; Funding Acquisition: C.N.M., J.C.; Resources: V.L., J.B.B., J.C.; Supervision: J.C.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Archambaud C, Salcedo SP, Lelouard H, Devilard E, de Bovis B, Van Rooijen N, Gorvel JP, Malissen B. Contrasting roles of macrophages and dendritic cells in controlling initial pulmonary Brucella infection. Eur J Immunol. 2010;40:3458–3471. doi: 10.1002/eji.201040497. [DOI] [PubMed] [Google Scholar]
- Atluri VL, Xavier MN, de Jong MF, den Hartigh AB, Tsolis RM. Interactions of the Human Pathogenic Brucella Species with Their Hosts. Annu Rev Microbiol. 2011;65:523–541. doi: 10.1146/annurev-micro-090110-102905. [DOI] [PubMed] [Google Scholar]
- Bailey Blackburn J, Pokrovskaya I, Fisher P, Ungar D, Lupashin VV. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Front Cell Dev Biol. 2016;4:23. doi: 10.3389/fcell.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med. 2003;198:545–556. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, Salcedo SP, Gorvel JP. Brucella coopts the small GTPase Sar1 for intracellular replication. Proc Natl Acad Sci USA. 2005;102:1673–1678. doi: 10.1073/pnas.0406873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerci DJ, Lorenzo MM, Sieira R, Gorvel JP, Ugalde RA. Essential role of the VirB machinery in the maturation of the Brucella abortus‐containing vacuole. Cell Microbiol. 2001;3:159–168. doi: 10.1046/j.1462-5822.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- Copin R, Vitry MA, Hanot Mambres D, Machelart A, De Trez C, Vanderwinden JM, Magez S, Akira S, Ryffel B, Carlier Y, Letesson JJ, Muraille E. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog. 2012;8:e1002575. doi: 10.1371/journal.ppat.1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Barsy M, Jamet A, Filopon D, Nicolas C, Laloux G, Rual JF, Muller A, Twizere JC, Nkengfac B, Vandenhaute J, Hill DE, Salcedo SP, Gorvel JP, Letesson JJ, De Bolle X. Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell Microbiol. 2011;13:1044–1058. [Google Scholar]
- De Jong MF, Sun YH, Den Hartigh AB, van Dijl JM, Tsolis RM. Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol Microbiol. 2008;70:1378–1396. doi: 10.1111/j.1365-2958.2008.06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhmer PH, Valguarnera E, Czibener C. Identification of a type IV secretion substrate of Brucella abortus that participates in the early stages of intracellular survival - Döhmer - 2013 - Cell. Microbiol. 2014;16:396–410. doi: 10.1111/cmi.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugier E, Salcedo SP, de Chastellier C, Pophillat M, Muller A, Arce-Gorvel V, Fourquet P, Gorvel JP. The Glyceraldehyde-3-Phosphate Dehydrogenase and the Small GTPase Rab 2 Are Crucial for Brucella Replication. PLoS Pathog. 2009;5:e1000487. doi: 10.1371/journal.ppat.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Ishibashi K, Itoh T. Large Scale Screening for Novel Rab Effectors Reveals Unexpected Broad Rab Binding Specificity. Mol Cell Prot. 2008;7:1031–10420. doi: 10.1074/mcp.M700569-MCP200. [DOI] [PubMed] [Google Scholar]
- Gordon DE, Bond LM, Sahlender DA, Peden AA. A targeted siRNA screen to identify SNAREs required for constitutive secretion in mammalian cells. Traffic. 2010;11:1191–1204. doi: 10.1111/j.1600-0854.2010.01087.x. [DOI] [PubMed] [Google Scholar]
- den Hartigh AB, Rolán HG, de Jong MF, Tsolis RM. VirB3 to VirB6 and VirB8 to VirB11, but not VirB7, are essential for mediating persistence of Brucella in the reticuloendothelial system. Journal of Bacteriology. 2008;190:4427–4436. doi: 10.1128/JB.00406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ines Marchesini M, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell Microbiol. 2011;13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Ibarra JA, Pérez-Rueda E, Yip CK, Steele-Mortimer O. Coiled-coil domains enhance the membrane association of Salmonella type III effectors. Cell Microbiol. 2011;13:1497–1517. doi: 10.1111/j.1462-5822.2011.01635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlyk T, Willett R, Pokrovskaya ID, Lupashin V. COG6 interacts with a subset of the Golgi SNAREs and is important for the Golgi complex integrity. Traffic. 2013;14:194–204. doi: 10.1111/tra.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JA, Yip CK, Walz T, Hughson FM. Molecular organization of the COG vesicle tethering complex. Nat Struct Mol Biol. 2010;17:1292–1297. doi: 10.1038/nsmb.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Dominska-Ngowe M, Dykxhoorn DM. Target silencing of components of the conserved oligomeric Golgi complex impairs HIV-1 replication. Virus Res. 2014;192:92–102. doi: 10.1016/j.virusres.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VJ, Sharma P, Kudlyk TA, Frost L, Rofe AP, Watson IJ, Duden R, Lowe M, Lupashin VV, Ungar D. Molecular insights into vesicle tethering at the Golgi by the conserved oligomeric Golgi (COG) complex and the golgin TATA element modulatory factor (TMF) J Biol Chem. 2013;288:4229–4240. doi: 10.1074/jbc.M112.426767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeni S, Child R, Ng TW, Kupko JJ, III, Wehrly TD, Porcella SF, Knodler LA, Celli J. Brucella Modulates Secretory Trafficking via Multiple Type IV Secretion Effector Proteins. PLoS Pathog. 2013;9:e1003556. doi: 10.1371/journal.ppat.1003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan D, Cazevieille C, Allardet-Servent A, Boschiroli ML, Bourg G, Foulongne V, Frutos P, Kulakov Y, Ramuz M. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol Microbiol. 1999;33:1210–1220. doi: 10.1046/j.1365-2958.1999.01569.x. [DOI] [PubMed] [Google Scholar]
- Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paixão TA, Roux CM, den Hartigh AB, Sankaran-Walters S, Dandekar S, Santos RL, Tsolis RM. Establishment of systemic Brucella melitensis infection through the digestive tract requires urease, the type IV secretion system, and lipopolysaccharide O antigen. Infect Immun. 2009;77:4197–4208. doi: 10.1128/IAI.00417-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerdá J, Moreno E, Sanguedolce V, Mege JL, Gorvel JP. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infect Immun. 1998;66:2387–2392. doi: 10.1128/iai.66.5.2387-2392.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrovskaya ID, Szwedo JW, Goodwin A, Lupashina TV, Nagarajan UM, Lupashin VV. Chlamydia trachomatis hijacks intra-Golgi COG complex-dependent vesicle trafficking pathway. Cell Microbiol. 2012;14:656–668. doi: 10.1111/j.1462-5822.2012.01747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011;21:1554–1569. doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin QM, Pei J, Ancona V, Shaw BD, Ficht TA, De Figueiredo P. RNAi Screen of Endoplasmic Reticulum–Associated Host Factors Reveals a Role for IRE1α in Supporting Brucella Replication. PLoS Pathog. 2008;4:e1000110. doi: 10.1371/journal.ppat.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestakova A, Suvorova E, Pavliv O, Khaidakova G, Lupashin V. Interaction of the conserved oligomeric Golgi complex with t-SNARE Syntaxin5a/Sed5 enhances intra-Golgi SNARE complex stability. J Cell Biol. 2007;179:1179–1192. doi: 10.1083/jcb.200705145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieira R, Comerci DJ, Sánchez DO, Ugalde RA. A Homologue of an Operon Required for DNA Transfer in Agrobacterium Is Required in Brucella abortus for Virulence and Intracellular Multiplication. J Bacteriol. 2000;182:4849–4855. doi: 10.1128/jb.182.17.4849-4855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EP, Miller CN, Child R, Cundiff JA, Celli J. Postreplication Roles of the Brucella VirB Type IV Secretion System Uncovered via Conditional Expression of the VirB11 ATPase. MBio. 2016;7:e01730–16. doi: 10.1128/mBio.01730-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Khan M, Magnani DD, Harms JS, Durward M, Radhakrishnan GK, Liu YP, Splitter GA. Brucella Induces an Unfolded Protein Response via TcpB That Supports Intracellular Replication in Macrophages. PLoS Pathog. 2013;9:e1003785. doi: 10.1371/journal.ppat.1003785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López-Otín C, Virgin HW, Celli J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T, Ng TW, Wehrly TD, Knodler LA, Celli J. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic. 2008;9:678–694. doi: 10.1111/j.1600-0854.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- Steet R, Kornfeld S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins. Mol Biol Cell. 2006;17:2312–2321. doi: 10.1091/mbc.E05-08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorova ES, Duden R, Lupashin VV. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J Cell Biol. 2002;157:631–643. doi: 10.1083/jcb.200111081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi Y, Imaoka K, Kataoka M, Uda A, Nakatsu D. Yip1A, a Novel Host Factor for the Activation of the IRE1 Pathway of the Unfolded Protein Response during Brucella Infection. PLoS Pathogens. 2015;11:e1004747. doi: 10.1371/journal.ppat.1004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale EJ. A Rab2 mutant with impaired GTPase activity stimulates vesicle formation from pre-Golgi intermediates. Mol Biol Cell. 1999;10:1837–1849. doi: 10.1091/mbc.10.6.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale EJ, Balch WE. Rab2 is essential for the maturation of pre-Golgi intermediates. J Biol Chem. 1996;271:29372–29379. doi: 10.1074/jbc.271.46.29372. [DOI] [PubMed] [Google Scholar]
- Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol. 1992;119:749–761. doi: 10.1083/jcb.119.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale EJ, Jackson MR. Rab2 protein enhances coatomer recruitment to pre-Golgi intermediates. J Biol Chem. 1998;273:17269–17277. doi: 10.1074/jbc.273.27.17269. [DOI] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Lupashin VV, Barlowe C, Waters MG. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J Cell Biol. 1998;141:1107–1119. doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Sapperstein SK, Chiang EC, Lupashin VV, Barlowe C, Waters MG. Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J Cell Biol. 1999;147:729–742. doi: 10.1083/jcb.147.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter DM, Paul KS, Waters MG. Purification and Characterization of a Novel 13 S Hetero-oligomeric Protein Complex That Stimulates in VitroGolgi Transport. J Biol Chem. 1998;273:29565–29576. doi: 10.1074/jbc.273.45.29565. [DOI] [PubMed] [Google Scholar]
- Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott-Schwartz J. Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol. 2001;155:557–570. doi: 10.1083/jcb.200107045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Blackburn JB, Climer L, Pokrovskaya I, Kudlyk T, Wang W, Lupashin V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Sci Rep. 2016;6:29139. doi: 10.1038/srep29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Ungar D, Lupashin V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem Cell Biol. 2013;140:271–283. doi: 10.1007/s00418-013-1117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J Cell Biol. 2005;168:747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.