

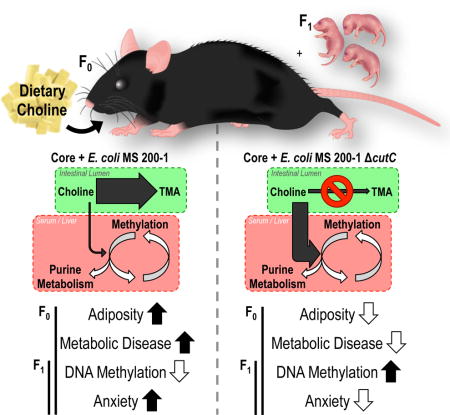
SUMMARY
Choline is an essential nutrient and methyl donor required for epigenetic regulation. Here, we assessed the impact of gut microbial choline metabolism on bacterial fitness and host biology by engineering a microbial community that lacks a single choline-utilizing enzyme. Our results indicate that choline-utilizing bacteria compete with the host for this nutrient, significantly impacting plasma and hepatic levels of methyl-donor metabolites and recapitulating biochemical signatures of choline deficiency. Mice harboring high levels of choline-consuming bacteria showed increased susceptibility to metabolic disease in the context of a high-fat diet. Furthermore, bacterially-induced reduction of methyl-donor availability influenced global DNA methylation patterns in both adult mice and their offspring and engendered behavioral alterations. Our results reveal an underappreciated effect of bacterial choline metabolism on host metabolism, epigenetics, and behavior. This work suggests that interpersonal differences in microbial metabolism should be considered when determining optimal nutrient intake requirements.
Keywords: Microbiome, Choline Deficiency, Epigenetics, Trimethylamine, Trimethylamine-N-oxide, Gnotobiotic
eTOC blurb
The gut microbiota is a dynamic metabolic organ associated with host health and disease phenotypes. Romano, Del Campo et al. report that choline-consuming gut bacteria reduce the bioavailability of this essential nutrient and deplete methyl-donor metabolites, resulting in alterations to host epigenetic programming and increased susceptibility to metabolic disease.
Choline is a quaternary amine required for many biological processes including maintenance of the structural integrity of cell membranes and support of cholinergic neurotransmission. Additionally, choline provides one-carbon units for the synthesis of the ubiquitous methyl donor S-adenosylmethionine (SAM). Long-term dietary choline deficiency results in substantial alterations to epigenetic regulation such as DNA methylation (Pogribny and Beland, 2009). Changes in DNA methylation have been associated with various human pathologies including cancer, aging, atherosclerosis, cognitive disabilities (such as depression, anxiety, schizophrenia, or Alzheimer’s disease), and autoimmune disorders (Pogribny and Beland, 2009). Furthermore, choline plays an important role in lipid metabolism, as it is required for the assembly/secretion of very low-density lipoproteins (VLDL) (Fungwe et al., 1992). Despite choline’s high abundance in common dietary constituents, it is estimated that only 10% of the US population consistently meets or exceeds the daily recommend intake of this essential nutrient established by the Institute of Medicine (Zeisel and da Costa, 2009).
The demand for choline increases during pregnancy and lactation: choline levels in amniotic fluid are 14-fold higher than in maternal plasma, and circulating levels of choline in infants are higher than in adults (Ilcol et al., 2002; McMahon and Farrell, 1985). During gestation, maternal choline deficiency reduces global DNA methylation in the fetal hippocampus and alters the development and function of cholinergic neurons that participate in learning, memory, and attention (Mellott et al., 2007; Zeisel, 2007). A recent study showed that perinatal phosphatidylcholine supplementation (above current standards) is associated with less social withdrawal and fewer attention-deficit problems in ~3 year-old children compared to an age-matched placebo group (Ross et al., 2016). Remarkably, it is estimated that 90–95% of pregnant women fail to meet the minimum recommended levels of choline intake (Brunst et al., 2014).
While choline does not compete with other nutrients for enterocyte transport, previous evidence suggest that gut microbes can limit its bioavailability (Romano et al., 2015). Gut bacterial choline metabolism generates trimethylamine (TMA) via a recently identified pathway (Craciun and Balskus, 2012). The choline utilization (cut) gene cluster encodes the glycyl radical enzyme choline TMA-lyase (CutC), which converts choline into TMA and acetaldehyde under anaerobic conditions. Culture-based and metagenomic studies have revealed a wide distribution of the cut pathway among bacteria and indicate it represents a major pathway for microbial choline utilization in the human intestine (Chen et al., 2016; Falony et al., 2015; Martínez-del Campo et al., 2015; Romano et al., 2015).
TMA produced in the intestinal lumen is absorbed into the bloodstream and further metabolized in the liver to trimethylamine-N-oxide (TMAO) by host flavin monooxygenase enzymes (Baker and Chaykin, 1962). Human studies have revealed that high serum levels of TMAO are positively associated with impaired renal function, non-alcoholic fatty liver disease (NAFLD), cardiovascular disease (CVD), and diabetes (Dambrova et al., 2016; Koeth et al., 2013; Tang et al., 2014; Wang et al., 2011). Additionally, a recent study in Malawian children showed linear growth failure and impaired cognitive function associated with low serum choline and high TMAO-to-choline ratios (Semba et al., 2016).
Despite recent progress made in characterizing the impact of TMAO on the host, there have not been studies that comprehensively evaluate the functional consequences of gut microbial ‘removal’ of choline in healthy or diseased states. Specifically, it is not clear whether choline consumption by gut bacteria impacts host methyl-donor availability and the multiple metabolic processes that depend on this nutrient. Here, we develop an experimental framework for addressing these questions by establishing a gnotobiotic mouse model harboring a synthetic gut bacterial community in which choline utilization can be manipulated. Using this model we discovered that choline-utilizing bacteria effectively compete with the host for this nutrient and lower bioavailability of methyl-donor metabolites. We then explored the potential implications of this finding on two health conditions that are highly sensitive to methyl-donor availability: high-fat diet induced metabolic disease (Craig, 2004) and pregnancy/early life (Zeisel, 2006). We found that bacterially induced depletion of metabolites involved one-carbon metabolism results in increased susceptibility to metabolic disease and alters epigenetic regulation across multiple tissues including the fetal brain.
RESULTS
Identification of a choline utilization gene cluster in E. coli MS 200-1, a human gastrointestinal isolate
Previous analyses uncovered cut gene clusters in a subset of species within the Enterobacteriaceae, including 94 E. coli strains (Martínez-del Campo et al., 2015). A more recent BLAST search identified an additional 164 E. coli strains that carry these genes. The large majority (~96%) of these cut gene cluster-containing isolates are from the human urogenital tract, gastrointestinal tract, or bloodstream (Fig. 1A). Only 4% of sequenced E. coli genomes have a cut gene cluster, indicating that these genes are part of this organism’s open and continuously evolving pangenome.
Fig. 1. In vitro characterization of E. coli MS 200-1 wild-type (WT) strain and variants bearing choline utilization (cut) gene cluster mutations.
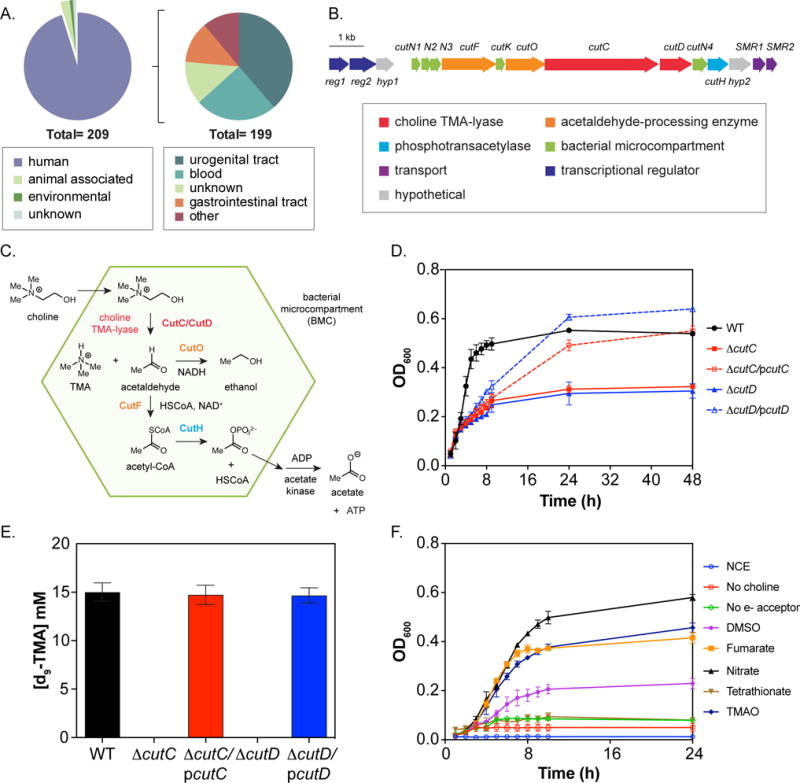
(A) Environmental distribution of sequenced cut gene cluster-containing E. coli genomes. (B) Content and organization of the cut gene cluster from E. coli MS 200-1. (C) Proposed model for anaerobic choline utilization. (D) Growth phenotype of WT, ΔcutC and ΔcutD strains on choline-containing minimal medium. Cells were grown by anaerobic respiration in NCE medium supplemented with 15 mM choline, 40 mM fumarate and 0.5 mM glucose to stimulate growth. (E) LC-MS quantification of d9-TMA produced by wild-type and cut variants in minimal medium containing 15 mM d9-choline, 40 mM fumarate and 0.5 mM glucose. Deletions were complemented by plasmid-based inducible expression of the corresponding wild-type gene (designated pcutC and pcutD) by addition of 1 μM IPTG. (F) Anaerobic respiration of choline by E. coli MS 200-1 utilizing different electron acceptors. Growth curves were performed in minimal media supplemented with 15 mM choline plus 40 mM of the indicated electron acceptor. All values are averages ± standard errors of the means (SEMs) of three independent experiments.
E. coli MS 200-1, a human gut strain that was isolated from the ileum of a patient with normal histology, was chosen for genetic characterization of the cut gene cluster due to its genetic tractability and ability to robustly colonize the small intestine, which is the primary site of choline absorption. This strain possesses a “type II” cut gene cluster (Fig. 1B), which contains fewer total genes compared to the “type I” cut gene cluster originally described in the δ-proteobacterium Desulfovibrio desulfuricans (Craciun and Balskus, 2012). Type II cut gene clusters appear to encode all the proteins required for anaerobic choline metabolism, including choline-trimethylamine lyase and its activase (CutC and CutD), five putative bacterial microcompartment (BMC) structural proteins (CutN1, CutN2, CutN3, CutN4, and CutK), a coenzyme A (CoA)-acylating aldehyde oxidoreductase (CutF), an alcohol dehydrogenase (CutO) and a phosphotransacetylase (CutH) (Fig. 1C). We inspected the genetic context of the cut gene clusters from different γ-Proteobacteria and identified additional genes potentially involved in choline utilization, including two genes encoding predicted transcriptional regulators (reg1 and reg2), two genes encoding putative small multidrug resistance proteins (SMR1 and SMR2), and two genes encoding conserved hypothetical proteins (hyp1 and hyp2). These genes are largely conserved in γ-Proteobacterial cut gene clusters but are absent from type I cut gene clusters found predominantly in Firmicutes, Actinobacteria and δ-Proteobacteria.
Three of these genes can be assigned roles in choline metabolism. Reg1 is transcriptional regulator from the TetR family that contains a BetI domain. BetI is a repressor of the bet (betaine) regulon, which encodes the enzymes that convert choline into glycine betaine. In the absence of choline, BetI represses the expression of the bet genes (Lamark et al., 1996). Similarly, Reg1 could be a transcriptional repressor of the cut pathway, which is induced when choline is present in the environment. However, the presence of a second transcriptional regulator (encoded by reg2) containing a helix-turn-helix DNA binding domain suggests that the regulation of the cut gene cluster could be more complex than the bet regulon. Small multidrug resistance (SMR) proteins act as transporters that confer resistance to quaternary cation compounds via proton motive force-energized efflux (Bay and Turner, 2012). EmrE, the best-characterized SMR protein member, transports a variety of drugs as well as choline and betaine, which are proposed to be its natural substrates. SMR1 and SMR2 could therefore encode cut pathway-specific transporters.
Genetic manipulation of choline-TMA lyase and its activase
To confirm the involvement of the cut gene cluster from E. coli MS 200-1 in anaerobic choline degradation, we constructed in-frame deletion mutants of the cutC and cutD genes, which encode choline-TMA lyase and its associated radical SAM activating protein. The ability of the resulting strains to grow using choline as the carbon source under anaerobic conditions with fumarate as a terminal electron acceptor was assessed. Both knockout mutants failed to grow on choline-containing minimal medium (Fig. 1D), establishing their essential role in anaerobic choline metabolism. Growth curve analyses showed the phenotypes described above could be restored by the individual expression of each wild-type gene from an IPTG inducible vector (Fig. 1D).
We used liquid chromatography-mass spectroscopy (LC-MS) assays to investigate whether the cutC and cutD mutants were impaired in their ability to produce TMA when incubated in choline-containing minimal media. As shown in Fig. 1E, conversion of d9-choline into d9-TMA was abolished in both knockouts and was restored by expression of the corresponding wild-type allele from a plasmid. Altogether these results validate the importance of cutC and cutD for anaerobic choline metabolism and TMA production in E. coli.
Respiratory electron acceptors support anaerobic choline growth in vitro
We examined whether various electron acceptors could support anaerobic growth on choline. Choline-containing minimal media supplemented with different electron acceptors was inoculated with the wild-type strain. Fumarate, nitrate, DMSO and TMAO were all able to support anaerobic growth on choline (Fig. 1F). In contrast, tetrathionate, the only electron acceptor used during anaerobic respiration of ethanolamine and propanediol (closely related pathways) in Salmonella enterica serotype Typhimurium, did not support anaerobic growth. This finding is supported by the absence of homologs of the ttr operon in the E. coli MS 200-1 genome and the presence of genes that encode nitrate reductases, S-oxide reductases, and N-oxide reductases. It has been recently demonstrated in mice that nitrate generated through the inflammatory response in the gut can be used by E. coli as a substrate for anaerobic respiration, allowing them to outcompete fermentative bacteria (Winter et al., 2013). We thus hypothesize that strains able to couple nitrate respiration with choline consumption may have a competitive advantage in the inflamed gut.
Choline utilization provides a moderate fitness advantage to E. coli MS 200-1 in vivo
To assess the impact of microbial choline utilization on bacterial colonization and host biology two groups of adult germ-free (GF) female C57BL/6 mice were colonized with the following microbial mixture: Bacteroides caccae, Bacteroides ovatus, Bacteroides thetaiotaomicron, Colinsella aerofaciens, and Eubacterium rectale. These organisms were selected because they are dominant members of the human gut and they do not produce TMA from choline (Qin et al., 2010; Romano et al., 2015). In addition to these five species (from here on referred to as the “core community”), the mixture contained either the wild type E. coli MS 200-1 strain (from here on referred to as “Choline Consuming” community, CC+) or the E. coli MS 200-1 ΔcutC strain, which is unable to consume choline and produce TMA (referred to as CC−). All mice were fed a purified diet containing a relatively high choline content (1% wt/wt choline; Envigo TD.140179) for two weeks after colonization. Although both E. coli strains colonized the intestinal tract, deletion of the cutC gene moderately but significantly impaired colonization compared to the wild type E. coli strain (Fig. 2A; 34.6±5.0% vs. 42.0±4.1% respectively). Additionally, the presence of a functional cut pathway negatively affected C. aerofaciens abundance, which colonized to significantly lower levels in the presence of the TMA-producing strain (Fig. 2A). These results suggest that choline metabolism provides a fitness advantage to E. coli and that choline consumption and/or TMA production impacts gut community structure. Analysis of serum samples from these mice confirmed that only mice colonized with the CC+ community accumulated TMAO (Fig. 2B). These mice also exhibited a 3-fold reduction in serum choline (Fig. 2C). These results suggest that bacterial choline consumption decreases choline bioavailability to the host.
Fig. 2. In vivo fitness and functional assessment of E. coli MS 200-1 choline utilization deficient mutants.
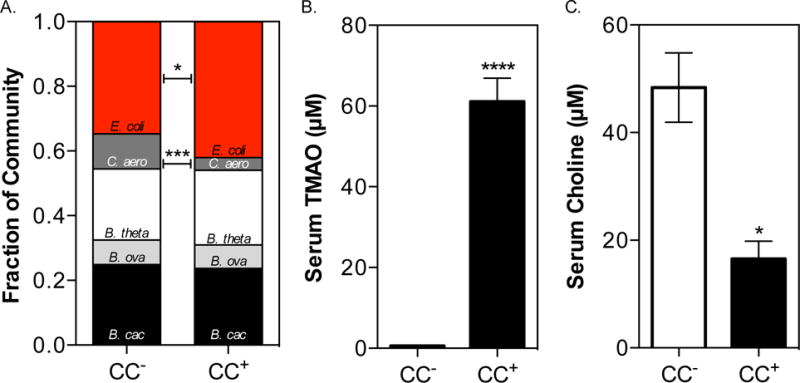
C57BL/6 females (5 animals in each group) were colonized with the following microbial core community: B. caccae, B. ovatus, B. thetaiotaomicron, C. aerofaciens, and E. rectale. In addition to these five species, mice were co-colonized with either the WT E. coli MS 200-1 strain (CC+) or the ΔcutC (choline trimethylamine-lyase) bearing variant (CC−). Mice were maintained for 2 weeks on a 1% (wt/wt) choline diet. (A) COmmunity PROfiling by Sequencing (COPRO-Seq) analysis of core community members and E. coli in cecal contents. (B) Serum levels of TMAO. (C) Serum levels of choline. All values are averages ± SEMs. Values that are significantly different (Student’s t-test) are indicated: *, P <0.05; ***, P <0.001; ****, P <0.0001.
Bacterial choline utilization recapitulates biochemical signatures of diet-induced choline deficiency
Choline is an important precursor for the synthesis of methionine and ultimately the methyl donor SAM. Furthermore, choline and SAM are metabolically linked with purine and creatine biosynthesis, as well as the transsulfuration pathway (Fig. 3A). We investigated whether the presence of intestinal microbial choline utilization altered serum and hepatic levels of metabolites involved in one-carbon, transsulfuration, and purine metabolism in the CC+ and CC− mice discussed above using untargeted metabolomics. We found that serum levels of metabolites involved in the generation of SAM, including choline, betaine, methionine, dimethylglycine, and methionine sulfoxide, were significantly reduced in the mice colonized with the CC+ community (Fig. 3B). Additionally, metabolites from the transsulfuration pathway, including cysteine and glutathione, were significantly increased while those associated with purine metabolism, including inosine, hypoxanthine, xanthine, kynurenine, and uric acid, were significantly reduced (Fig. 3C, D). Synthesis of creatine requires a large amount of methyl groups (Mudd and Poole, 1975), and creatine supplementation prevents the hepatic consequences of a choline-deficient diet (Deminice et al., 2015). However, we found that creatine levels are higher in serum of CC+ colonized mice compared with the CC− group (Fig. 3B). While choline deficiency may impair creatine synthesis, impaired renal function can significantly increase its concentration in serum. Interestingly, TMAO, which is increased in the CC+ community (Fig. 2B), is associated with renal insufficiency, chronic kidney disease, and necrosis (Tang et al., 2014).
Fig. 3. Analysis of one-carbon, transsulfuration, and purine metabolism.
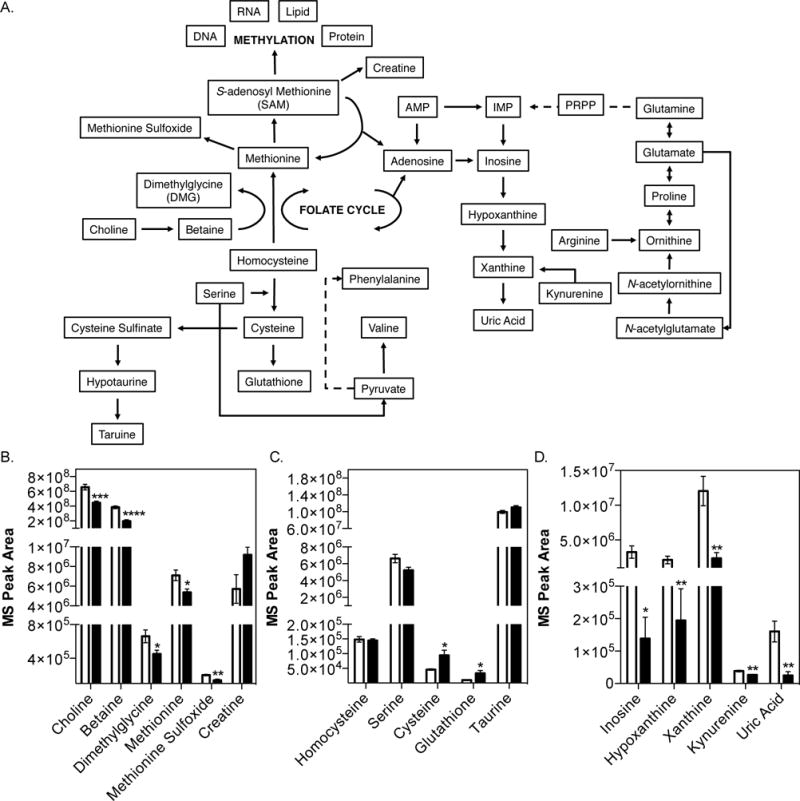
Measurements of serum metabolites from adult female mice colonized with either the CC− (white bars) or CC+ (black bars) community maintained on a 1% choline diet was conducted using an uHPLC-MS/MS (5 animals in each experimental group). (A) Choline metabolism pathway schematic. Serum levels of metabolites involved in (B) one-carbon metabolism, (C) transsulfuration pathway, and (D) purine metabolism. All values are averages ± SEMs. Values that were significantly different (Student’s t-test) are indicated: *, P <0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001.
Analyses of liver metabolites revealed similar differences between the two groups of mice, including lower levels of betaine, methionine, and purine metabolites in mice colonized with the CC+ community (Fig. S1A, B). CDP-choline, an intermediate in the de novo production of phosphatidylcholine from choline (Kennedy and Weiss, 1956), was also detected at lower levels in the CC+ mice (Fig. S1C). During choline deficiency hepatocytes overproduce reactive oxygen species (ROS) due to abnormal mitochondrial function and inhibition of complex I, which can lead to the signaling of apoptosis (Zeisel, 2012). Mitochondrial ROS production stimulates the transsulfuration pathway and early release of internal glutathione stores (Bhandary et al., 2012). We quantified metabolites associated with the transsulfuration pathway (Fig. S1D) and found significantly higher levels of glutathione in the livers of mice colonized with the CC+ community (Fig. S1E). Moreover, differential expression of genes associated with ROS production and apoptosis was observed in the livers of mice colonized with the CC+ community (Table S1). As mentioned above, choline deficiency is known to impair mitochondrial function by specifically inhibiting complex I. We observed significantly reduced expression of Ndufb2, a complex I NADH dehydrogenase (Table S1). Reduced Ndufb2 expression was accompanied by a simultaneous increase in NADH levels (Fig. S1F).
The molecular and biochemical signatures elicited by bacterial choline utilization are similar to those caused by choline deficiency. We further investigated how the serum levels of choline in CC+ mice compared to those of CC+ mice fed a choline-deficient diet (Envigo TD.88052). We found that choline levels in mice colonized with the CC+ community maintained on a 1% choline diet did not differ significantly from those in mice colonized with the same community maintained on a choline-deficient diet. Both groups of mice showed significantly lower levels of choline compared to mice colonized with the CC− community fed a 1% choline diet (Fig. 4A). Mice colonized with the CC+ community maintained on a choline-supplemented vs. a choline-deficient diet exhibited no differences in the abundance of metabolites involved in the generation of SAM (choline, betaine, methionine, dimethylglycine, and methionine sulfoxide) and most metabolites involved in the transsulfuration pathway (Fig. 4B, C). However, the metabolomes from both treatment groups were not identical; for example metabolites associated with purine metabolism (i.e. inosine, hypoxanthine, xanthine, kynurenine, and uric acid) were detected at lower levels in mice maintained on the choline deficient diet compared to the CC+ mice maintained on a 1% choline diet (Fig. S2). Altogether these results suggest that while gut microbial choline metabolism can impact nutrient bioavailability in ways that are consistent with dietary choline deficiency, the alterations to the global metabolome are not as stark as those imparted by a choline-depleted diet.
Fig. 4. Analysis of one-carbon and purine metabolism in serum from mice maintained on a choline-supplemented diet vs. choline-deficient diet.
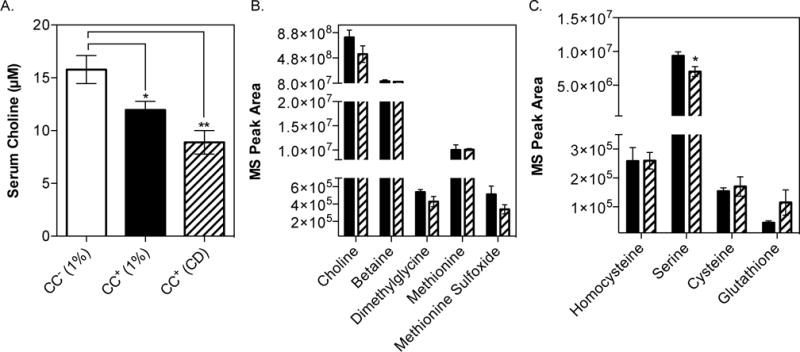
C57BL/6 females were colonized with the CC+ community and maintained for 2 weeks on either a 1% (wt/wt) choline-supplemented diet or choline-deficient diet. Mice were fasted for 4 h prior to sacrifice and serum collected. Serum metabolome analysis was conducted using uHPLC-MS/MS (5 animals in each experimental group). (A) Circulating levels of choline in serum. Serum levels of metabolites involved in (B) one-carbon metabolism and (C) transsulfuration pathway. All values are averages ± SEMs. Values that were significantly different (Student’s t-test) are indicated: *, P <0.05; **, P <0.01.
Bacterial choline metabolism alters global DNA methylation and exacerbates diet-induced metabolic disease
Many enzymes that impart epigenetic modifications, such as those that add and remove methyl groups to DNA and histones, are highly sensitive to intracellular metabolite levels (Keating and El-Osta, 2015). Extended periods of dietary methyl donor deficiency can not only alter epigenetic profiles but also promote the development of metabolic disease (Pogribny and Beland, 2009). Previous work suggests that chronic high-fat (HF) feeding exacerbates methyl donor deficiencies (Craig, 2004). We tested the contribution of microbial choline metabolism to the development of diet-induced metabolic disease in mice sustained on a HF diet. Groups of mice colonized with the CC+ and CC− community were maintained on a HF (42% Kcal) diet supplemented with 1% choline (Envigo TD.150782) for 8 weeks. Global DNA methylation assays revealed that mice colonized with the CC+ community had lower levels of DNA methylation across multiple tissues (brain, heart, liver, and colon) relative to mice colonized with the CC− community (Fig. 5A). Additionally, these mice showed significantly higher levels of TMAO (Fig. 5B), inguinal fat accumulation (Fig. 5D), and circulating leptin levels (Fig. 5E), while having similar body weight (Fig 5C). Similar adiposity phenotypes as a result of microbial choline metabolism were observed two weeks after colonization on a 1% choline diet (normal fat) (Fig. S3).
Fig. 5. Impact of sustained microbial choline consumption on metabolic disease and global DNA methylation profiles.
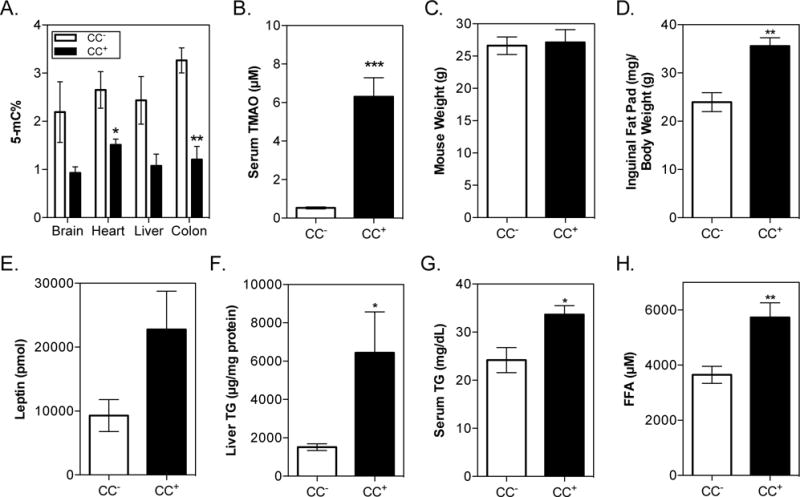
C57BL/6 females (6 animals in each experimental group) were colonized with either CC+ or CC− community and maintained for 8 weeks on a HF (42% Kcal) + 1% (wt/wt) choline diet. Mice were fasted for 8 h prior to sacrifice. (A) Global DNA methylation. DNA was extracted and normalized as detailed in the Experimental Procedures. DNA methylation was determined using a colorimetric ELISA kit for brain, heart, liver, and colon. (B) Serum TMAO. (C) Mouse weight. (D) Inguinal fat pad mass normalized to body weight. (E) Leptin levels in serum. (F) Hepatic TG levels normalized to protein concentration. (G) Serum TG levels. (H) Circulating FFA levels. All values are averages ± SEMs. Values that were significantly different (Student’s t-test) are indicated: *, P <0.05, **, P <0.01, ***, P <0.001.
Enlargement of adipose tissue increases the release of free fatty acids (FFAs) into circulation and impairs FFA clearance (Björntorp et al., 2009). Circulating FFAs are taken up by the liver where they are either oxidized to produce energy or re-esterified into TG (Boden et al., 2001). On a HF diet there is a large demand for phosphatidylcholine (PtdCho), which can be synthesized from free choline (Kennedy and Weiss, 1956). Hepatic PtdCho is required for assembly and secretion of very low-density lipoproteins (VLDL), and impairment of VLDL assembly/secretion contributes to the induction of hepatic steatosis (Fungwe et al., 1992). When PtdCho synthesis is impeded by reduced choline bioavailability, TG, a constituent of VLDL, accumulates in the liver. Induction of hepatic steatosis also increases circulating TG by a variety of mechanisms (Choi and Ginsberg, 2011). We found that hepatic and plasma TG levels (Fig. 5F, G), as well as circulating FFA levels (Fig. 5H), were elevated in the CC+ group. While both TMAO accumulation and choline deficiency have been linked with NAFLD development, only TMAO has been previously associated with increases in visceral fat mass (Randrianarisoa et al., 2016). Altogether these results show that microbial choline utilization exacerbates metabolic disease and the combined effects of limited choline availability and TMAO accumulation may synergize to promote adipogenesis (Fig. S4).
Maternal gut microbial choline metabolism alters DNA methylation in the brains of developing neonates and increases anxiety
There is a high demand for choline during pregnancy. Choline is required during fetal development for regulating stem cell proliferation, affects neural tube formation, and influences lifelong memory function (Zeisel, 2006). The results presented above show that microbial choline metabolism lowers choline and methyl donor bioavailability to the host. We assessed whether gut microbial choline utilization influences DNA methylation in mothers and offspring. Male and female GF C57BL/6 mice were colonized with either the CC− or CC+ community described above and maintained on a 1% choline diet (normal fat content) for 4 weeks prior to the establishment of mating pairs. Pregnant mothers were sacrificed and pups collected by C-section when neonates were at the E16–E17.5 developmental stages (i.e. days post fertilization, birth occurs at E18.5). TMAO (Fig. 6A) was detected in plasma collected from mothers colonized with the CC+ community and fetal tissue (liver) from their pups (Fig. 6B). This finding is consistent with human data showing detectable levels of TMAO in the first urine passed by neonates (Foxall et al., 1995). Plasma levels of choline (Fig. 6C), and DNA methylation patterns (Fig. 6D) in the mothers were comparable to non-pregnant females colonized with the same communities (Fig. 2C and Fig. S3I). Interestingly, global DNA methylation patterns in both pregnant and non-pregnant females were increased in the CC+ compared to CC− colonized animals. While this differs from the hypomethylation observed in the CC+ colonized animals consuming HF+1% choline diet (Fig. 5A), it is in line with previous reports in which female mice showed increased global methylation in response to methyl-donor deficiency (Nohara et al., 2011). These findings highlight the complex metabolic interactions that modulate epigenetic regulation, particularly the different methyl donor demands imposed by different diets (HF vs. low-fat diet). Additionally, we detected a significant reduction in DNA methylation levels in the brains, but not the livers, of neonates collected from CC+ mothers (Fig. 6E). However, DNA methylation patterns in the liver appear to be impacted differently as a function of sex (Fig. 6E). This result is consistent with previous work showing sexual dimorphism in global DNA methylation profiles caused by methyl-donor deficiency (Nohara et al., 2011). Differences in global methylation patterns between tissues may reflect different demands for methyl donors during development. Altogether, these results show that choline utilization status of the intestinal microbiota in parents can affect maternal methyl donor availability and DNA methylation profiles as well as the offspring’s epigenetic status in utero.
Fig. 6. Intergenerational effects of gut microbial choline consumption.
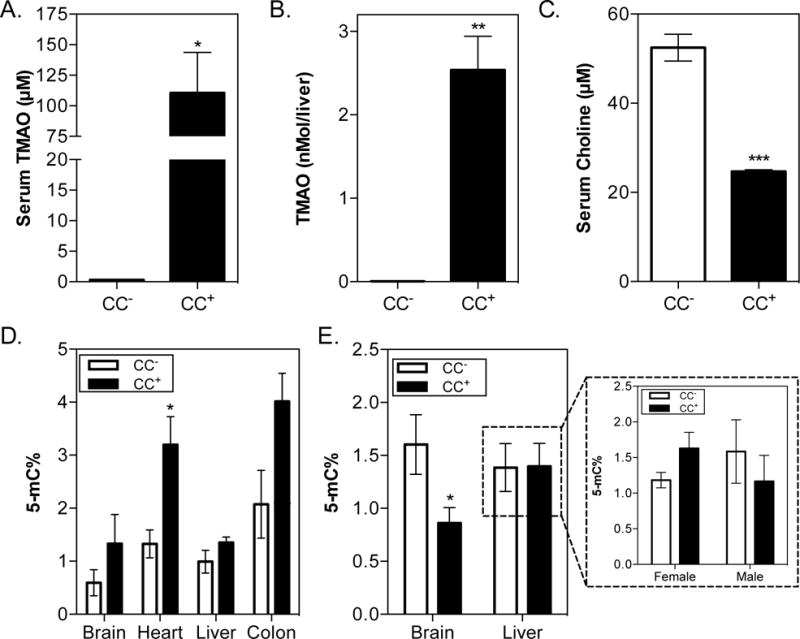
C57BL/6 males and females (3 animals per sex) were colonized with either the CC+ or CC− community and maintained for 4 weeks on a 1% (wt/wt) choline diet. Mating pairs were formed (3 pairs per community) and females were monitored for vaginal plug formation. Females were sacrificed 17 days after visual confirmation of a vaginal plug/pregnancy and neonates were collected via C-section. (A) Maternal serum TMAO levels. (B) Fetal liver TMAO levels. (C) Maternal serum choline levels. (D) Global DNA methylation of mothers. (E) Global DNA methylation of F1 offspring. Neonates were sexed by PCR and their DNA extracted from the Brain and livers of 10 neonates for each community (5 female and 5 male) and normalized. DNA methylation was determined using a colorimetric ELISA kit. All values are averages ± SEMs. Values that were significantly different (Student’s t-test) are indicated: *, P <0.05, ***, P <0.001.
Previous studies have shown that offspring from mothers supplemented with choline during the later half of pregnancy avoid any overt signs of age-associated cognitive decline (Zeisel and da Costa, 2009). Potential mechanisms for cognitive-decline associated with suboptimal choline intake include reduced sphingomyelin abundance (a choline lipid derivative which is the main lipid in myelin sheaths) (Brody et al., 1987), reduced acetylcholine levels (Picciotto et al., 2012), and/or altered epigenetic regulation, including DNA methylation, which is known to play a pivotal role in complex brain functions (Wang et al., 2016). We tested whether changes in methyl donor levels in parents were associated with behavioral modifications in young adult animals that were both indirectly (in utero) and directly (after birth) exposed to gut microbial choline consumption. For these experiments, we used ApoE−/− mice because of their genetic predisposition for altered behavior. Specifically, ApoE-deficient mice have cholinergic deficits which impair learning and memory as well as alter fear perception (Gordon et al., 1995). Previous studies showed that ApoE-deficient mice were unable to learn new tasks, had impaired spatial memory/reasoning, and developed unusual repetitive behaviors (Oitzl et al., 1997).
Male and female GF ApoE−/− mice were colonized with either the CC− or CC+ community and maintained on a 1% choline diet (normal fat content) for 4 weeks prior to the establishment of mating trios. Pregnancies were carried to term and pups were weaned 20 days after birth onto the same diet the parents were consuming. During nursing, parents colonized with the CC+ community exhibited increased levels of infanticide (Fig. 7A). F1 offspring displayed circulating TMAO levels equivalent to colonized adults harboring the same communities (Fig. 7B). Behavior (anxiety) was assessed in F1 mice using a marble-burying assay at 8 weeks of age. Pups born to mothers colonized with the CC+ community buried significantly more marbles than their CC− counterparts, regardless of sex (Fig. 7C). Additionally, ApoE−/− mice (F0) colonized with the CC+ community during adulthood displayed excessive barbering behaviors not observed in the CC− control group (Fig. S5). Barbering is considered an abnormal repetitive behavior, and increased barbering has been observed in mouse models of anxiety and depressive-like behavior (Pereda et al., 2015). Altogether these results suggest that methyl-donor depletion induced by gut microbes alters epigenetic regulation and behavior both in parents and offspring.
Fig. 7. Bacterially-induced methyl-donor deficiency increases anxious behaviors.
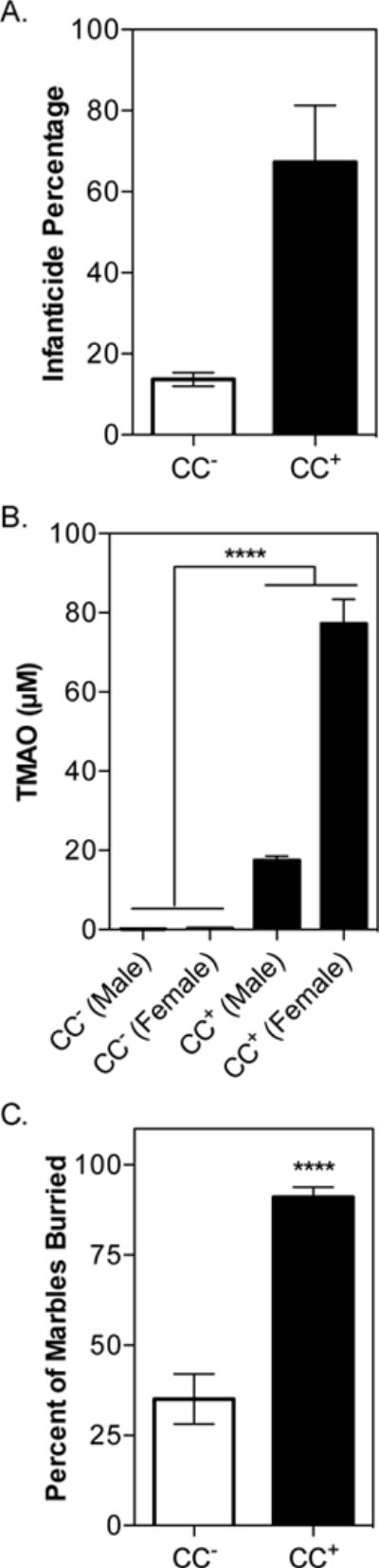
ApoE−/− males and females were colonized with either the CC+ or CC− community and maintained for 4 weeks on a 1% (wt/wt) choline diet. Mating trios were formed (2 per community) and mothers carried pregnancies to term. (A) Parental infanticide. Pup survival was monitored during nursing. Bars represent average percentage of pups killed by infanticide over the course of 2 independent litters by 4 mothers/2 fathers per community. (B) F1 serum TMAO levels. Community composition can be found in Fig. S6. (C) Percentage of marbles buried by offspring (male and female) of GF ApoE−/− colonized with either the CC+ or CC− community (n=11–20/community). All values are averages ± SEMs. Values that were significantly different (Student’s t-test) are indicated: ****, P <0.0001.
DISCUSSION
This work links the presence of a single bacterial gene present in the human gut (cutC) with: (i) changes in bacterial fitness; (ii) the metabolic fate of the essential nutrient choline; (iii) modifications in the host epigenome; (iv) susceptibility to metabolic disease and alterations in behavior. We show that harboring gut bacteria capable of transforming choline into TMA not only increases plasma levels of TMAO, but also decreases the bioavailability of choline and downstream metabolites involved in one-carbon metabolism. To date the most definitive studies linking TMAO with the development of disease have used oral TMAO administration to mice as a method for increasing levels in circulation. However, TMAO production in vivo is linked to microbial consumption of specific substrates, including choline (Romano et al., 2015). Our results suggest that choline-consuming TMA-producing gut microbes can significantly impact health not only through TMAO accumulation but also by competing with the host for this essential nutrient, eliciting a quasi-choline deficient state. This previously ignored aspect of microbial choline metabolism could have major implications on health and development, especially during pregnancy.
Epigenetic mechanisms provide a direct link between the environment (e.g., diet) and regulation of gene expression. While DNA methylation is in part regulated by non-modifiable genetic risk factors (e.g., single-nucleotide polymorphisms in one-carbon metabolism enzymes) it is profoundly affected by modifiable dietary factors. Methylation reactions depend heavily on a steady input of methyl donor precursors, including choline, folate, and betaine, which are obtained through our daily diet (Pogribny and Beland, 2009). We found that microbial choline utilization affected DNA methylation patterns across multiple tissues in adult mice while simultaneously increasing adiposity (Fig. 5A, D and Fig. S3). Previous studies have shown that disruption of epigenetic mechanisms significantly impact the development of metabolic disease by increasing oxidative stress, reducing chromosome stability and promoting the development of obesity, insulin resistance, and vascular dysfunction (Campión et al., 2009; Lavebratt et al., 2012; Martinez et al., 2014; Wang et al., 2012). Interestingly TMAO levels have also been positively associated with visceral fat mass (Randrianarisoa et al., 2016). Altogether, this suggests that microbial choline metabolism exacerbates diet-induced obesity by altering the host epigenome and gene expression.
We also explored the impact of gut microbial choline metabolism during pregnancy, a life-period characterized by an exceptionally high demand for methyl-donors. Maternal deficiency in dietary choline intake reduces global DNA methylation in the hippocampus in utero and impacts the development and function of cholinergic neurons that participate in learning, memory and attention (Mellott et al., 2007; Zeisel, 2007). Consistent with these previous observations, our findings (Fig. 6E) suggest that maternal gut microbial metabolism of choline alters its bioavailability in the developing fetus, which in turn affects the offspring’s brain epigenome and behavior.
This study illustrates the value of using gnotobiotic mice colonized with engineered synthetic microbial communities for dissecting the role(s) of individual gut microbial metabolic pathways. A potential drawback of this approach may be that effects of a single species, gene, or pathway may be overestimated due to an increased representation in a less competitive environment. Nonetheless, it should be noted that: (i) high levels of choline utilizing bacteria may not be required to efficiently compete with the host for choline (Romano et al., 2015), (ii) single species often represent a large fraction of the human gut microbiome, particularly during dysbiosis (Qin et al., 2010) and; (iii) this last point is particularly relevant for species that can use molecules produced during inflammation (e.g., nitrate) to support choline consumption (Fig. 1F) (Winter et al., 2013). Furthermore, human studies have revealed large interpersonal differences in the amount of TMAO generated from a uniform bolus of dietary choline, implying that some gut microbiomes convert choline to TMA more efficiently than others (Miller et al., 2014). Altogether this evidence suggests that in addition to diet and host genetics the gut microbiota is an important factor modulating choline bioavailability and that microbial metabolic capabilities should be examined on an individual basis when establishing nutritional requirements for optimal health and development.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Federico Rey (ferey@wisce.edu).
Experimental Model and Subject Details
Bacterial strains and growth conditions
All strains and plasmids used in this study are listed in the Key Resources Table. E. coli strains and derivatives were grown on LB, BHI or no-carbon-E (NCE) medium (Vogel and Bonner, 1956). When needed, antibiotics were added to the media at the following concentrations: ampicillin (100 μg/ml), chloramphenicol (25 μg/ml), kanamycin (50 μg/ml), or tetracycline (25 μg/ml).
Construction of cut null mutants
Non-polar deletion strains of cutC and cutD were generated using the lambda Red recombinase system (Datsenko and Wanner, 2000). Genes were replaced with a chloramphenicol resistance marker. PCR products were amplified using the template plasmid pKD3 and primers listed in the Table S2. Following amplification products were digested with DpnI and gel-purified. Electrocompetent cells of E. coli MS 200-1 carrying the Red recombinase expression plasmid pKD119 (see the Key Resources Table) were transformed with the desired PCR product. After transformation, chloramphenicol-resistant and tetracycline-sensitive recombinants were selected at 37 °C. Gene disruptions were confirmed by PCR using primers flanking the targeted region.
Gnotobiotic studies
All experiments involving mice were performed using protocols approved by the University of Wisconsin-Madison Animal Care and Use Committee. Strains used to colonize mice were grown as monocultures on Mega Medium agar plates anaerobically for 48 to 72 h at 37°C. Single colonies were then inoculated into 3 ml of Mega Medium and grown anaerobically for 24 h at 37°C. After 24 h, strains belonging to the same treatment group were combined in an equal volume ratio in a Hungate tube. C57BL/6 germ-free mice (wild-type and ApoE−/−) were inoculated by oral gavage with ~0.2 ml of mixed bacterial culture inside the gnotobiotic isolator (Romano et al., 2015). Mice were maintained on the experimental diets (see Key Resources Table) for two weeks after colonization unless otherwise noted. Unless noted all experiments were performed in female mice. Animals used in acute experiments were colonized at 7 weeks of age and euthanized at 9 weeks of age, while those used in sustained experiments were colonized at 6 weeks of age and euthanized at 14 weeks of age. Mating pairs were colonized around 6 weeks of age and housed by sex for 4 weeks on a 1% choline diet prior to mating. Male and female F1 pups collected by C-section (from ~14-week-old females) were sacrificed at E16–17.5. F1 animals (females and males) used in behavior assays were 8 weeks of age. Prior to sacrifice mice were fasted for 4 h in the regular diet and 8 h in the High Fat+1% choline diet, unless noted. Mice were anesthetized with 1–5% inhalant isoflurane supplied in oxygen. Tissue was immediately collected, flash frozen, and stored at −80 °C.
Method Details
Complementation studies
The cutC and cutD genes were PCR amplified using genomic DNA from strain E. coli MS 200-1 as the template (primers listed Table S2). The resulting products were cloned into the EcoRI and XbaI sites of the pEXT22 vector (Dykxhoorn et al., 1996). Plasmids were transformed into the respective mutants by electroporation. The growth of these strains was assessed using the same conditions detailed in “Growth studies”, but adding 1 μM of IPTG to the medium to induce gene expression.
Growth studies
To evaluate the phenotype of the wild type E. coli MS 200-1 and the cutC and cutD null mutants, strains were grown in anoxic NCE medium supplemented with 15 mM choline, 40 mM fumarate, 1 mM MgSO4, 0.1% casaminoacids, 0.5 mM glucose, and 1% vitamin and trace mineral solutions (American Type Culture Collection). To evaluate the growth using different electron acceptors 40 mM nitrate, DMSO, TMAO, or tetrathionate were added instead of fumarate, and glucose was omitted from the media. Medium was sparged with N2 and was dispensed inside an anaerobic chamber (atmosphere of 95% nitrogen and 5% hydrogen) into Hungate tubes. Tubes containing 5 ml of medium were inoculated with 50 μl of an overnight culture and incubated at 37 °C for 48 h. Growth was monitored as the increase in the absorbance at 600 nm in a Genesys 20 spectrophotometer (Thermofisher Scientific).
Quantitation of TMA production from choline
Strains were grown anaerobically on NCE plus fumarate medium containing 15 mM (trimethyl-d9)-choline (Cambridge Isotope Laboratories, Tewksbury, MA), 1 μM of IPTG and 0.5 mM glucose in order to quantify TMA produced by the wild-type and the cutC and cutD knock-out strains under these conditions. Three 0.2-ml replicates were inoculated with 2 μl of an overnight culture in a 96-well plate sealed with aluminum foil and grown anaerobically for 48 h at 37 °C. Samples were diluted 30,000-fold into buffer A (95% acetonitrile and 5% 100 mM ammonium formate). The concentration of d9-TMA in culture medium was determined using LC-MS. LC-MS/MS analysis was performed on an Agilent 6410 Triple Quadrupole LC/MS instrument (Agilent Technologies, Wilmington, DE). The LC analysis of d9-TMA was performed in positive mode, using a Kinetex (Phenomenex) HILIC column (2.6 μm, 30 mm × 2.1 mm, 100 Å), preceded by a SecurityGuard ULTRA Holder (Phenomenex) The LC conditions were: 0% B for 2.5 min, a gradient increasing to 100% B over 5 min, 100% B for 2.5 min, and 0% B for 2.5 min (solvent A = 95% acetonitrile and 5% 100 mM ammonium formate, solvent B = 50% acetonitrile, 40% water and 10% 50 mM ammonium formate). The flow rate was maintained at 0.8 ml/min for each run. Run time per sample was 12.5 min. The injection volume was 3 μl for both standards and samples. Samples and blanks were introduced via an electrospray ionization (ESI) source. The mass spectrometers were operated in multiple reaction monitoring (MRM) mode. The capillary voltage was set to 4.0 kV and the fragmentor voltage to 110 V. The drying gas temperature was maintained at 200 °C with a flow rate of 10 l/min and a nebulizer pressure of 45 psi. The precursor-product ion pairs used in MRM mode were: m/z 69.1→m/z 51. The collision energies for the precursor-product ion pairs were 21 V. MS1 and MS2 resolution was set to unit, the time filter width used was 0.07 min, and the ΔEMV (Electron Multiplier Voltage) was 400 V. Data analysis was performed with Mass Hunter Workstation Data Acquisition software (Agilent Technologies).
Community Profiling by Sequencing
Bacterial communities resulting from inoculation of germ-free animals were analyzed according to published methods (McNulty et al., 2011; Romano et al., 2015). DNA was extracted from cecal samples according to published bead-beating procedures (Turnbaugh et al., 2009). In short, fecal samples were resuspended in a solution containing 500 μl of 2× extraction buffer [200 mM Tris (pH 8.0), 200 mM NaCl, 20 mM EDTA], 210 μl of 20% SDS, 500 μl phenol:chloroform:isoamyl alcohol (pH 7.9, 25:24:1) and 500 μl of 0.1-mm diameter zirconia/silica beads. Cells were mechanically disrupted using a bead beater (BioSpec Products, Barlesville, OK) for 3 min at room temperature. The aqueous layer was removed and DNA precipitated using 600μl isopropanol and 60μl 3M Na-acetate. Pellets were dried with ethanol and resuspended in TE. NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel, Bethlehem, PA) was used to remove contaminants. Isolated DNA was stored at −80°C until downstream processing.
Libraries were prepared according to a slightly modified version of the protocol accompanying the Illumina Genomic DNA Sample Prep Kit. Briefly, total bacterial gDNA was sonicated in 0.5 ml tubes in a BioRuptor XL water bath sonicator (Total Time: 20 min, Water Temp: 4°C, Power Setter: High, Time ON: 30 sec, Time OFF: 30 sec), cleaned up and concentrated using a Macherey-Nagel PCR Purification column. DNA was blunted and poly-A tailed. The A-tailed molecules were ligated to the relevant barcoded Illumina adapter sequence. Adaptered-DNA was then size-selected (~200 bp) by agarose gel electrophoresis. Fragments of the appropriate size were PCR amplified and purified, after which the purified PCR products were given to the University of Wisconsin-Madison Biotechnology Center for quality assurance and library sequencing following the manufacturer’s protocols. Sequences were demultiplexed by 7 bp barcode, requiring an exact match (sequences without a barcode match were excluded from the analysis). After demultiplexing, all sequences were trimmed to 35 bases to eliminate low-quality bases at the ends of each read and to allow for analysis using Bowtie as a part of the COPROseq pipeline (https://github.com/DanishKhan14/aligner_RL). Sequences were aligned to the reference genomes of the 6 bacteria included in the study using Bowtie. Only reads which mapped uniquely to the reference genomes were used for abundance analysis. Raw counts were normalized based on the genome size of each organism included in the analysis. The proportional representation of each organism in the analysis was determined by dividing its normalized counts within a sample by the total normalized counts for all organisms within that sample.
qRT-PCR
Primers used for qRT-PCR are listed in the Table S2. cDNA was prepared using ~1 μg total RNA extracted as described below in the RNAseq subsection. 1 μl cDNA was used in each 10 μl SYBR Green (BioRad) reaction. Plates were run on a BioRad CFX96 instrument. Fold-change was calculated using standard Delta-Delta Ct calculations with β-actin as an internal control.
RNAseq library prep
RNA was extracted from frozen livers by TRIzol extraction and then further cleaned using the Qiagen RNeasy Mini Kit and quantified using a Nanodrop 2000 spectrophotometer. A single-end cDNA library was prepared using the Illumina TruSeq stranded mRNA Sample Preparation kit (RS-122-2101) according to the manufacturer’s specifications and sequenced at the UW-Madison Biotechnology Center. Analysis details can be found below in RNAseq Analysis.
RNAseq analysis
Samples were multiplexed, normalized, and pooled prior to sequencing on a HiSeq2000 at the University of Wisconsin-Madison Biotechnology Center. Sequencing reads were processed and filtered using the FastX Toolkit (Hannon Lab). The first 10 bp of each read were removed to eliminate GC bias at the beginning of each read using the FastX Trimmer. The 6 bp Illumina adapter sequences were then clipped using the FastX Clipper and a quality filter was applied to eliminate any reads with a quality score <30 as determined by the FastX Quality Filter. Reads were then aligned to the mm9 mouse reference genome constructed to include only annotated genes (NM_RefSeqs) by Bowtie2 alignment (Langmead and Salzberg, 2012). Seed length was set to 28 and allowed 2 mismatches per seed. Gene expression was then calculated using RSEM with a forward probability of 0.0 (for stranded libraries). Differential expression (DE) was calculated using EBSeq with a false discovery rate (FDR) = 0.1 (Leng et al., 2013; Li and Dewey, 2011). DE genes were curated by hand for functional clustering analysis.
Serum and hepatic metabolite extraction
Serum samples used for TMAO and choline measurements were prepared for analysis by precipitating proteins with 4 volumes of ice-cold methanol spiked with 2.5 μM deuterium-labeled choline and TMAO internal standards. Samples were centrifuged at 18,213 × g at 4°C for 3 min. The recovered supernatants were diluted 1:1 in uHPLC-grade water prior to screening. Serum samples for metabolome study were prepared by precipitating 50 μl of serum with 4 volumes of ice-cold methanol. Precipitate solutions were kept at −20°C for 20 min and then centrifuged at 18,000 × g for 10 min at 4°C. Supernatant was removed and the protein pellet was resuspended in 1ml of 40:40:20 methanol:acetonitrile:water and allowed to incubate on ice for 10 min. The resuspension was centrifuged at 18,000 × g for 10 min at 4°C and the resulting supernatant pooled with the previously removed supernatant. Collected supernatant was dried under N2 gas and resuspended in 0.5 ml HPLC grade water. Hepatic metabolomes were extracted from 50 mg of homogenized tissue in 1 ml pre-chilled 80:20 methanol:water on dry ice for 5 min. Extractions were centrifuged at 18,000 × g for 5 min at 4°C. Supernatant was removed and the tissue pellet was resuspended in 0.8 ml of 40:40:20 methanol:acetonitrile:water and allowed to incubate on ice for 5 min. The resuspension was centrifuged at 18,000 × g for 5 min at 4°C and resulting supernatant was combined with the first extract. The extraction with 0.8 ml of 40:40:20 methanol:acetonitrile:water was repeated and combined with the pooled first and second extraction. Pooled extract was centrifuged at 18,000 × g for 5 min at 4°C to further remove any remaining insoluble particles. The supernatant was dried under N2 gas and resuspended in HPLC grade water at a ratio of 150 μl/5 mg tissue.
uHPLC/MS/MS metabolome profiling
After sample preparation, identification and quantitation of TMAO and choline were performed using a uHPLC (Thermo Scientific/Dionex 3000) coupled to a high-resolution mass spectrometer (Thermo Scientific Q Exactive). Liquid chromatography separation was achieved on a Dikma Bio-Bond C4 column (150 mm by 2.1 mm; 3-μm particle size) using a 7-min isocratic gradient (50:50 methanol [MeOH]–water, 5 mM ammonium formate, and 0.1% formic acid). A heated electrospray ionization interface, working in positive mode, was used to direct column eluent to the mass spectrometer. Quantitation of TMAO and d9-TMAO was performed via targeted MS/MS using the following paired masses of parent ions and fragments: TMAO (76.0762 and 58.0659) and d9-TMAO (85.1318 and 68.1301). Quantitation of choline and d9-choline was performed in full-MS scan mode by monitoring their exact masses: 104.1075 and 113.1631, respectively (Romano et al., 2015).
Serum and hepatic metabolomes were measured using a C18 method previously published (Pisithkul et al., 2015). Samples were analyzed using an HPLC-tandem MS (HPLC-MS/MS) system consisting of a Thermo Scientific/Dionex UHPLC coupled by electrospray ionization (ESI) (negative mode) to a hybrid quadrupole– high-resolution mass spectrometer (Q Exactive Orbitrap; Thermo Scientific) operated in full scan mode for detection of targeted compounds based on their accurate masses. Liquid chromatography (LC) separation was achieved using an Acquity UPLC BEH C18 column (2.1- by 100-mm column, 1.7-μm particle size). Solvent A was 97:3 water:methanol with 10 mM tributylamine (TBA) and 10 mM acetic acid, pH 8.2; solvent B was 100% methanol. The total run time was 25 min with the following gradient: 0 min, 5% B; 2.5 min, 5% B; 5 min, 20% B; 7.5 min, 20% B; 13 min, 55% B; 15.5 min, 95% B; 18.5 min, 95% B; 19 min, 5% B; and 25 min, 5% B. Metabolite peaks were identified using the Metabolomics Analysis and Visualization Engine (MAVEN) (Melamud et al., 2010).
Marble-burying assay
8-week old mice were placed in naïve, freshly autoclaved cages with an array of 25 autoclaved marbles. After 30 min mice were removed and two independent investigators (one of them blinded to the treatment) determined the number of marbles buried ≥ 50%. Independent counts did not ever differ by more than 2 marbles. Cages were maintained in a biological safety cabinet for the duration of the experiment with visual barriers between replicate cages.
Neonate sexing
Neonates were sexed using PCR. DNA was extracted from tail snips collected at the time of sacrifice by incubation at 98 °C in 75 μl of 25 mM NaOH/0.2 mM EDTA followed by the addition of 75 μl of 40 mM Tris HCl (pH 5.5) (Wang and Storm, 2006). 1 μl of total genomic DNA was amplified with the primers located in Table S2. PCR reactions were performed in a final volume of 25 μl using GoTaq Green Master Mix (Promega) with 0.2 μM of the above-mentioned primers. PCR parameters are as follows: initial denaturation at 94 °C for 2 min, 35 cycles with 94 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s, followed by final elongation at 72 °C for 5 min. PCR products were visualized on a 2% agarose gel in 1× TAE with SYBR Safe DNA gel stain (Invitrogen) using the transillumination setting on the FOTO/Analyst FX (FotoDyne). Males were identified as having a single PCR product of 280 bp, females were identified as having two PCR products, one at 685 bp and the other at 480 bp (McFarlane et al., 2013).
Hepatic triglyceride quantification
Liver TG was quantified following the Bligh and Dyer extraction method (Bligh and Dyer, 1959). 10–30 mg frozen liver tissue was homogenized in PBS (a 40 μl of 1× PBS/mg tissue). An aliquot of homogenate was diluted (1:100) in water and 5 μl used to measure protein using the Bradford Assay (BioRad; Cat.# 500-0201). 50 μl of the remaining homogenate were mixed with 550 μl of PBS and 3 ml of ice cold 2:1 MeOH:chloroform in a glass Hungate tube with Teflon lined screw cap. Tubes were vortexed vigorously for 1 minute and then incubated overnight at −20°C. 500 μl ice-cold chloroform and 1 ml ice-cold water were added and tubes vortexed for 1 minute. Organic and aqueous layers were separated and the bottom chloroform layer was transferred into a new tube. The organic extract was dried under N2. 10 μl of chloroform were added to the tubes and vortexed for 5 seconds before 90 μl of 10% Triton X-100 (in 100% isopropanol) were added. Tubes were then vortexed for 5 seconds (or until no visible residue was left on the sides of the tubes). Lipid calibrator (Wako Lipid Calibrator; Cat.# 464-01601) was diluted with 1 ml of water. Standards for the calibration curve were made by mixing different volumes of the resuspended calibrator with 10 μl chloroform, 24 μl 37.5% Triton X-100 (in 100% isopropanol), and the corresponding volume of water to total 100 μl. 50 μl of each standard and sample was mixed with 900 μl Wako R1 Type M (Wako R1 Type M; Cat.# 461-08992), vortexed, and incubated at 37°C for 5 minutes. After 5 minutes, 300 μl Wako R2 Type M (Wako R1 Type M; Cat.# 461-09092) was added to each sample, vortexed, and incubated at 37°C for 5 minutes. Samples were vortexed until they looked clear. 200 μl aliquots of each sample were transferred (triplicates) into a 96 well plate and the absorbance measured at 595nm. Triglyceride content was expressed as μg of TG per milligram of protein. Number of biological replicates is indicated in the respective figure legends.
Serum triglyceride quantification
Serum TG were quantified using the L-type Triglyceride M kit from Wako Diagnostics (Richmond, VA) in line with manufacturer’s instructions for microplate applications. In short, 12μl serum sample were mixed with 270 μl Wako R1 Type M. Samples were vortexed and incubated at 37°C for 5 minutes. Following incubation 90 μl of Wako R2 Type M was added and samples were vortexed and incubated at 37°C for 5 minutes. Samples were vortexed until clear. 115 μl were pipetted in triplicate into at flat bottom 96-well plate and the absorbance measured at 595nm. Lipid calibrator was resuspended in water to a final concentration of 300 mg/dL and serially diluted to create standard curve. 12μl of each standard was processed as detailed above to create a standard curve. Number of biological replicates is indicated in respective figure legends.
DNA global methylation assay
Global methylation was measured using MethylFlash Global DNA Methylation ELISA Easy Kit from EpiGentek (Farmingdale, NY). DNA was extracted from tissue using the bead beating method detailed above. In short, tissue (half of the brain, ~25mg of heart, ~100mg of liver, and half of the distal colon) were suspended in a solution containing 500 μl of 2× extraction buffer, 210 μl of 20% SDS, 500 μl phenol:chloroform:isoamyl alcohol, 500 μl of 0.1-mm diameter zirconia/silica beads, and one 3.2-mm diameter stainless steel bead. Cells were mechanically disrupted using a bead beater for 3 min at room temperature. The aqueous layer (~450μl) was removed and DNA precipitated using 600μl isopropanol and 60μl 3M Na-acetate. Pellets were dried with ethanol and resuspended in TE. NucleoSpin Gel and PCR Clean-up Kit was used to remove contaminants. Isolated DNA was stored at −80°C until downstream processing. DNA concentration was calculated using 3 replicate readings on the NanoDrop Spectrophotometer ND-100 (Thermofisher Scientific) and diluted to a final concentration of 100ng/3μl. Using 100ng (3μl) of diluted DNA, global 5mC was measured directly according to manufacturer’s instructions provided with the kit. All standards were measured in duplicate while all samples were measured once due to well limitations in the ELISA plate. Number of biological replicates is indicated in the respective figure legends.
Free Fatty Acids assay
Serum FFA levels were measured using a colorimetric Free Fatty Acid Assay Kit (Cat. # STA-618) from Cell Biolabs Inc. (San Diego, CA) according to manufacturer’s instructions provided with the kit. Briefly, flash frozen serum was diluted (1:10) in 1× Assay Buffer for the assay. 10 μl of each sample and standard was then mixed with 200 μl of 1× Enzyme Mixture A and incubated in a 96-well plate for 30 minutes at 37°C in the dark. After incubation, 100 μl of the prepared Detection Enzyme Mixture were added and samples were incubated for 10 minutes at 37°C in the dark. Absorbance was measured at 540nm. Three technical replicates were run for each sample and standard. Number of biological replicates is indicated in the respective figure legends.
Leptin assay
Serum leptin levels were measured using the Leptin (mouse) ELISA Kit (Cat. # ADI-900-019A) from Enzo (Farmingdale, NY) according to manufacturer’s instructions provided with the kit. Briefly, samples were diluted (1:32) in Assay Buffer. 100 μl of each sample, standard, and blank were added to the ELISA plate (provided with the kit) and incubated for 1 h with shaking at room-temperature. Wells were washed 3× and 100 μl of antibody were added to each well (except blanks) and the plate was incubated for 1h with shaking at room-temperature. Following incubation, wells were washed 3×, 100 μl of blue conjugate were added to each well (except the blank) and the plate was incubated for 30 minutes at room temperature with shaking. Following incubation, wells were washed 3× and 100μl of substrate solution was added to each well. The plate was incubated for 30 minutes with shaking at room temperature. After incubation, a 100 μl aliquot of stop solution was added and absorbance measured at 450nm. All standards were measured in duplicate while all samples were measured once due to well limitations in the ELISA plate. Number of biological replicates is indicated in the respective figure legends.
Community profiling by 16S rRNA gene sequencing
Community composition was determined by amplicon sequencing of the variable 4 (V4) region of the 16S rRNA gene (Kozich et al., 2013) using the Illumina MiSeq platform (San Diego, CA). PCR was performed using primers for the variable 4 (V4) region of the bacterial 16S rRNA gene (Kozich et al., 2013). PCR reactions contained 12.5 ng DNA, 10 μM each primer, 12.5 μl 2× HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, USA), and water to 25 μl. Cycling conditions were 95 °C for 3 min, then 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, and finally 72 °C for 5 min. PCR products were purified by gel extraction from a 1% low-melt agarose gel using a ZR-96 Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA). Individual samples were quantified by Qubit Fluorometer (Invitrogen, Carlsbad, CA, USA) and equimolar pooled. The pool plus 5% PhiX control DNA was sequenced with the MiSeq 2×250 v2 kit (Illumina, San Diego, CA, USA) using custom sequencing primers at the University of Wisconsin – Madison Biotechnologuy Center (Kozich et al., 2013). Sequences were analyzed in mothur (Schloss et al., 2009) following recommended procedures (Kozich et al., 2013). Sequences were clustered into 97% operational taxonomic units (OTUs) using OptiClust (Westcott and Schloss, 2017) and classified to a custom database containing the species used in this study: Bacteroides caccae (AAVM02000001), Bacteroides ovatus (AAXF02000018), Bacteroides thetaiotaomicron (AE015928), Collinsella aerofaciens (AAVN02000007), Eubacterium rectale (L34627), and a number of Escherichia coli MS strains as the strain used (MS 200-1) was not available (ADTK01000014, ADTL01000003, ADTP01000052, ADTR01000016, ADTX01000014, ADUA01000035, ADUG01000003, ADWQ01000120, ADWR01000036, ADWS01000032, ADWT01000015, ADWU01000043, ADWV01000069).
Quantification and Statistical Analysis
The number of mice per group is indicated in corresponding figure legends. No formal sample-size estimation was performed. Number of mice used for different assays is based on previous experience and published literature. Significance was assessed using unpaired two-tailed Student’s t-test or ANOVA (as annotated in the corresponding figure legends) in GraphPad Prism. In all figures, the mean value is visually depicted ± SEMs. P values correlate with symbols as follows: *, P <0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001. Mice were randomly assigned into experimental groups after matching for age and gender. Investigators remained unblinded to group assignments throughout unless noted. Differential expression of genes was completed using EBseq as described in the “RNAseq analysis” subsection of Methods Details. There were not subjects/data points excluded for statistical analyses/reporting.
Data and Software Availability
The accession number for RNA sequencing data at NCBI GEO is: GSE100983.
The accession number for COPROseq and 16S data, deposited to the Sequence Read Archive, is: BioProject PRJNA393700 and BioSamples SAMN07346941, SAMN07346942, SAMN07346943, SAMN07346944, SAMN07346945, SAMN07346946, SAMN07346947, SAMN07346948, SAMN07346949, SAMN07346950, SAMN07346951, SAMN07346952, SAMN07346953, SAMN07346954, SAMN07346955, SAMN07346956, SAMN07346957, SAMN07346958, SAMN07346959, SAMN07346960, SAMN07346961, SAMN07346962.
COPROseq Pipeline can be found at: https://github.com/DanishKhan14/aligner_RL
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Bacteroides caccae | ATCC | 43185 |
| Bacteroides ovatus | ATCC | 8483 |
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | 29148 |
| Collinsella aerofaciens | ATCC | 25986 |
| Eubacterium rectale | ATCC | 33656 |
| Escherichia coli | BEI | MS 200-1 |
| EMC | This study | MS 200-1 derivative, ΔcutC |
| EMD | This study | MS 200-1 derivative, ΔcutD |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Triton X | Sigma | Cat. # T8787-50ML |
| Trizol | Ambion | Cat. # 15596026 |
| RNase Zap | Thermo Scientific | Cat. # AM9780 |
| D9-Choline | Cambridge Isotope Laboratories Inc. | Cat. # DLM-549-1 |
| D9-TMAO | Cambridge Isotope Laboratories Inc. | Cat. # DLM-4779-PK |
| Phenol:Chloroform:Isoamyl Alcohol (pH 7.9, 25:24:1) | Invitrogen | Cat. # 15593-049 |
| HotStart ReadyMix | KAPA Biosystems | Cat. # 07958935001 Also know as KK2602 |
| SYBR Green Supermix | BioRad | Cat. # 172-5271 |
| T4 DNA Ligase | NEB | Cat. # M0202M |
| T4 Polymerase | NEB | Cat. # M0203L |
| T4 PNK | NEB | Cat. # M0201L |
| Taq Polymerase | Invitrogen | Cat. # 18038-042 |
| GoTaq Green Master Mix (2X) | Promega | Cat. # M712B |
| SYBR Safe DNA Gel Stain | Invitrogen | Cat. # S33102 |
| DpnI | NEB | Cat. # R0176S |
| Critical Commercial Assays | ||
| Leptin (mouse) ELISA Kit | Enzo | Cat. # ADI-900-019A |
| Free Fatty Acid Assay Kit (Colorimetric) | Cell Biolabs Inc. | Cat. # STA-618 |
| Illumina TruSeq stranded mRNA Sample Preparation Kit | Illumina | Cat. # RS-122-2101 |
| MethylFlash Global DNA Methylation ELISA Easy Kit | EpiGentek | Cat. # P-1030-48 Cat. # P-1030-96 |
| L-type Triglyceride M Kit | Wako Diagnostics | Cat. # 461-08992 Cat. # 461-09092 Cat. # 464-01601 |
| NucleoSpin Gel and PCR Clean-up Kit | Macherey-Nagel | Cat. # 740609.250 |
| ZR-96 Zymoclean Gel DNA Recovery Kit | Zymo Reserach | Cat. # D4021 |
| RNeasy Mini Kit | Qiagen | Cat. # 74104 |
| Bradford Protein Assay Kit | BioRad | Cat. # 500-0201 |
| Deposited Data | ||
| RNAseq raw and processed reads (GSE100983) | GEO | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100983 |
| Community COPROseq and 16S rRNA gene raw reads BioProject: PRJNA393700 BioSamples: SAMN07346941 SAMN07346942 SAMN07346943 SAMN07346944 SAMN07346945 SAMN07346946 SAMN07346947 SAMN07346948 SAMN07346949 SAMN07346950 SAMN07346951 SAMN07346952 SAMN07346953 SAMN07346954 SAMN07346955 SAMN07346956 SAMN07346957 SAMN07346958 SAMN07346959 SAMN07346960 SAMN07346961 SAMN07346962 |
SRA | https://www.ncbi.nlm.nih.gov/bioproject/PRJNA393700 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus C57BL/6 | Germ-free mice raised in the gnotobiotic facility at the University of Wisconsin - Madison | N/A |
| Mus musculus C57BL/6 ApoE−/− | Germ-free mice raised in the gnotobiotic facility at the University of Wisconsin - Madison | N/A |
| Oligonucleotides | ||
| Oligonucleotides can be found in Table S2 | ||
| Recombinant DNA | ||
| pKD119 | CGSC | CGSC#: 7990 |
| pKD3 | CGSC | CGSC#: 7631 |
| pEXT22 | CGSC | CGSC#: 12327 |
| pcutC (pEXT22 derivative carrying cutC) | This study | N/A |
| pcutD (pEXT22 derivative carrying cutD) | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| EBSeq | Leng et al., 2013 | https://www.biostat.wisc.edu/~kendzior/EBSEQ/ |
| RSEM | Li, B., and Dewey, C.N., 2011 | https://deweylab.github.io/RSEM/ |
| COPROseq | This study | https://github.com/DanishKhan14/aligner_RL |
| FASTX Toolkit | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Mouthr | Schloss et al., 2009 | https://www.mothur.org/ |
| MAVEN | Melamud et al., 2010 | http://genomics-pubs.princeton.edu/mzroll/index.php |
| Other | ||
| 1 % Choline Diet | Envigo (Madison, WI) | TD.140179 |
| Choline Deficient Diet | Envigo (Madison, WI) | TD.88052 |
| High Fat (42 % Kcal) + 1 % Choline Diet | Envigo (Madison, WI) | TD.150782 |
| Bio-Bond C4 3 μm 150 × 2.1 mm | Dikma | Cat. # 84413 |
| ACQUITY UPLC BEH C18 Column, 130 Å , 1.7 μm, 2.1 mm × 100 mm | Waters | Cat. # 186002352 |
| Kinetex HILIC column (2.6 μm, 30 mm × 2.1 mm, 100 Å ) | Phenomenex | Cat. # 00A-4461-AN |
Supplementary Material
Highlights.
Gut bacteria compete with the host for choline, decreasing bioavailability.
Microbial choline degradation depletes methyl-donor metabolites.
Microbial choline utilization alters in utero epigenetic programming of the brain.
Mice with choline-consuming gut microbiota display altered behavior.
Acknowledgments
The authors thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing sequencing and support services, and the University of Wisconsin Center for High Throughput Computing (CHTC) in the Department of Computer Sciences for providing computational resources, support, and assistance. Our deepest gratitude to Kimberly Dill-McFarland and Leticia Reyes for experimental assistance in response to reviewer comments. We also thank David Stevenson for technical support, Danish A. Khan and Kimberly A. Krautkramer for assistance with bioinformatic analyses and Robert Kerby for helpful comments during the course of these studies and Fredrik Bäckhed for supplying germ-free ApoE−/− mice. The project described was supported in part by NIH DK108259 (F.E.R.), a Smith Family Award for Biomedical Research (E.P.B), a Packard Fellowship for Science and Engineering (E.P.B), and a Blavatnik Biomedical Accelerator Award (E.P.B). This material is based upon work that is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture WIS01910, Hatch WIS01901.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, K.A.R, A.M.d.C, E.P.B, and F.E.R; Methodology, K.A.R, A.M.d.C, K.K, E.I.V, D.A.N, E.P.B, and F.E.R; Formal Analysis, K.A.R and A.M.d.C; Investigation, K.A.R, A.M.d.C, K.K, and C.L.C; Resources, A.M.d.C, C.L.C, E.I.V, and D.A.N; Data Curation, K.A.R; Writing – Original Draft, K.A.R, A.M.d.C, E.P.B, and F.E.R; Writing – Review & Editing, K.A.R, A.M.d.C, K.K, C.L.C, E.I.V, D.A.N, E.P.B, and F.E.R; Visualization, K.A.R, A.M.d.C, K.K, and C.L.C; Supervision, D.A.N, E.P.B, F.E.R; Funding Acquisition, D.A.N, E.P.B and F.E.R.
References
- Ahima RS, Flier JS. Adipose Tissue as an Endocrine Organ. Trends Endocrinol Metab. 2000;11:327–332. doi: 10.1016/s1043-2760(00)00301-5. [DOI] [PubMed] [Google Scholar]
- Baker JR, Chaykin S. The biosynthesis of trimethylamine-N-oxide. J Biol Chem. 1962;237:1309–1313. [PubMed] [Google Scholar]
- Bay DC, Turner RJ. Small Multidrug Resistance Protein EmrE Reduces Host pH and Osmotic Tolerance to Metabolic Quaternary Cation Osmoprotectants. J Bacteriol. 2012;194:5941–5948. doi: 10.1128/JB.00666-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandary B, Marahatta A, Kim HR, Chae HJ. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases. Int J Mol Sci. 2012;14:434–456. doi: 10.3390/ijms14010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björntorp P, Bergman H, Varnauskas E. PLASMA FREE FATTY ACID TURNOVER RATE IN OBESITY. Acta Med Scand. 2009;185:351–356. doi: 10.1111/j.0954-6820.1969.tb07347.x. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A RAPID METHOD OF TOTAL LIPID EXTRACTION AND PURIFICATION. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Boden G, Lebed B, Schatz M, Homko C, Lemieux S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. 2001;50:1612–1617. doi: 10.2337/diabetes.50.7.1612. [DOI] [PubMed] [Google Scholar]
- Brody BA, Kinney HC, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. I. An autopsy study of myelination. J Neuropathol Exp Neurol. 1987;46:283–301. doi: 10.1097/00005072-198705000-00005. [DOI] [PubMed] [Google Scholar]
- Brunst KJ, Wright RO, DiGioia K, Enlow MB, Fernandez H, Wright RJ, Kannan S. Racial/ethnic and sociodemographic factors associated with micronutrient intakes and inadequacies among pregnant women in an urban US population. Public Health Nutr. 2014;17:1960–1970. doi: 10.1017/S1368980013003224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campión J, Milagro FI, Martínez JA. Individuality and epigenetics in obesity. Obes Rev. 2009;10:383–392. doi: 10.1111/j.1467-789X.2009.00595.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Jameson E, Doxey AC, Airs R, Purdy KJ, Murrell JC. Metagenomic data-mining reveals contrasting microbial populations responsible for trimethylamine formation in human gut and marine ecosystems. Microb Genomics. 2016;2 doi: 10.1099/mgen.0.000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22:353–363. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig SAS. Betaine in human nutrition. Am J Clin Nutr. 2004;80:539–549. doi: 10.1093/ajcn/80.3.539. [DOI] [PubMed] [Google Scholar]
- Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Hartmane D, Pugovics O, Erglis A, Liepinsh E. Diabetes is Associated with Higher Trimethylamine N-oxide Plasma Levels. Exp Clin Endocrinol Diabetes. 2016;124:251–256. doi: 10.1055/s-0035-1569330. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deminice R, de Castro GSF, Francisco LV, da Silva LECM, Cardoso JFR, Frajacomo FTT, Teodoro BG, dos Reis Silveira L, Jordao AA. Creatine supplementation prevents fatty liver in rats fed choline-deficient diet: a burden of one-carbon and fatty acid metabolism. J Nutr Biochem. 2015;26:391–397. doi: 10.1016/j.jnutbio.2014.11.014. [DOI] [PubMed] [Google Scholar]
- Dykxhoorn DM, St Pierre R, Linn T. A set of compatible tac promoter expression vectors. Gene. 1996;177:133–136. doi: 10.1016/0378-1119(96)00289-2. [DOI] [PubMed] [Google Scholar]
- Falony G, Vieira-Silva S, Raes J. Microbiology Meets Big Data: The Case of Gut Microbiota–Derived Trimethylamine. Annu Rev Microbiol. 2015;69:305–321. doi: 10.1146/annurev-micro-091014-104422. [DOI] [PubMed] [Google Scholar]
- Foxall PJ, Bewley S, Neild GH, Rodeck CH, Nicholson JK. Analysis of fetal and neonatal urine using proton nuclear magnetic resonance spectroscopy. Arch Dis Child Fetal Neonatal Ed. 1995;73:F153–F157. doi: 10.1136/fn.73.3.f153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fungwe TV, Cagen L, Wilcox HG, Heimberg M. Regulation of hepatic secretion of very low density lipoprotein by dietary cholesterol. J Lipid Res. 1992;33:179–191. [PubMed] [Google Scholar]
- Gordon I, Grauer E, Genis I, Sehayek E, Michaelson DM. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci Lett. 1995;199:1–4. doi: 10.1016/0304-3940(95)12006-p. [DOI] [PubMed] [Google Scholar]
- Huang W, Dedousis N, Bandi A, Lopaschuk GD, O’Doherty RM. Liver Triglyceride Secretion and Lipid Oxidative Metabolism Are Rapidly Altered by Leptin in Vivo. Endocrinology. 2006;147:1480–1487. doi: 10.1210/en.2005-0731. [DOI] [PubMed] [Google Scholar]
- Ilcol YO, Uncu G, Ulus IH. Free and Phospholipid-Bound Choline Concentrations in Serum during Pregnancy, after Delivery and in Newborns. Arch Physiol Biochem. 2002;110:393–399. doi: 10.1076/apab.110.5.393.11832. [DOI] [PubMed] [Google Scholar]
- Keating ST, El-Osta A. Epigenetics and Metabolism. Circ Res. 2015;116:715–736. doi: 10.1161/CIRCRESAHA.116.303936. [DOI] [PubMed] [Google Scholar]
- Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013 doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Røkenes TP, McDougall J, Strøm AR. The complex bet promoters of Escherichia coli: regulation by oxygen (ArcA), choline (BetI), and osmotic stress. J Bacteriol. 1996;178:1655–1662. doi: 10.1128/jb.178.6.1655-1662.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavebratt C, Almgren M, Ekström TJ. Epigenetic regulation in obesity. Int J Obes. 2012;36:757–765. doi: 10.1038/ijo.2011.178. [DOI] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, Haag JD, Gould MN, Stewart RM, Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JA, Milagro FI, Claycombe KJ, Schalinske KL. Epigenetics in Adipose Tissue, Obesity, Weight Loss, and Diabetes. Adv Nutr Int Rev J. 2014;5:71–81. doi: 10.3945/an.113.004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. Characterization and Detection of a Widely Distributed Gene Cluster That Predicts Anaerobic Choline Utilization by Human Gut Bacteria. mBio. 2015;6:e00042–15. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane L, Truong V, Palmer JS, Wilhelm D. Novel PCR Assay for Determining the Genetic Sex of Mice. Sex Dev. 2013;7:207–211. doi: 10.1159/000348677. [DOI] [PubMed] [Google Scholar]
- McMahon KE, Farrell PM. Measurement of free choline concentrations in maternal and neonatal blood by micropyrolysis gas chromatography. Clin Chim Acta Int J Clin Chem. 1985;149:1–12. doi: 10.1016/0009-8981(85)90267-0. [DOI] [PubMed] [Google Scholar]
- McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. The Impact of a Consortium of Fermented Milk Strains on the Gut Microbiome of Gnotobiotic Mice and Monozygotic Twins. Sci Transl Med. 2011;3:106ra106–106ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD. Metabolomic Analysis and Visualization Engine for LC–MS Data. Anal Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellott TJ, Kowall NW, Lopez-Coviella I, Blusztajn JK. Prenatal choline deficiency increases choline transporter expression in the septum and hippocampus during postnatal development and in adulthood in rats. Brain Res. 2007;1151:1–11. doi: 10.1016/j.brainres.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Corbin KD, da Costa KA, Zhang S, Zhao X, Galanko JA, Blevins T, Bennett BJ, O’Connor A, Zeisel SH. Effect of egg ingestion on trimethylamine-N-oxide production in humans: a randomized, controlled, dose-response study. Am J Clin Nutr. 2014;100:778–786. doi: 10.3945/ajcn.114.087692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SH, Poole JR. Labile methyl balances for normal humans on various dietary regimens. Metabolism. 1975;24:721–735. doi: 10.1016/0026-0495(75)90040-2. [DOI] [PubMed] [Google Scholar]
- Nohara K, Baba T, Murai H, Kobayashi Y, Suzuki T, Tateishi Y, Matsumoto M, Nishimura N, Sano T. Global DNA methylation in the mouse liver is affected by methyl deficiency and arsenic in a sex-dependent manner. Arch Toxicol. 2011;85:653–661. doi: 10.1007/s00204-010-0611-z. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Mulder M, Lucassen PJ, Havekes LM, Grootendorst J, de Kloet ER. Severe learning deficits in apolipoprotein E-knockout mice in a water maze task. Brain Res. 1997;752:189–196. doi: 10.1016/s0006-8993(96)01448-5. [DOI] [PubMed] [Google Scholar]
- Pekkala S, Munukka E, Kong L, Pöllänen E, Autio R, Roos C, Wiklund P, Fischer-Posovszky P, Wabitsch M, Alen M, et al. Toll-like receptor 5 in obesity: The role of gut microbiota and adipose tissue inflammation: Gut Microbiota, TLR5, Inflammation, and Obesity. Obesity. 2015;23:581–590. doi: 10.1002/oby.20993. [DOI] [PubMed] [Google Scholar]
- Pereda D, Pardo MR, Morales Y, Dominguez N, Arnau MR, Borges R. Mice lacking chromogranins exhibit increased aggressive and depression-like behaviour. Behav Brain Res. 2015;278:98–106. doi: 10.1016/j.bbr.2014.09.022. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron. 2012;76:116–129. doi: 10.1016/j.neuron.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisithkul T, Jacobson TB, O’Brien TJ, Stevenson DM, Amador-Noguez D. Phenolic Amides Are Potent Inhibitors of De Novo Nucleotide Biosynthesis. Appl Environ Microbiol. 2015;81:5761–5772. doi: 10.1128/AEM.01324-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci. 2009;66:2249–2261. doi: 10.1007/s00018-009-0015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price-Carter M, Tingey J, Bobik TA, Roth JR. The Alternative Electron Acceptor Tetrathionate Supports B12-Dependent Anaerobic Growth of Salmonella enterica Serovar Typhimurium on Ethanolamine or 1,2-Propanediol. J Bacteriol. 2001;183:2463–2475. doi: 10.1128/JB.183.8.2463-2475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randrianarisoa E, Lehn-Stefan A, Wang X, Hoene M, Peter A, Heinzmann SS, Zhao X, Königsrainer I, Königsrainer A, Balletshofer B, et al. Relationship of Serum Trimethylamine N-Oxide (TMAO) Levels with early Atherosclerosis in Humans. Sci Rep. 2016;6:26745. doi: 10.1038/srep26745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio. 2015;6:e02481–14. doi: 10.1128/mBio.02481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RG, Hunter SK, Hoffman MC, McCarthy L, Chambers BM, Law AJ, Leonard S, Zerbe GO, Freedman R. Perinatal Phosphatidylcholine Supplementation and Early Childhood Behavior Problems: Evidence for CHRNA7 Moderation. Am J Psychiatry. 2016;173:509–516. doi: 10.1176/appi.ajp.2015.15091188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba RD, Zhang P, Gonzalez-Freire M, Moaddel R, Trehan I, Maleta KM, Ordiz MI, Ferrucci L, Manary MJ. The association of serum choline with linear growth failure in young children from rural Malawi. Am J Clin Nutr. 2016;104:191–197. doi: 10.3945/ajcn.115.129684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersbaek R, Nielsen R, Mandrup S. PPARγ in adipocyte differentiation and metabolism-Novel insights from genome-wide studies. FEBS Lett. 2010;584:3242–3249. doi: 10.1016/j.febslet.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Tang WW, Wang Z, Kennedy DJ, Wu Y, Buffa J, Agatisa-Boyle B, Li XS, Levison BS, Hazen SL. Gut Microbiota-Dependent Trimethylamine N-oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ Res. 2014 doi: 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. J Biol Chem. 1956;218:97–106. [PubMed] [Google Scholar]
- Wang Z, Storm D. Extraction of DNA from mouse tails. BioTechniques. 2006;41:410–412. doi: 10.2144/000112255. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu R, Liu L, Chowdhury R, Barzilai N, Tan J, Rossetti L. The effect of leptin on Lep expression is tissue-specific and nutritionally regulated. Nat Med. 1999;5:895–899. doi: 10.1038/11335. [DOI] [PubMed] [Google Scholar]
- Wang J, Wu Z, Li D, Li N, Dindot SV, Satterfield MC, Bazer FW, Wu G. Nutrition, Epigenetics, and Metabolic Syndrome. Antioxid Redox Signal. 2012;17:282–301. doi: 10.1089/ars.2011.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Surzenko N, Friday WB, Zeisel SH. Maternal dietary intake of choline in mice regulates development of the cerebral cortex in the offspring. FASEB J. 2016;30:1566–1578. doi: 10.1096/fj.15-282426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott SL, Schloss PD. OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere. 2017;2 doi: 10.1128/mSphereDirect.00073-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. Host-Derived Nitrate Boosts Growth of E. coli in the Inflamed Gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Choline: Critical Role During Fetal Development and Dietary Requirements in Adults. Annu Rev Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Gene response elements, genetic polymorphisms and epigenetics influence the human dietary requirement for choline. IUBMB Life. 2007;59:380–387. doi: 10.1080/15216540701468954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Dietary choline deficiency causes DNA strand breaks and alters epigenetic marks on DNA and histones. Mutat Res Mol Mech Mutagen. 2012;733:34–38. doi: 10.1016/j.mrfmmm.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH, da Costa KA. Choline: an essential nutrient for public health. Nutr Rev. 2009;67:615–623. doi: 10.1111/j.1753-4887.2009.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.