

SUMMARY
CRISPR loci are a cluster of repeats separated by short “spacer” sequences derived from prokaryotic viruses and plasmids that determine the targets of the host’s CRISPR-Cas immune response against its invaders. For type I and II CRISPR-Cas systems, single-nucleotide mutations in the seed or protospacer adjacent motif (PAM) of the target sequence cause immune failure and allow viral escape. This is overcome by the acquisition of multiple spacers that target the same invader. Here we show that targeting by the Staphylococcus epidermidis type III-A CRISPR-Cas system does not require PAM or seed sequences, and thus prevents viral escape via single-nucleotide substitutions. Instead, viral escapers can only arise through complete target deletion. Our work shows that, as opposed to type I and II systems, the relaxed specificity of type III CRISPR-Cas targeting provides robust immune responses that can lead to viral extinction with a single spacer targeting an essential phage sequence.
eTOC BLURP
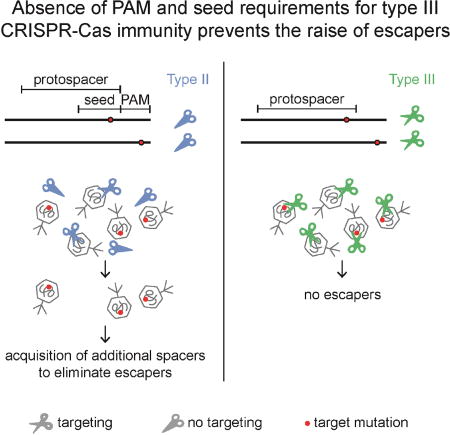
Exploring the target specificity of type III-A CRISPR-Cas systems, Pyenson et al. find that most point mutations in the target region still allow robust immunity. As a consequence, viral escape from the type III-A CRISPR-Cas immune response requires the full deletion of the target, which is a very rare event
INTRODUCTION
Clustered, regularly interspaced, short, palindromic repeats (CRISPR) loci and their associated (cas) genes provide protection against viral (Barrangou et al., 2007) and plasmid (Marraffini and Sontheimer, 2008) infection by capturing short DNA sequences from these invaders and integrating them into the CRISPR locus of the prokaryotic host (Barrangou et al., 2007). These sequences, known as spacers, specify the targets of the CRISPR immune response. Depending on their cas gene content, CRISPR-Cas systems can be classified into six different types (I–VI) (Makarova et al., 2015). In all types, the spacer sequence is transcribed and processed into a short RNA guide known as the CRISPR RNA (crRNA). How the crRNA guide directs the detection and cleavage of its complementary target depends on the type of CRISPR-Cas system. During type I and II CRISPR-Cas targeting a PAM, a conserved short sequence flanking the region of complementarity between the crRNA and the target DNA (also known as the protospacer), is required for target recognition and cleavage (Gasiunas et al., 2012; Jinek et al., 2012; Mojica et al., 2009; Semenova et al., 2011). In addition, the target sequence immediately adjacent to the PAM, 6–8 nucleotides known as the target “seed”, must be fully complementary to the crRNA guide (Jinek et al., 2012; Semenova et al., 2011; Wiedenheft et al., 2011). As a consequence of these strict targeting requirements, type I and II CRISPR-Cas systems fail to provide immunity against mutant bacteriophages, present in the viral population, carrying single-nucleotide mutations in the seed or PAM (Deveau et al., 2008; Semenova et al., 2011). To prevent the takeover of these viral escape mutants, the bacterial host population must acquire multiple spacers to ensure the extinction of the virus (van Houte et al., 2016).
Type III CRISPR-Cas systems have a unique molecular mechanism which requires target transcription for both immunity (Deng et al., 2013; Elmore et al., 2016; Goldberg et al., 2014) and DNA degradation (Samai et al., 2015). The type III Cas10 effector complex uses the crRNA guide to interact with a complementary sequence in the target transcript, an interaction that triggers both the DNase and RNase activities of the complex (Elmore et al., 2016; Estrella et al., 2016; Kazlauskiene et al., 2016), and results in the degradation of both the viral target DNA and its transcripts (Samai et al., 2015). Analysis of the sequence requirements for type III targeting has shown that only mutations that create homology between flanking and repeat sequence upstream of the protospacer and spacer, respectively, can abrogate immunity (Han et al., 2017; Kazlauskiene et al., 2016; Marraffini and Sontheimer, 2010; Samai et al., 2015; Zebec et al., 2014). These results have argued against the presence of a PAM for type III targeting. In addition, it has been shown that different type III systems tolerate mutations in the protospacer, both in vivo (Goldberg et al., 2014; Maniv et al., 2016; Peng et al., 2015) and in vitro in the RNA target (Kazlauskiene et al., 2016; Staals et al., 2014), suggesting the absence of a critical seed sequence for targeting. In spite of these studies, a thorough test for the presence of PAM and seed sequences, and/or any other sequence requirements for an efficient type III CRISPR-Cas immune response has not been performed.
Here we constructed plasmid libraries containing mutations in the CNPH82 phage target sequence of the type III-A CRISPR-Cas system of Staphylococcus epidermidis RP62a (Marraffini and Sontheimer, 2008). We either mutagenized the upstream flanking sequence of this target to look for the presence of a PAM or the protospacer itself to look for a seed sequence. We found that the great majority of mutations still enabled type III-A targeting, unequivocally demonstrating that these systems do not require either PAM or seed sequences for efficient immunity. In contrast, type III-A CRISPR-Cas systems display a broad target specificity. When tested in the context of phage infection, this relaxed targeting mechanism prevented the evasion of type III-A CRISPR-Cas immunity through target point mutations. Instead, viral escape mutants harbor target deletions and are very infrequent. When the target is located in a gene essential for phage propagation, escape is impossible and type III CRISPR-Cas immunity results in the complete destruction of the viral population. Our work shows that, as opposed to type I and II systems which require the acquisition of multiple spacer sequences to prevent viral escape and persistence (van Houte et al., 2016), the relaxed specificity of type III CRISPR-Cas targeting provides a robust immune response that can lead to viral extinction with a single spacer sequence.
RESULTS
Type III-A CRISPR-Cas targeting tolerates sequence changes upstream of the protospacer
To fully understand the sequence requirements for the S. epidermidis type III-A CRISPR-Cas immunity (Marraffini and Sontheimer, 2008), we performed a comprehensive mutational analysis of one of its targets. Specifically, we decided to determine the number and position of target mutations that enable escape from this system. To do this, we introduced the target sequence (the 35-nt protospacer and 10-nt of each flanking sequence) from phage CNPH82 (Daniel et al., 2007) matching spacer 2 (spc2) from the S. epidermidis CRISPR locus (Fig. S1A) into a transcribed region of the Gram-positive vector pLZ12 (Perez-Casal et al., 1991). We then created target plasmid libraries containing different numbers and combinations of protospacer mutations which were introduced via transformation into staphylococci. We first developed this method to comprehensively determine the PAM and seed sequence requirements of the Streptococcus pyogenes type II-A CRISPR-Cas system (Jiang et al., 2013a) and was subsequently used to study type I targeting in detail (Fineran et al., 2014). To obtain the number of transformants necessary to cover all the sequences present in the target libraries we heterologously expressed the type III-A CRISPR-cas locus of S. epidermidis RP62a (a strain with a low transformation efficiency) in Staphylococcus aureus OS2 (a strain with high transformation efficiency (Schneewind et al., 1992); 104–105 colonies per transformation) using the staphylococcal vector pC194 (Hatoum-Aslan et al., 2013).
We started by looking for the presence of a PAM sequence flanking the CNPH82 target. It is known that the sequence flanking the 5’ end of the protospacer affects type III-A CRISPR-Cas immunity. Homology between the repeat sequence preceding the spacer (known as the “tag” sequence in the crRNA) and the corresponding flanking sequence of the target (known as the “anti-tag”), prevents immunity (Marraffini and Sontheimer, 2010) and DNA cleavage, but not RNA cleavage (Kazlauskiene et al., 2016; Samai et al., 2015). In addition, the type III-B Cmr complex produced by Pyrococcus furiosus requires the presence of a short motif named rPAM (RNA prosotspacer-adjacent motif) (Elmore et al., 2016) in the target RNA for efficient DNA degradation. Therefore we decided to perform a comprehensive analysis of the mutations in the 5’ end target flanking sequence that result in the failure of type III-A CRISPR-Cas immunity. To achieve this we generated a plasmid target library harboring all possible nucleotide combinations at positions −1 to −5 upstream of the spc2 target (Fig. 1A). This library was transformed into staphylococci carrying either a targeting, wild-type, type III-A CRISPR-Cas system (WT-CRISPR) or a non-targeting, mutated version of this locus that lacks the CRISPR array (ΔCRISPR) (Fig. S1B). For comparison, we also transformed an empty plasmid (no target control) and a plasmid harboring the wild-type spc2 target. As expected, the transformation efficiency for the latter is reduced from > 106 transformants/µg DNA in ΔCRISPR cells, to ∼ 103 transformants/µg DNA in WT-CRISPR staphylococci (Fig. 1B). We have previously characterized these transformants (Jiang et al., 2013b) and found that they harbor defective CRISPR-Cas loci, and therefore constitute the background of our assays. Surprisingly, transformation of the plasmid library resulted in a reduction of more than 2 orders of magnitude in the transformation efficiency, suggesting that type III-A immunity is robust against most target flanking sequences. To further characterize this sequence flexibility, we collected ∼ 100,000 cells from each transformation experiment, plasmid DNA was extracted and the target amplified via PCR for next-generation sequencing (Fig. S1B). For every target variant a transformation score was calculated as the normalized reads of a particular sequence or group of sequences in WT-CRISPR transformants relative to the normalized reads for the same sequences obtained in ΔCRISPR cells. Therefore a score lower than 1 indicates targeting whereas a score equal or higher than 1 shows that the target sequence was enriched in WT-CRISPR cells due to lack of plasmid destruction. It is important to note that the 5’ flanking sequence library transformation into WT-CRISPR cells yielded much less than 100,000 transformants, likely because most sequences are still targeted (this was also the case for some of our other libraries, see below). Therefore several transformations were performed to reach the desired number of cells (100,000). As a consequence, the sequencing reads for the targets of background escape mutants also increased considerably, leading to an inflation of the transformation score of targeted sequences to values close to 1. Notwithstanding this caveat we could still distinguish targeted from non-targeted sequences based on the transformation score, which is much higher than 1 for the latter. We used the transformation scores first to determine whether the wild-type flanking sequence (CTTCG, present in the CNPH82 phage spc2 target) contains a functional PAM that, similar to the case of type I and II systems, will enable type III-A CRISPR escape if mutated. All the 32 permutations generated by the introduction of mutations in the CTTCG sequence displayed a score lower or close to 1 (Fig. 1C and Supplementary Data File), similar to the non-mutated sequence (and much lower than the GAGAN positive control, see below), indicating that all sequences are similarly targeted and that there is no particular motif within the CTTCG flanking sequence, such as a PAM, essential for immunity.
Fig. 1. Type III-A CRISPR-Cas targeting tolerates sequence changes upstream of the protospacer.

(A) Sequences flanking the spc2 spacer and upstream of the wild-type randomized library target. N, any nucleotide. (B) Transformation efficiencies for different pTargets into cells containing the pWT-CRISPR or pΔCRISPR plasmids. The mean ± SD of 3 independent experiments are reported. (C) Transformation scores for library sequences with all possible deviations from the wild-type target flanking sequence (CTTCG, in red, is a positive control for type III-A targeting). Dots represent any nucleotide but the wild-type in that position. GAGAN (in yellow; N is any nucleotide) is a no targeting, negative control. (D) Transformation scores for all different flanking sequence variants present in the library, in decreasing order. The CRISPR repeat sequence and the wild-type target flanking sequences are identified as negative and positive controls for type III-A CRISPR-Cas immunity. The sequences that allowed the highest level of escape are shown in the yellow box. (E) Nucleotide enrichments were analyzed for all variants of the pTarget library. Normalized depletion counts for each position and nucleotide were compared for WT-CRISPR and ΔCRISPR samples and used to calculate a Z-score. Any nucleotide at each position with a Z-score above 1 is considered significant. See also Fig. S1.
Previously it was shown that matches at positions −2, −3 and −4 between the tag and anti-tag sequences prevented type III-A CRISPR-Cas immunity in S. epidermidis (Marraffini and Sontheimer, 2010). However, in this early study only a few mutant flanking sequences were tested. We used our library of mutations to investigate the full spectrum of target flanking sequences that will enable escape from type III-A CRISPR-Cas immunity. To do this we plotted the transformation score for every flanking sequence in the library in descending order (Fig. 1D and Supplementary Data File). The score for most variants was lower than or close to 1, showing again that most flanking sequences can license type III-A targeting. A small group of variants displayed scores higher than 1 and therefore represent the flanking sequences that can abolish type III CRISPR-Cas immunity. As expected from previous reports (Marraffini and Sontheimer, 2010), we found among these the perfectly matching anti-tag sequence (AGAAC) as well as sequences with partial matches to the tag. Interestingly, the type III-A CRISPR-Cas response was best disrupted by targets containing a GAGAN flanking sequence. This sequence was also found to be highly enriched after calculating the Z-score for every nucleotide in each of the five positions of the randomized library (Fig. 1E), which measures the number of standard deviations that a particular nucleotide is enriched or depleted in the WT-CRISPR transformants. Although GAGAN does not seem to have matches to the AGAAC tag, we realized that there is still substantial homology if one nucleotide gap is allowed (Fig. S2A). We performed transformation assays to corroborate that such flanking sequence abrogates type III-A CRISPR-Cas immunity (Fig. S2B). These experiments demonstrate that most flanking sequences allow type III-A CRISPR-Cas targeting; only those exhibiting substantial homology to the repeat sequence flanking the spacer can prevent it.
To further demonstrate this we mutated the repeat sequence upstream of spc2, from AGAAC to TCTAC (Fig. 2A) and transformed the plasmid library into ΔCRISPR and WT-CRISPR cells harboring this modified CRISPR array (Fig. 2B). If our conclusions are correct then the non-targeted sequences of the library should match this new repeat tag sequence. As was the case in the previous experiment, no single or multiple mutations of the wild-type flanking sequence allowed escape (Fig. 2C and Supplementary Data File). More importantly, in addition to the perfectly matching TCTAC anti-tag, the top group of escape mutant sequences was GTCNN (Fig. 2D and Supplementary Data File) and the same sequence was found to be highly enriched after calculating the Z-score for every nucleotide in each of the five positions of the randomized library (Fig. 2E). Similarly to the results above this sequence is homologous to the modified repeat tag if a gap is allowed in the tag/anti-tag alignment (Extended Data Fig. 2c) and completely abrogates type III-A CRISPR-Cas immunity in a transformation assay (Extended Data Fig. 2d). Altogether, these results establish that the S. epidermidis type III-A CRISPR-Cas system does not require a PAM flanking the target to effectively destroy it. Quite the opposite to type I and II systems, in which any sequence harboring PAM mutations allows for escape, only a select set of flanking sequences, those having homology with the CRISPR repeat tag, allow the evasion of type III-A CRISPR-Cas immunity.
Fig. 2. The terminal sequence of the CRISPR repeat determines the target flanking sequences that escape type III-A immunity.

(A) Sequences flanking the spc2* spacer, a mutant repeat sequence (TCTAC) upstream of spc2, and upstream of the wild-type randomized library target. N, any nucleotide. (B) Transformation efficiencies for different pTargets into cells containing the pWT-CRISPR(spc2*) or pΔCRISPR plasmids. The mean ± SD of 3 independent experiments are reported. (C) Transformation scores for library sequences with all possible deviations from the wild-type target flanking sequence (CTTCG, in red, is a positive control for type III-A targeting). Dots represent any nucleotide but the wild-type in that position. GTCNN (in yellow; N is any nucleotide) is a no targeting, negative control. (D) Transformation scores for all different flanking sequence variants present in the library, in decreasing order. The mutated spc2* CRISPR repeat sequence and the wild-type target flanking sequences are identified as negative and positive controls for type III-A CRISPR-Cas immunity. The sequences that allowed the highest level of escape are shown in the yellow box. (E) Nucleotide enrichments were analyzed for all variants of the pTarget library. Normalized depletion counts for each position and nucleotide were compared for WT-CRISPR and ΔCRISPR samples and used to calculate a Z-score. Any nucleotide at each position with a Z-score above 1 is considered significant. See also Fig. S3.
Type III-A CRISPR-Cas targeting tolerates sequence changes within the protospacer
Next we used our library transformation assay (Fig S1B) to perform a comprehensive analysis of the tolerance of type III-A CRISPR-Cas systems for changes in the protospacer sequence. All the combinations of possible mutations result in more than 1021 different sequences (435, 4 different nucleotides in each of the 35 different protospacer positions) and it is impossible to test given the number of colonies obtained in a transformation experiment (100,000). Even considering only two nucleotides per protospacer position (a wild-type or mutant nucleotide in each position) the number of combinations is still too high (235 > 1010). Therefore we decided to explore the sequence requirements for type III-A CRISPR escape across the first (positions 1–10), second (11–20) and last (26–35) 10-nt regions of the protospacer (Fig. 3A). We tested all the combinations that result from having either the wild-type or its Watson-Crick complementary nucleotide in every position of each region, i.e. we tested 210 (1024) different sequences for each 10-nucleotide target library. Comparison of the transformation efficiency of each target library with wild-type and no-target control plasmids (Fig. 3B) indicated that while most mutations in the last 10-nt of the target sequence did not affect type III CRISPR-Cas immunity, mutations in the first and second 10-nt regions enabled some degree of CRISPR escape. To determine if mutations in particular target positions enable escape from type III CRISPR-Cas immunity, we obtained the transformation scores for each library sequence (Supplementary Data File) and examined the scores of targets with a single nucleotide change in each of the 10 positions analyzed. These scores were compared to those of a wild-type target (without mutations) or of a completely mutant target (all 10 positions of the region mutated). Variants with a single substitution in any of the first or second 10 positions of the target had similar scores to the wild-type control and a significantly reduced score than the fully mutated target (Figs. 3C–E and Supplementary Data File). This data demonstrates that, as suggested by previous studies (Goldberg et al., 2014; Maniv et al., 2016; Millen et al., 2012), single-nucleotide substitutions do not impair type III-A CRISPR-Cas immunity. We next examined the data to determine if the accumulation of protospacer mutations enabled escape and binned the reads for each target variant from the library based on the number of substitutions in each 10-nt region (Figs. 3F–H and Supplementary Data File). We found that at least four (Fig. 3F) and six (Fig. 3G) substitutions are required within the first and second 10-nt protospacer region, respectively, to allow some level of escape from type III-A CRISPR-Cas immunity. As deduced from the transformation efficiency of the library harboring mutations in the last 10-nt region of the protospacer, both single (Fig. 3E and Supplementary Data File) and multiple (Fig. 3H and Supplementary Data File) nucleotide substitutions still allow complete targeting in all cases. This is line with findings from our previous mutagenesis of this protospacer region (Maniv et al., 2016) and agrees with data showing the presence of shorter crRNA species that lack part of the 3’ end sequence due to guide RNA maturation (Hatoum-Aslan et al., 2011) (Fig. S1A).
Fig. 3. Type III-A CRISPR-Cas targeting tolerates sequence changes within the protospacer.

(A) Construction of target libraries harboring mutations in the first (1–10), second (11–21) or last (26–35) 10-nucleotide region of the spc2 protospacer W: A or T, S: C or G. (B) Transformation efficiencies for pTargets containing protospacer libraries in positions (1–10), (11–20) and (26–35) into cells containing the pWT-CRISPR or pΔCRISPR plasmids. Transformation efficiencies of plasmids harboring either no target or a wild-type target are shown as controls. The mean ± SD of 3 independent experiments are reported. (C-E) Transformation score for sequences containing only a single mutation in each of the 10-nucleotide region. The score for the fully wild-type (WT) or fully mutated sequences are shown as controls of type III-A targeting. (F-H) Transformation score for sequences containing increasing number of mutations in each of the 10-nucleotide region. The score for the fully wild-type (0 mutations) or fully mutated (10 mutations) sequences are shown as controls of type III-A targeting. See also Fig. S3.
To rule out the possibility that specific single-nucleotide mutations not covered in our protospacer (1–10) library could disrupt type III-A CRISPR-Cas immunity, we completely randomized the first 5 positions (TAGTA) of the protospacer (N5, Fig. 4A). As it was the case for the (1–10) library, transformation of the N5 library resulted in an intermediate level of targeting (Fig. 4B), suggesting that many sequences still allow targeting. Most of the 32 permutations generated by the introduction of mutations in the TAGTA sequence displayed a score lower or close to 1 (Fig. 4C and Supplementary Data File), similar to the non-mutated sequence, indicating that most sequences are similarly targeted and that the TAGTA does not constitute a seed sequence essential for immunity. Corroborating this observation, a cumulative plot (Fig. 4D and Supplementary Data File) showed that almost any mismatch combination is tolerated in the first five nucleotides of the protospacer sequence, with only the accumulation of 4–5 mutations displaying some decrease in immunity. Results obtained through analysis of both libraries containing different mutations in the first 5 and 10 nucleotides of the protospacer were validated in a transformation assay of targets harboring either 5 or 10 substitutions in this region (Fig. S3). Altogether these results show that, in contrast to type I and II CRISPR targeting, multiple- but not single-nucleotide mutations in the protospacer allow escape from type III-A CRISPR-Cas immunity.
Fig. 4. Accumulation of mismatches in the first 5 nucleotides of the protospacer abrogate type III-A CRISPR-Cas immunity.

(A) pTarget library containing the first 5 nucleotides of the protospacer completely randomized (N5). The wild-type target and the spc2 sequences are also shown. (B) Transformation scores for library sequences with all possible deviations from the wild-type target (TAGTA). Dots represent any nucleotide but the wild-type in that position. (C) Transformation efficiencies for different pTargets (no target, wild-type target, N5 target) into cells containing the pWT-CRISPR or pΔCRISPR plasmids. The mean ± SD of 3 independent experiments are reported. (D) Transformation scores for library target variants containing an increasing number of mismatches to the wild-type target.
Deletion of target region enables viral escape from type III-A CRISPR-Cas targeting
The previous results highlight the high tolerance to target mutations of type III-A CRISPR-Cas systems. Because phage populations almost invariably harbor target mutations in the region targeted by a given spacer, the less flexible targeting rules of type I and II systems facilitate phage escape from CRISPR immunity (Deveau et al., 2008; Semenova et al., 2011). We decided to compare how phages escape type II and type III CRISPR-Cas targeting, to test the hypothesis that the more flexible targeting of the latter would result in less escape. We used staphylococci harboring a vector with either the S. pyogenes type II-A system (lacking the cas1, cas2 and csn2 genes that are required for the acquisition of new spacers) (Heler et al., 2015) or the S. epidermidis type III-A (Hatoum-Aslan et al., 2013) CRISPR-cas locus and infected them with the lytic phage ϕNM4γ4 (Goldberg et al., 2014). Western blot of Cas9 and Csm4 indicated similar levels of expression for both CRISPR loci (Fig. S4A). To be able to compare targeting by these systems, each was programmed with spacers targeting the same phage sequences (Fig. 5A). In addition, we decided to target two different regions of the phage: an intergenic region downstream of gp33, a gene encoding a non-essential dUTPase (Frigols et al., 2015), and a coding region in gp47, a gene producing an essential major phage capsid protein. A fresh preparation of ϕNM4γ4 was plated on an indicator strain lacking CRISPR to measure the total number of plaque-forming units (pfu) of the phage stock, and in each of the four staphylococcal strains harboring CRISPR loci (Fig. 5B). For both type II-A and type III-A targeting, more escapers were obtained when the non-essential intergenic region of the phage was targeted. However, whereas about 1 phage in 105 bypassed type II-A CRISPR-Cas immunity, the count of type III-A escapers resulted two orders of magnitude lower. When the essential gp47 gene was targeted, about 1 phage in 109 escaped type II-A immunity but plaques were not detected in the presence of type III-A targeting (1 in 1010 is the limit of detection of this assay). These results corroborate our hypothesis that the flexibility of type III CRISPR-Cas targeting leads to a very low frequency of escape. We decided to determine the nature of the mutations that allow escape from type II-A and III-A systems. First we analyzed whether phages escaping one CRISPR system were also able to escape the other (Fig. S4B). Interestingly, while the mutations that enabled escape from type II-A targeting did not allow for escape of type III-A immunity, type III-A escaper phages contained mutations that also bypassed type II-A CRISPR-Cas immunity. To determine the nature of the escaper mutations we sequenced the target locus on these 10 escapers after PCR amplification. Escapers of type II-A CRISPR-Cas immunity invariably contained a mutation in one of the G bases of the PAM, both for the gp33 (Fig. 5C) and gp47 (Fig. 5D) targets. In contrast, PCR amplification of the non-essential gp33 gene region from type III-A escaper phages revealed the presence of target deletions (Fig. 5E). Deletions ranged from ∼ 700 to 2500 bp (Fig. 5F) and eliminated the full target sequence from the escaper genomes. These results are in agreement with our data demonstrating the absence of seed or PAM sequences for type III-A CRISPR-Cas targeting, which would have allowed phage to survive through single point mutations in these regions. As a consequence of this, the most frequent event that can lead to escape from type III-A CRISPR-Cas immunity is the deletion of the target sequence from the viral genome.
Fig. 5. Deletion of target region enables viral escape from type III-A CRISPR-Cas targeting.

(A) Sequences of the targets on the phage ϕNM4γ4 genome chosen for this study. Light and dark blue lines: type III-A targets. Light and dark green lines: type II-A targets. (B) Frequency of escape of ϕNM4γ4 from the different CRISPR-Cas targeting shown in (A). The mean ± SD of 3 independent experiments are reported. (C) PCR amplification and sequencing of the gp33 target of phages that escape type II-A immunity. Mutations in the PAM are shown in light green. (D) PCR amplification and sequencing of the gp47 target of phages that escape type II-A immunity. Mutations in the PAM are shown in dark green. (E) PCR amplification of the gp33 target of phages that escape type III-A immunity. (F) Mapping of the different deletions (A–F) spanning the gp33 target of phages that escape type III-A immunity. The coordinates of each deletion on the ϕNM4γ4 genome are reported. See also Fig. S4.
Type III-A CRISPR-Cas targeting results in viral extinction
The above results also suggest that type III-A CRISPR-Cas immunity is impossible to escape if the target is located in an indispensable gene that cannot tolerate deletions. As opposed to the type I and II CRISPR-Cas immune response, which requires the acquisition of multiple spacer sequences to completely destroy the infecting phage (van Houte et al., 2016), a single type III-A spacer targeting such an essential region could drive viral extinction. To test this, we infected staphylococci carrying the type II-A or type III-A CRISPR-Cas system, targeting either the non-essential gp33 gene or the indispensable gp47 region (Fig. 5A), with ϕNM4γ4 phage at a multiplicity of infection (MOI) of 20. Every four hours the surviving phage present in the different culture supernatants were enumerated by counting the total pfu (formed on an indicator, non-CRISPR, strain) and the escaper pfu (formed on staphylococci carrying the targeting CRISPR-cas locus). Also every four hours, the surviving bacteria were measured and the supernatant phage was used to re-infect a fresh culture. Initial immunity against the majority of the phage population as well as phage adsorption resulted in a marked decrease in the total phage count (Fig. S5A). By 12 hours, however, when the non-essential gp33 gene was targeted both by type II-A or type III-A CRISPR-Cas systems, total phage numbers recovered to a high titer, largely due to the rise of the phage escape mutant population. As a reflection of the escaper increase, the bacterial host population was killed and decreased to undetectable levels (Fig. S5B). Similar results were obtained when the essential gp47 gene was the subject of type II-A CRISPR-Cas immunity (Figs. 6A–B). In contrast, when staphylococcal hosts were equipped with a type III-A CRISPR-Cas system targeting the gp47 gene, the absence of phage escape mutants prevented the resurgence of the phage population, which was driven to extinction (Fig. 6A). As a result of this, the type III-A bacterial hosts were able to survive phage killing (Fig. 6B).
Fig. 6. Type III-A CRISPR-Cas targeting results in viral extinction.

(A) ϕNM4γ4 phage titers measured as pfu/ml every 4 hours after infection of cells carrying type II-A (dark green) or type III-A (dark blue) systems targeting the gp47 essential gene. Total phage titers (dotted line) were calculated after plating samples on non-CRISPR strain RN4220. Escaper phage titers (full line) were calculated by plating samples on CRISPR-immune bacterial strains. (B) Bacterial growth measured as OD600 every 4 hours after ϕNM4γ4 infection of cells carrying type II-A (dark green) or type III-A (dark blue) systems targeting the gp47 essential gene. Growth measurements of non-infected cells (dotted lines) are shown as controls. Results are representative of three different experiments. See also Fig. S5.
Next we tested whether the level of host protection provided by a single type III spacer would be possible with the type II-A CRISPR-Cas system if the bacterial population contained multiple different spacers. To do this we picked ten random targets in the ϕNM4γ4 genome (Fig. 7A). The immunity of a mixed population harboring five of these spacers was not enough to drive the phage to extinction (Fig. 7B) and preserve the bacterial host (Fig. 7C). Only the simultaneous targeting by ten spacers led to a significant decrease of the phage titers, although not to a complete clearing as was the case for the single type III-A spacer (Fig. 7D), and to the survival of the infected staphylococci (Fig. 7E). Altogether these results show that, due to the detrimental effects of deletions within essential phage genes, the targeting of such sequences by the S. epidermidis type III-A CRISPR-Cas system leads to the extinction of the virus.
Fig. 7. Diversity of spacers in the host population enables a robust type II-A CRISPR-Cas immune response.

(A) Genome of the staphylococcal phage ϕNM4γ4. The position of the targets used in this study (matching the top or bottom strand) is shown. The green arrow represents the central promoter of this phage. (B) ϕNM4γ4 phage titers measured as pfu/ml every 4 hours after infection of cells carrying type II-A CRISPR-Cas system targeting five different genomic regions (A–E). Total phage titers (black line) were calculated after plating samples on non-CRISPR strain RN4220. Escaper phage titers (colored lines) were calculated by plating samples on each of the five different CRISPR-immune bacterial strains. (C) Bacterial growth measured as OD600 every 4 hours after ϕNM4γ4 infection of cells carrying type II-A CRISPR-Cas system targeting five different genomic regions (A–E). Growth measurements of non-infected cells (dotted lines) are shown as controls. (D) same as (B) but after infection of cells carrying type II-A CRISPR-Cas system targeting ten different genomic regions (A-I and gp33). (E) same as (C) but following infection of cells carrying type II-A CRISPR-Cas system targeting ten different genomic regions (A-I and gp33). Results are representative of three different experiments.
DISCUSSION
Our results show that type III-A CRISPR-Cas immunity in staphylococci does not require the presence of specific sequence elements in the target region and therefore phages and other invaders cannot bypass targeting through the introduction of single nucleotide mutations. We showed that the accumulation of mutations within the protospacer sequence are necessary to abrogate targeting and that these mutations are not clustered in any particular sector of the protospacer. We found that four to eight mutations, depending on the spc2 target region, noticeably weaken the immune response mediated by this spacer. The exact number of cumulative mutations that are required to abrogate immunity most likely depend on the spacer sequence, since it has been shown that the details of the sequence requirements for targeting are spacer-specific not only for type III (Maniv et al., 2016) but also for type I (Xue et al., 2015) and type II (Hsu et al., 2013) systems. Regardless of this, our results clearly established that the S. epidermidis type III-A CRISPR-Cas system does not rely on the presence of a protospacer seed sequence for targeting. Likewise, our experiments failed to identify any PAM sequence required for immunity. Instead, through the investigation of the complete flanking sequence space and the alteration of the repeat sequence, we definitively established that lack of homology between the 5’ end repeat sequence preceding the targeting spacer and the 5’ end flanking sequence of the protospacer is a fundamental requisite for targeting. As opposed to the limited number of flanking sequences previously tested (Marraffini and Sontheimer, 2010), our high throughput experiments allowed us to precisely determine the homology requirements for target tolerance, establishing that the presence of a gap in the alignment of flanking sequences is the most efficient configuration to prevent targeting. Given that type III systems recognize their targets at the RNA level (Elmore et al., 2016; Estrella et al., 2016; Kazlauskiene et al., 2016), we speculate that both the effect of this gap and of the accumulation of mutations in the protospacer region impact type III-A CRISPR-Cas immunity by affecting the dsRNA interaction between the crRNA and the protospacer RNA during targeting. In particular, since the structural analysis of the type III-B complex of Thermus thermophilus revealed that the Cas10 subunit is in close proximity to the crRNA tag (Taylor et al., 2015), it is possible that the gap in the tag/anti-tag RNA interaction directly affects the DNase activity of this subunit. In vivo, in our studies and otherwise, mutations on the DNA will always appear in the RNA that results from its transcription and therefore the effects of the mutations in RNA vs. DNA degradation cannot be separated. In vitro studies that looked exclusively at crRNA-guided RNA cleavage have shown that many mutations on the target RNA can still allow cleavage (Kazlauskiene et al., 2016; Staals et al., 2014), further suggesting that the tolerance to target mutations that we describe here is determined through crRNA:protospacer RNA interactions. Future structural analysis aimed at elucidating the molecular mechanism behind the activation of DNA degradation through RNA recognition in Cas10 complexes will be required to completely understand how mutations affect type III-A immunity at the molecular level.
The type III-B Cmr complex produced by Pyrococcus furiosus requires the presence of a short motif named rPAM (RNA prosotspacer-adjacent motif) (Elmore et al., 2016) in the target RNA for DNA degradation. In our study however, we were unable to find such motif either within target libraries or phages escapers that could avoid type III-A immunity by mutating it. One explanation for this discrepancy could be related to the cooccurrence of the type III-B with two type I loci (I–A and I–G) in P. furiosus. These systems not only have all the same repeat sequence, but also have similar PAMs to each other and to the rPAM of the type III-B targets (Elmore et al., 2015). Since there is evidence that type III-B systems can utilize type I spacers in Sulfolobus islandicus (Deng et al., 2013), it is possible that the P. furiosus type III-B system has evolved to work with the type I spacers and therefore prefers similar flanking motifs to the type I targeting machinery. Future work will determine if co-occurrence of type I and III systems results in an rPAM requirement in hosts harboring both systems.
More importantly, we found that the relaxed targeting rules of type the III-A CRISPR-Cas system have a crucial impact in the nature of the immunity it provides to the host. First, it poses a challenge for the interrogation of the sequence space by the Cas10 complex. In both type I and II systems, the initial recognition of short sequences such as the PAM (∼2 bp) and seed (∼6–8 bp) reduces the complexity of the search space and allows for rapid target finding (Redding et al., 2015; Sternberg et al., 2014); this would not be feasible during type III-A immunity. It is possible that the unique type III targeting mechanism, which recognizes the RNA transcript and not on the DNA (Elmore et al., 2016; Estrella et al., 2016; Kazlauskiene et al., 2016) could facilitate the scanning of the sequence space for the presence of targets. Second, because the presence of many mismatches between a given spacer and its target still license type III-A CRISPR-Cas immunity, the likelihood of having a chromosomal hit for a viral-derived spacer is much higher than in the case of type I and type II targeting, and this could lead to higher levels of toxicity of type III systems. Indeed, the results of pairwise competition experiments show that the S. epidermidis type III-A system, but not the S. pyogenes type II-A, generates a fitness cost for the staphylococcal host (Fig. S5C). Third, we hypothesize that the absence of seed and PAM sequence requirements for targeting may affect the ability of type III-CRISPR-cas loci to acquire new spacers. Besides a single report showing spacer incorporation by a rare type III-U system (Silas et al., 2016), the expansion of the spacer repertoire upon infection has been elusive for the most common and best studied type III-A and III-B CRISPR loci. Since it is unnecessary for these systems to acquire multiple spacers to effectively neutralize a phage, they may have evolved to display a much lower frequency of spacer acquisition. The possibility of self-targeting toxicity mentioned above could also favor the counter-selection of highly active spacer acquisition in type III systems. In addition, single nucleotide escape mutations in the seed or PAM sequences of type I CRISPR-Cas targets promotes a highly efficient mode of spacer acquisition known as “priming” (Datsenko et al., 2012) that relies on a low level of cleavage of the mutated target (Künne et al., 2016; Semenova et al., 2016). Our results suggest that spacer acquisition could not be boosted by such a priming mechanism during type III CRISPR-Cas immunity, since the most common phage escape mutants completely lack the protospacer sequence.
The fourth, and possibly most relevant, consequence of the relaxed sequence requirements of type III-A CRISPR-Cas targeting, is that it provides the host with a much more robust immune response capable of limiting the viral escape rate. To evade type III-A targeting, the phage population must contain individuals that have acquired many mutations, either in the protospacer region or in the 5’ flanking sequence, and for the latter these mutations would have to specifically generate homology with the CRISPR repeat. We believe that such accumulation of mutations is very infrequent, hence the only mechanism of type III-A evasion that we detected was the full deletion of the target region. However, phages have evolved some of the most compact genomes (Brüssow and Hendrix, 2002) where almost every gene plays an important, if not essential, role in the viral infectious cycle and therefore escape from type III-A CRISPR-Cas immunity through target deletion would often be impossible. In addition, the flexibility of type III-A CRISPR-Cas targeting results in a robust immune response not only against phages harboring a perfect target but also other related phages that have a conserved but mutated target sequence. This contrasts with type I and II CRISPR-Cas immunity, which can be evaded frequently by viruses containing single-nucleotide mutations in the seed or PAM sequences (Barrangou et al., 2007; Deveau et al., 2008; Semenova et al., 2011). To circumvent this deficiency, the type I and II CRISPR-Cas immune responses require the acquisition of multiple spacers to prevent escape and effectively defend the host population (van Houte et al., 2016). In contrast, type III-A CRISPR-Cas systems should be able to rely on fewer spacer sequences targeting essential regions of the phage genome to drive the infecting virus to extinction. In summary, our results show that in addition to a distinctive targeting mechanism that results in the cleavage of both DNA and its transcripts, type III CRISPR-Cas immunity also displays a unique high level of targeting flexibility that prevents the emergence and evolution of mutant phage escapers during the host-virus arms race.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luciano Marraffini (marraffini@rockefeller.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and growth conditions
Cultivation of S. aureus RN4220 (Kreiswirth et al., 1983) or OS2 (Schneewind et al., 1992) and E. coli strains were carried out in tryptic soy broth (TSB) or LB liquid media (BD) respectively at 37°C. S. aureus media were supplemented with chloramphenicol at 10 µg/ml or spectinomycin at 250 µg/ml to maintain pCRISPR or pTarget plasmids, respectively. E. coli media were supplemented with spectinomycin at 50 µg/ml for pTarget plasmid maintenance.
METHOD DETAILS
Construction of pTarget plasmids
The sequences of all primers used in this study are listed in Table S1. pTarget was constructed by adding the spc2 protospacer found in phage CNPH82 (Daniel et al., 2007) along with its 10-nt upstream and downstream flanking sequences into the shuttle vector pLZ12 (Perez-Casal et al., 1991). The phage target was appended to plasmid specific primers NP99 and NP100. The vector backbone was amplified using the primer pairs NP25-NP99 and NP24-NP100 and joined using Gibson assembly (Gibson et al., 2009). pTarget(1–5) or pTarget(1–10), pTarget was amplified with primer pairs NP218-NP25 and NP219-NP24 or NP220-NP25 and NP221-NP24, respectively, and the two amplicons were joined using Gibson assembly. pTarget(WT anti-tag) or pTarget(mutant anti-tag), pTarget was amplified with primer pairs NP189-NP25 and NP190-NP24 or NP197-NP25 and NP198-NP24, respectively, and the two amplicons were joined using Gibson assembly. pTarget(WT anti-tag w/gap) or pTarget(mutant anti-tag w/gap), pTarget was amplified with primer pairs NP191-NP25 and NP192-NP24 or NP199-NP25 and NP200-NP24, respectively, and the two amplicons were joined using Gibson assembly.
To generate the pTarget libraries the wild-type pTarget plasmid was first amplified without mutagenesis to generate an insert and a vector template for subsequent PCRs. This eliminated high background levels of wild-type pTarget plasmids in the library. We used 0.05 ng of pTarget plasmid to amplify the target region with primers NP32 and NP67 and the pTarget backbone with NP62 and NP25. The products were gel-extracted, purified and 0.05 ng of each amplicon was used as template. To construct the target library plasmids pTarget(−1–5), pTarget(N5), pTarget(1–10), pTarget(11–20) and pTarget(26–35), the NP32/NP67 template was amplified using primer pairs NP24-NP101, NP24-NP103, NP24-NP103, NP24-NP106 and NP24-NP108, respectively; and the NP62/NP25 backbone was amplified with primer pairs NP25-NP102, NP25-NP104, NP25-NP105, NP25-NP107 and NP25-NP109, respectively. The two amplicons were joined using Gibson assembly.
Construction of pCRISPR plasmids
pWT-CRISPR and pΔCRISPR were obtained previously (Hatoum-Aslan et al., 2013). pCRISPR(spc2* tag) was generated by synthesis of a CRISPR array containing a mutated second repeat (pNP61; see Supplementary Sequences File). Synthetic DNA was amplified with primers L56-L65 and the pΔCRISPR backbone was amplified with primers L66-L55. Two-piece Gibson assembly was used to join the two amplicons.
To introduce ϕNM4γ4 spacers into the type III pCRISPR plasmid we used digested pGG79-F (Goldberg et al., 2014) with BsaI and ligated annealed oligonucleotides with BsaI-compatible ends. Oligonucleotides NP137-NP138 and NP135-NP136 were used to introduce the gp33 (pNP55) and gp47 (pNP54) spacers, respectively. The same approach was used to introduce ϕNM4γ4 spacers into the type II pCRISPR plasmid, using the vector pDB114, which lacks cas1, cas2, and csn2 genes and is therefore unable to acquire new spacers. Plasmid pNP53 was made with oligos PN87-PN88 and harbors the gp33 spacer. Plasmid pRH249 was made with oligos H346-H347 and harbors the gp47 spacer. Plasmid pRH79 was made with oligos H29-H30 and harbors spacer A. Plasmid pRH297 was made with oligos H488-H489 and harbors spacer B. Plasmid pRH294 was made with oligos H482-H483 and harbors spacer C. Plasmid pRH292 was made with oligos H478-H479 and harbors spacer D. Plasmid pRH293 was made with oligos H480-H481 and harbors spacer E. Plasmid pNP72 was made with oligos H117-H118 and harbors spacer F. Plasmid pNP92 was made with oligos PN71-PN72 and harbors spacer G. Plasmid pRH299 was made with oligos H494-H495 and harbors spacer H. Plasmid pNP73 was made with oligos H433-H434 and harbors spacer I.
To add hexa-histidyl tags into the Cas proteins for Western blot experiments, two plasmids were used. A Csm4 version harboring an hexa-histidyl tag was, pAH79 previously generated and validated (Hatoum-Aslan et al., 2013). To add the tag to the C-terminus of Cas9, plasmid pNP114 was constructed by amplifying the backbone pDB114 (Bikard et al., 2014) with primer pair L409-NP343 and the insert cas9 gene from pKW07 (Heler et al., 2015) with L410-NP342. The two fragments were joined with Gibson Assembly.
Library transformation and DNA-seq
After Gibson assembly, library plasmids were introduced via electroporation into NEB 5-alpha Electrocompetent E.coli using a GenePulser Xcell (BioRad) with the following parameters: 1.7 kV, 200 Omega, and 25 µF, 1 mm. Immediately following electroporation, 1ml of SOC media was added and cells were recovered for 1 hour at 37°C. The number of transformants were quantified u sing a serial dilution and plating on LB agar containing 50 µg/ml spectinomycin. Transformants were stored at 4°C overnight and based on the serial dilution colony count, 100,000 colonies were plated on selective LB agar, with ∼ 33,000 cfu plated per 150 mm diameter plate. Colonies were pooled by scraping the plate with 2 ml LB and pellets were frozen at −20°C prior to plasmid library extraction using the QIAprep Spin Miniprep Kit. Library plasmids (1 µg DNA) were introduced into S. aureus OS2 competent cells [harboring pWT-CRISPR, pΔCRISPR or pCRISPR(spc2* tag)] via electroporation. After recovery and serial dilution on tryptic soy agar (TSA) plates containing 10 µg/ml chloramphenicol and 250 µg/ml spectinomycin, transformants were stored at 4°C overnight. Based on the serial dilution colony count, 100,000 colonies were plated on 150 mm plates; with a maximum of ∼ 33,000 colonies per plate. A total of 100,000 colonies were collected for each strain. Multiple transformations for cells harboring pWT-CRISPR or pCRISPR(spc2* tag) were made to reach this number. Colony collection was achieved adding 2 ml of TSB on top of the transformant plates and scrapping the colonies into the media. The pellet for each library in each strain was frozen as −20°C and then thawed to perform DNA extraction. Target library plasmids were extracted from S. aureus transformants using QIAprep Spin Miniprep Kit and the target region was amplified by PCR using different primer pairs (this introduced unique sequences in each PCR product, which served as identifiers to distinguish the origin of reads after next-generation sequencing of the PCR products) as follows: pWT-CRISPR/pTarget(−1–5), NP61-NP65; pWT-CRISPR/pTarget(N5), NP63-NP67; pWT-CRISPR/pTarget(1–10), NP120-NP65; pWT-CRISPR/pTarget(11–20), NP122-NP67; pWT-CRISPR/pTarget(26–35), NP124-NP65; pΔCRISPR/pTarget(−1–5), NP62-NP66; pΔCRISPR/pTarget(N5), NP64-NP68; pΔCRISPR/pTarget(1–10), NP121-NP66; pΔCRISPR/pTarget(11–20), NP123-NP68; pΔCRISPR/pTarget(26–35), NP127-NP68; pCRISPR(spc2* tag)/pTarget(−1–5), NP121-NP68. Amplicons were given to the Rockefeller University Genomics Core for library preparation and Illumina Hi-Seq Sequencing. The data was analyzed using Python: first verifying the presence of the target and flanking region outside of the mutagenized region to identify the sample. Next, the frequency of each target or flank variant was calculated for each library.
Western blot analysis
To extract total protein from staphylococci harboring pAH73 or pNP114, cells were grown up to OD600 of ∼1.0 and collected by centrifugation. The pellet was resuspended in 50 µl of Cell Lysis Buffer [25 mM Hepes, 150 mM KCl, 5% (v/v) glycerol, 20% sucrose] with 5 µl of 5 mg/ml Lysostaphin. After incubation at 37 C for 30 minutes, 50 µl of BugBuster Master Mix (EMD Millipore) was added. To ensure that equal protein concentrations were analyzed, the total protein concentration was measured by the Bradford assay (Bradford, 1976). Samples were mixed 1:1 with 2X Laemlli Sample Buffer and run on a Mini Protean 4%-20% TX gel for 30 minutes at 200 V. Transfer to a PVDF membrane was done using an iBlot Gel Transfer device (Thermofisher). The membrane was blocked for 1 hour at room temperature in PBS-T (0.05% Tween 20) containing 5 % milk and then then incubated overnight at 4 C with 1:5000 dilution of THE™ His-tag antibody (Genescript). The membrane was incubated with secondary goat HRP antibody for 1 hour at room temperature. The membrane was visualized by incubating with HRP substrates and imaging for chemiluminescence.
Quantification and characterization of bacteriophage escapers
To evaluate the presence of ϕNM4γ4 bacteriophage (Goldberg et al., 2014) escapers present in a phage stock population, overnight cultures were launched by inoculating a single colony of S. aureus RN4220 harboring either pNP53, pNP54, pNP55, pRH249 or without any plasmid into TSB. 100µl of each overnight culture was diluted into 5 ml of TSA (top agar) supplemented with 5 mM CaCl2 (required for phage adsorption) and plated on a bed of TSA. After top agar solidification, serial dilutions of a concentrated phage stock were plated on the top agar and incubated overnight at 37 °C to count plaques the next day. If plaques were not detected the experiment was repeated by mixing 100 µl (calculated to contain 20 billion pfu) of phage stock with 100 µl of the overnight culture into 5 ml of top agar. The frequency of escape was calculated as the number of plaques obtained for each pCRISPR-containing strain over the number of plaques obtained in the no plasmid control. To check whether escapers of a given CRISPR-Cas system were able to bypass targeting by a different system and/or spacer, 10 escaper plaques were isolated and resuspended on 20 µl of TSB. 2 µl of the resuspension were plated on top agar TSA containing staphylococci with different CRISPR-Cas systems.
Target sequencing of bacteriophage escapers
Ten escaper plaques were isolated and resuspended on 20 µl of TSB and incubated at 37 °C for 30 minutes. 1 µl of the phage mixture was used as template for PCR amplification. For type II escapers, primers GG215-GG211 and PN95-PN96 were used to amplify the gp47 and gp33 targets, respectively. For type III escapers of gp33 targeting primers NP183-NP184 were used to detect the deletions. PCR products were submitted to Sanger sequencing.
Time course of bacteriophage escape
Overnight cultures of S. aureus RN4220 harboring either pNP53, pNP54, pNP55 or pRH249 were diluted to an optical density value at 600 nm (OD600) of 0.2 in 3.5 ml of TSB supplemented with 5 mM CaCl2 and 10 µg/ml chloramphenicol. The culture was grown at 37 °C for 4 hours without phage, re-dilute d to OD600 of 0.2 and divided into two 2.5 ml samples. Phage at an MOI of 20 was added to one of these aliquots (0 hours post-infection). An aliquot was taken to measure the pfu/ml as described above (both in the pCRISPR cells, to count the escaper phages, and in the RN4220 cells, to count the total phage present) as well as the OD600 of the infected cells. The non-infected culture was used to replenish the cells used in the infected cultures every four hours and avoid the raise of non-CRISPR bacteriophage-resistant mutants. After 4 hours of growth at 37 °C the pfu/ml and OD 600 were measured again (4 hours post-infection). Both samples were centrifuged and the supernatant of the phage-infected sample was mixed with the pelleted, non-infected cells to reach an OD600 of 0.2. A second aliquot of non-infected staphylococci was also resuspended to OD600 of 0.2. After 4 hours of growth at 37 °C the procedure was repeated to obtain the 8 hours post-infection pfu/ml and OD600 values and then five more times to record the 28 hours post-infection data.
Pairwise competition analysis
Overnight cultures of pE194 (Horinouchi and Weisblum, 1982b), pC194 (Horinouchi and Weisblum, 1982a), pWJ40 (Heler et al., 2015), and pWJ30β (Hatoum-Aslan et al., 2013) were diluted to the same OD600. An equal proportion of pE194 (erythromycin-resistant) was mixed with either pC194 (vector control, chloramphenicol-resistant), pWJ40 (harboring the S. pyogenes type II-A CRISPR-cas locus, chloramphenicol-resistant), or pWJ30β (harboring the S. epidermidis type III-A CRISPR-cas locus, chloramphenicol-resistant) and diluted 1:1000 in BHI, in triplicate. For each passage, overnight cultures were diluted 1:1000 in fresh BHI. Culture proportions were quantified at indicated passages by plating and counting colonies on plain, chloramphenicol, or erythromycin-containing TSA plates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis of Deep Sequencing Data
For each target, transformation scores were defined as the ratio of normalized reads of a sequence in WT-CRISPR to ΔCRISPR cells. To analyze the impact of each position in the PAM sequence, transformation scores for each nucleotide type were summed at each position. A normalized score was then computed by applying a standard Z-transformation across all positions and nucleotides. Any nucleotide with a Z-score greater than 1 was considered to be significantly enriched. For each of the pTarget libraries, generalized linear models were fit to the depletion scores and position significance was assessed using the glmnet package in R. Significant positions were considered to be those with p<0.01.
DATA AND SOFTWARE AVAILABILITY
Not applicable.
Supplementary Material
Supplementary data file. Related to Figs. 1–4. Number of reads and calculated transformation score for each of the sequences in the different target libraries analyzed in this study.
Type III CRISPR-Cas immunity does not require PAM or seed sequence motifs
Escape from type III immunity requires complete deletion of the target sequence
Type III targeting of an essential phage gene leads to phage extinction
The targeting flexibility of type III systems provides a robust immune response
Acknowledgments
We thank Robert Heler for pRH plasmids and Gregory Goldberg for pGG plasmids. We would like to acknowledge The Rockefeller University Genomics Resource Center core facility for performing next-generation sequencing. AV is supported by the Arnold. O Beckman Postdoctoral Fellowship. KG is supported by the Tri-Institutional Training Program in Computational Biology and Medicine (via NIH training grant T32GM083937) and by the PhRMA Foundation Pre-Doctoral Informatics Fellowship. OE is supported by NIH grants R01CA194547 and U24CA210989. LAM is supported by the Rita Allen Scholars Program, an Irma T. Hirschl Award, a Sinsheimer Foundation Award, a Burroughs Wellcome Fund PATH award, an NIH Director’s New Innovator Award (1DP2AI104556-01) and a HHMI-Simons Faculty Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS. NP and LAM conceived the study and designed experiments. NP executed the experimental work except the pairwise competition assay, which was performed by AV. KG and OE analyzed NGS data. LAM and NP wrote the paper with the help of AV, KG and OE.
References
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, Marraffini LA. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 2014;32:1146–1150. doi: 10.1038/nbt.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brüssow H, Hendrix RW. Phage genomics: small is beautiful. Cell. 2002;108:13–16. doi: 10.1016/s0092-8674(01)00637-7. [DOI] [PubMed] [Google Scholar]
- Daniel A, Bonnen PE, Fischetti VA. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J Bacteriol. 2007;189:2086–2100. doi: 10.1128/JB.01637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- Deng L, Garrett RA, Shah SA, Peng X, She Q. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol. 2013;87:1088–1099. doi: 10.1111/mmi.12152. [DOI] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore J, Deighan T, Westpheling J, Terns RM, Terns MP. DNA targeting by the type I–G and type I–A CRISPR-Cas systems of Pyrococcus furiosus. Nucleic Acids Res. 2015;43:10353–10363. doi: 10.1093/nar/gkv1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore JR, Sheppard NF, Ramia N, Deighan T, Li H, Terns RM, Terns MP. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 2016;30:447–459. doi: 10.1101/gad.272153.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella MA, Kuo FT, Bailey S. RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 2016;30:460–470. doi: 10.1101/gad.273722.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Gerritzen MJ, Suarez-Diez M, Kunne T, Boekhorst J, van Hijum SA, Staals RH, Brouns SJ. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci USA. 2014;111:E1629–1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigols B, Quiles-Puchalt N, Mir-Sanchis I, Donderis J, Elena SF, Buckling A, Novick RP, Marina A, Penades JR. Virus satellites drive viral evolution and ecology. PLoS Genet. 2015;11:e1005609. doi: 10.1371/journal.pgen.1005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109:E2579–2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, Marraffini LA. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014;514:633–637. doi: 10.1038/nature13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Li Y, Deng L, Feng M, Peng W, Hallstrom S, Zhang J, Peng N, Liang YX, White MF, et al. A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017;45:1983–1993. doi: 10.1093/nar/gkw1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum-Aslan A, Maniv I, Marraffini LA. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci USA. 2011;108:21218–21222. doi: 10.1073/pnas.1112832108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum-Aslan A, Samai P, Maniv I, Jiang W, Marraffini LA. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J Biol Chem. 2013;288:27888–27897. doi: 10.1074/jbc.M113.499244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, Marraffini LA. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015;519:199–202. doi: 10.1038/nature14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi S, Weisblum B. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J Bacteriol. 1982a;150:815–825. doi: 10.1128/jb.150.2.815-825.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi S, Weisblum B. Nucleotide sequence and functional map of pE194, a plasmid that specifies inducible resistance to macrolide, lincosamide, and streptogramin type B antibodies. J Bacteriol. 1982b;150:804–814. doi: 10.1128/jb.150.2.804-814.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013a;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Maniv I, Arain F, Wang Y, Levin BR, Marraffini LA. Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 2013b;9:e1003844. doi: 10.1371/journal.pgen.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M, Tamulaitis G, Kostiuk G, Venclovas C, Siksnys V. Spatiotemporal control of type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol Cell. 2016;62:295–306. doi: 10.1016/j.molcel.2016.03.024. [DOI] [PubMed] [Google Scholar]
- Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- Künne T, Kieper SN, Bannenberg JW, Vogel AI, Miellet WR, Klein M, Depken M, Suarez-Diez M, Brouns SJ. Cas3-derived target DNA degradation fragments fuel primed CRISPR adaptation. Mol Cell. 2016 doi: 10.1016/j.molcel.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniv I, Jiang W, Bikard D, Marraffini LA. Impact of different target sequences on Type III CRISPR-Cas immunity. J Bacteriol. 2016;198:941–950. doi: 10.1128/JB.00897-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millen AM, Horvath P, Boyaval P, Romero DA. Mobile CRISPR/Cas-mediated bacteriophage resistance in Lactococcus lactis. PLoS One. 2012;7:e51663. doi: 10.1371/journal.pone.0051663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- Peng W, Feng M, Feng X, Liang YX, She Q. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015;43:406–417. doi: 10.1093/nar/gku1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Casal J, Caparon MG, Scott JR. Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J Bacteriol. 1991;173:2617–2624. doi: 10.1128/jb.173.8.2617-2624.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Sternberg SH, Marshall M, Gibb B, Bhat P, Guegler CK, Wiedenheft B, Doudna JA, Greene EC. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell. 2015;163:854–865. doi: 10.1016/j.cell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, Marraffini LA. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneewind O, Model P, Fischetti VA. Sorting of protein A to the staphylococcal cell wall. Cell. 1992;70:267–281. doi: 10.1016/0092-8674(92)90101-h. [DOI] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Savitskaya E, Musharova O, Strotskaya A, Vorontsova D, Datsenko KA, Logacheva MD, Severinov K. Highly efficient primed spacer acquisition from targets destroyed by the Escherichia coli type I–E CRISPR-Cas interfering complex. Proc Natl Acad Sci U S A. 2016;113:7626–7631. doi: 10.1073/pnas.1602639113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silas S, Mohr G, Sidote DJ, Markham LM, Sanchez-Amat A, Bhaya D, Lambowitz AM, Fire AZ. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016;351:aad4234. doi: 10.1126/science.aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals RH, Zhu Y, Taylor DW, Kornfeld JE, Sharma K, Barendregt A, Koehorst JJ, Vlot M, Neupane N, Varossieau K, et al. RNA Targeting by the Type III-A CRISPR-Cas Csm Complex of Thermus thermophilus. Mol Cell. 2014;56:518–530. doi: 10.1016/j.molcel.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DW, Zhu Y, Staals RH, Kornfeld JE, Shinkai A, van der Oost J, Nogales E, Doudna JA. Structural biology. Structures of the CRISPR-Cmr complex reveal mode of RNA target positioning. Science. 2015;348:581–585. doi: 10.1126/science.aaa4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houte S, Ekroth AK, Broniewski JM, Chabas H, Ashby B, Bondy-Denomy J, Gandon S, Boots M, Paterson S, Buckling A, et al. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature. 2016;532:385–388. doi: 10.1038/nature17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, van Duijn E, Bultema J, Waghmare S, Zhou K, Barendregt A, Westphal W, Heck A, Boekema E, Dickman M, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci USA. 2011;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Seetharam AS, Musharova O, Severinov K, Brouns SJ, Severin AJ, Sashital DG. CRISPR interference and priming varies with individual spacer sequences. Nucleic Acids Res. 2015;43:10831–10847. doi: 10.1093/nar/gkv1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebec Z, Manica A, Zhang J, White MF, Schleper C. CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2014;42:5280–5288. doi: 10.1093/nar/gku161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data file. Related to Figs. 1–4. Number of reads and calculated transformation score for each of the sequences in the different target libraries analyzed in this study.