

Abstract
The article takes up the problem of deficiency of molecular marker, which could illustrate molecular variability as well as phylogenetic relation within the genus of Stipa L. (Poaceae). Researches made so far hadn’t delivered sufficient information about relationships between particular taxa from the genus of Stipa. In the present study, we analyzed variability and phylogenetic informativeness of nuclear ribosomal DNA in six species from the genus against five other species from Poaceae including a division of this region into functional elements and domains. Our results showed that the intergenic spacer region, and especially its part adjacent to 26 S nrDNA, is a molecular marker giving a real chance for a phylogeny reconstruction of Stipa. The region seems to be the most phylogenetically informative for Stipa from all the chloroplast and nuclear markers tested so far. Comparative analysis of nrDNA repeat units from Stipa to other representatives of Poaceae showed that their structure does not deviate from the general scheme. However, the rate of evolution within the inter-repeats in the IGS region is extremely high and therefore it predestines the region for phylogenetic analyses of Stipa at genus level or in shallower taxonomic scale.
Introduction
The tribe Stipeae comprised over 580 species, common or dominant in open grassland and steppes, with species diversity in temperate regions of Asia, Europe, Africa, Australia and America1. Currently, based on morphological and molecular data, the species belonging to the tribe were segregated into several genera2–5, with Stipa L., Nasella (Trin.) E.Desf., Achnatherum P.Beauv., Austrostipa S.W.L.Jacobs & Everett, Jarava Ruiz & Pav., Piptatherum P.Beauv. and Piptochaetium J.Presl. being the richest in species4. Phylogenetic studies, based on cpDNA and ITS data have shown, that some of genera within Stipeae like, Jarava, Achnatherum, Piptochaetium, Austrostipa, Celtica F.M.Vazquez & Barkworth, Patis Ohwi, Hesperostipa (M.K.Elias) Barkworth, constituted well resolved and highly credible clades1, 3–7. However there is still some groups e.g. within Ptilagrostis Griseb, Piptatherum P.Beauv., Nasella, Achnatcherum and especially in Stipa s. str., in which phylogenetic relationships remined unresolved3–5. With over 160 species (Nobis npbl.) native to Asia, Europe and north Africa, Stipa s. str. is currently the richest in species genus within Stipeae. However, in the light of existing researches using molecular method, only small group of Himalayan species comprised Stipa capillacea Keng, S. regeliana Hack., S. purpurea Griseb., S. penicillata Hand.-Mazz. and S. roborowskyi Roshev. are separated from remaining Stipa species representing almost all distinguished sections within the genus3, 4. There is distinctly a lack of molecular method, that could explain both, molecular variability between selected taxa as well as phylogenetic relation within the genus Stipa. Thus, the aim of our paper was to find a molecular marker representing a sufficient level of informativeness to play a role of phylogenetic marker for Stipa.
In our research, we focused on the repeat units of nuclear RNA genes (nrDNA). In higher plants nrDNA is usually organized in long tandem repeats forming within the chromosomes the nucleolar organizing regions (NOR)8. The nrDNA repeat units comprise DNA coding for RNA subunits and non-coding regions, which are widely applied in plant phylogenetics9–11. Moreover, the region provides great potential to study historical or recent hybridization as well as introgression events. It is because of the fact that in case of reticulate evolution individual nrDNA copies are not homogenized immediately. For this reason, multiple divergent rDNA copies, representing different ribotypes, originating from orthologues and paralogues, can be simultaneously present in a nuclear genome11–13. Phylogenetic marker of such properties would be especially useful in studies on Stipa, since hybrid origin of some species representing the genus have been reported14–17.
The present study is the first report on complete sequence of nrDNA in Stipa and in the tribe Stipeae. The analysis of nrDNA structure in Stipa spp. was carried out in comparison with nrDNA sequences of Brachypodium distachyon (L.) P.Beauv., Oryza sativa L., Setaria italica (L.) P.Beauv., Sorghum bicolor (L.) Moench and Zea mays L., which constitute all the available data on complete nrDNA units published for the Poaceae family.
Results
Structural organization of nrDNA
In the examined species nrDNA repeat units followed a common organizational scheme (Fig. 1). A single unite comprised genes coding for 18 S, 5.8 S and 25 S nrRNA and three non-coding regions: Internal Transcribed Spacer 1 (ITS1), Internal Transcribed Spacer 2 (ITS2) and Intergenic Spacer (IGS). An exception to this scheme was Oryza sativa, where due to the order of functional elements and an occurrence of only one nrDNA unit in the analyzed chromosome, ITS2 could not be distinguished.
Figure 1.

nrDNA unit in Stipa. Structural organization scheme of the nulear-encoded nrDNA repeat unit in Stipa.
The analyzed IGS regions consisted of three functional elements: 3′ External Transcribed Spacer (3′ETS), Non-Transcribed Spacer) NTS and 5′ External Transcribed Spacer 5′ETS (Fig. 1). 3′ETS is a short nucleotide sequence lying downstream 3′ end of 25 S nrRNA gene and ending with transcription termination site (TTS). The exact location of pyrimidine reach motif characteristic for TTS is presented in Supplementary Table S1. In IGS of Stipa we found one putative TTS, while in each of the other species studied two putative TTSs were detected. On the other end of IGS the 5′ETS region is located. Its beginning is determined by a transcription initiation site (TIS) while its end is placed upstream 5′ end of 18 S nrDNA. In the analyzed sequences, the number of putative TIS ranged from one in Zea mays to four in Brachypodium distachyon (Supplementary Table S2). Between 3′ETS and 5′ETS a non-transcribed spacer (NTS) was located demonstrating high variability in sequence length, since only within one genus (Stipa) NTS length ranged from 1453 bp (S. caucasica) to 2347 bp (S. magnifica).
Variability of rDNA in Stipa
Sequence analysis comprising six species of the genus Stipa revealed a low level of genetic variation in the ITS1 and ITS2 regions (Table 1). Pairwise identity within the analyzed set of sequences amounted to 98.2% in ITS1 and 93% in ITS2. The contribution of variable (V) and parsimony-informative (Pi) sites in ITS1 reached only 4.52% and 1.36% respectively. Within the ITS2 seven variable characters (V = 3.38%) were found including only one parsimony informative site (Pi = 0.48%). This indicates that ITS2 was even more conservative than a complete rDNA unit. Compared to both ITS regions, IGS was characterized by a significantly higher level of genetic variation with V = 9.99%, Pi = 3.14% and 6% of singleton sites (S). However, the variation within the IGS was not homogenous, as evidenced by comparing 5′ETS with 3′ETS + NTS. Whilst the region 5′ETS was more conservative, at a similar level as was detected in the ITS1, 3′ETS analyzed together with NTS was the most variable fragment of the whole rDNA. For 3′ETS + NTS pairwise identity amounted to 65.9%, whereas the contribution of Pi sites reached 3.65% out of 11.49% of variable characters.
Table 1.
Comparison of domains within rDNA. Characteristics of selected functional elements and domains within rDNA from Stipa spp. In the case of species where more than one putative TIS was found, the variant with the shortest 5′ETS was considered.
| rDNA | ITS1 | ITS2 | IGS | 3′ETS + NTS | 5′ETS | |
|---|---|---|---|---|---|---|
| Alignment length, bp | 9114 | 221 | 207 | 3314 | 2628 | 686 |
| Sequence length, bp | 7991–8897 | 221 | 205–207 | 2193–3098 | 1513–2414 | 677–684 |
| Pairwise identity, % | 91.0% | 98.2% | 93.0% | 73.4% | 65.9% | 97.6% |
| Variable characters (V) | 362 (3.97%) | 10 (4.52%) | 7 (3.38%) | 331 (9.99%) | 302 (11.49%) | 29 (4.23%) |
| Parsimony-informative sites (Pi) | 113 (1.24%) | 3 (1.36%) | 1 (0.48%) | 104 (3.14%) | 96 (3.65%) | 8 (1.17%) |
| Singleton sites (S) | 221 (2.42%) | 7 (3.17%) | 6 (2.90%) | 199 (6%) | 178 (6.77%) | 21 (3.06%) |
Phylogenetic informativeness
Functional elements and domains distinguished within the rDNA repeat unit were analyzed for the profile of phylogenetic informativeness (PI). A set of data comprising Stipa spp. and Brachypodium distachyon was analyzed separately for: the complete rDNA unit, ITS1, ITS2, the whole ITS region including 5.8 S rDNA, 5′ETS and for 3′ETS together with NTS. The analysis of net PI profiles, relating the overall informativeness of the nucleotide sequence, confirmed very low phylogenetic signal carried by ITS1 and ITS2 parsed both individually and together as the ITS (Fig. 2A). A low level of net PI was also detected in the 5′ETS region. The highest values of net PI were obtained for the complete rDNA unit and only slightly lower for the 3′ETS + NTS fragment. However, within the peak and on its right site PI profiles for rDNA and 3′ETS + NTS were overlapping (Fig. 2A). In turn, considering the length of the nucleotide sequence, significantly stronger phylogenetic signal was reported for 3′ETS + NTS (Fig. 2B). Thus, concerning both the results of net and per site PI, the most informative phylogenetic marker for Stipa was the fragment of IGS including 3′ETS and NTS.
Figure 2.
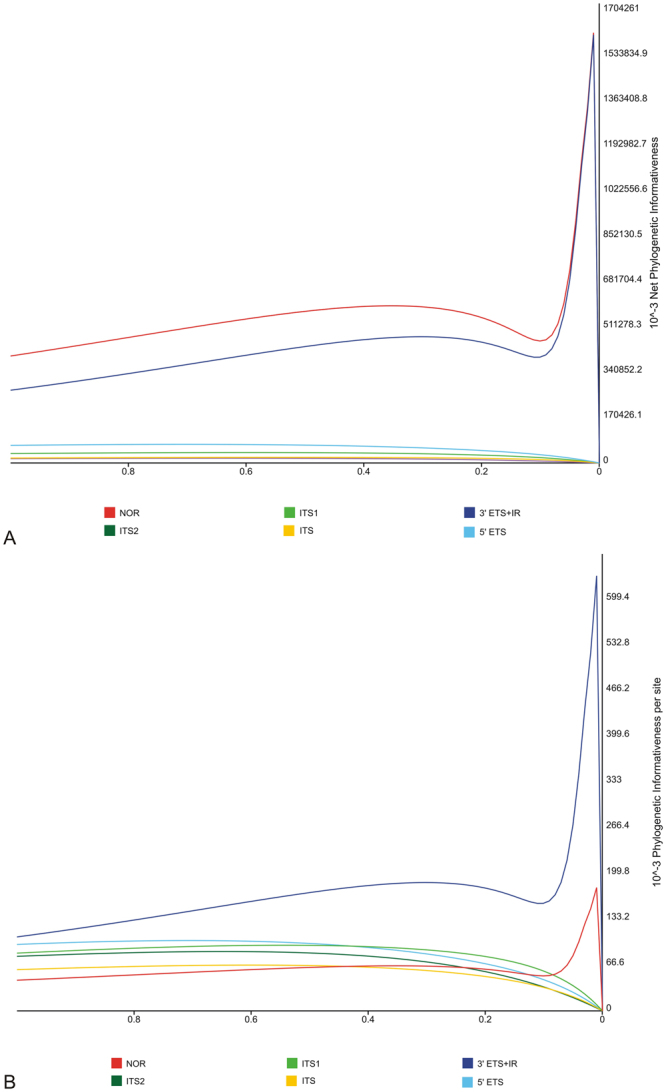
Phylogenetic informativeness. Profile of (A) net and (B) per site phylogenetic informativeness for selected regions within rDNA. The time axis was calibrated to the ultrametrisized ML tree calculated for the complete rDNA unit. An evolutionary time of 1.0 corresponds to the root of the tree and the value of 0 to its tips.
The result was confirmed by the analysis of cladograms calculated separately for the complete ITS and for the 3′ETS + NTS region derived with Maximum Parsimony (MP) method (Fig. 3). The phylogenetic tree based on ITS sequence variation consists of weakly supported clades with all the bootstrap values below 60%. On the contrary, credibility of clades within the tree based on the 3′ETS + NTS region reached up to 100%. Only the clade comprising Stipa lipskyi and S. narynica was supported with 50% bootstrap. Furthermore, the compared cladograms significantly differed in respect of location of S. caucasica. The phylogenetic reconstruction derived from ITS indicated a close relationship of S. caucasica and S. lipskyi (Fig. 3A), while the analysis of sequence variation within 3′ETS + NTS placed these species into separate phylogenetic lines with a high degree of certainty (Fig. 3B).
Figure 3.

Comparison of ITS and 3′ETS + NTS resolving power. Consensus cladograms derived from MP analysis of (A) the complete ITS and (B) 3′ETS + NTS region. Bootstrap values lower than 60% are hidden.
To verify applicability of 3′ETS + NTS as a phylogenetic marker for Stipa we performed the MP analysis on a set of sequences comprising 36 Stipa spp. (Supplementary Table S1) and Achnatherum chingii (Hitchc.) Keng as an outgroup. The length of analyzed sequences ranged from 314 bp in Stipa bungeana Trin. ex Bunge to 925 bp in S. holosericea Trin. The alignment had a total length of 1238 bp, contained 326 Pi sites and was characterized by a 67.5% sequence pairwise identity. The bootstrap consensus tree inferred from 500 replicates derived from MP analysis (Fig. 4) is well-resolved and fully consistent with the most parsimonious tree (1427 steps, consistency index = 0.559659) and with results of Bayesian inference (BI) analysis.
Figure 4.

3′ETS + NTS -based cladogram. The 50% majority-rule bootstrap consensus tree inferred from 500 replicates derived from MP analysis of 3′ETS + NTS region. Bootstrap values >50% above, Bayesian of P > 0.95 below branches.
Inter-repeat region
For a detailed cognition of IGS variability, sequence self-comparison was carried out using dot-plot analysis (see Supplementary Figs S5–S15). The analysis revealed a higher level of sequence conservativeness in the beginning and the end of each IGS and a presence of inter-repeats (IR) region located between them. An identification of number and length of the repetitive sequence motifs occurring in Stipa allowed for defining five different IRs ranging from 28 to 162 bp (Table 2). There was a single nucleotide polymorphism (SNP) observed between particular IR copies within a species. The number of IR copies was highly variable between the species. For example, IR S1 (91 bp in length) was found in only three repeats in S. pennata and S. caucasica however in S. narynica it was present in 12 copies (Fig. 5.), what strongly affected the length of the entire IGS. Similarly, inter-repeat S5 occurred in two copies in S. caucasica, S. orientalis and S. pennata whilst eight repeats were found in S. narynica.
Table 2.
Inter-repeats. Characteristics of repeated regions defined for the species analyzed in the study.
| Species | Inter-repeat | E-value | Sites per species | Width, bp |
|---|---|---|---|---|
| Stipa spp. | S1 | 1.5e-738 | 3–12 | 91 |
| S2 | 6.4e-696 | 2 | 162 | |
| S3 | 3.8e-636 | 2–5 | 77 | |
| S4 | 8.4e-189 | 2 | 55 | |
| S5 | 2.4e-168 | 2–8 | 28 | |
| Brachypodium distachyon | B1 | 8.7e-031 | 4 | 78 |
| B2 | 5.3e-016 | 5 | 39 | |
| B3 | 3.2e-005 | 5 | 39 | |
| Oryza sativa | O1 | 7.8e-069 | 4 | 173 |
| O2 | 2.1e-012 | 5 | 57 | |
| Setaria italica | Si1 | 1.6e-057 | 5 | 77 |
| Si2 | 1.4e-008 | 5 | 25 | |
| Sorghum bicolor | Sb1 | 1.1e-075 | 3 | 212 |
| Sb2 | 2.2e-063 | 8 | 69 | |
| Sb3 | 4.7e-051 | 5 | 69 | |
| Sb4 | 2.1e-030 | 8 | 48 | |
| Sb5 | 3.2e-012 | 5 | 25 | |
| Zea mays | Z1 | 8.7e-177 | 10 | 192 |
Figure 5.

Structure of the IGS of nrRNA in Stipa. Blue line – plot of GC content, green line – plot of AT content; green bar – best global reconstruction of CpG islands. Colored bars – repeated regions.
The comparison if IGS regions indicated an extremely high level of sequence diversity within the IR region. The simplest inter-repeat pattern was found in Zea mays, where only one type of IR occurred in 10 copies, each 192 bp in length. A simple scheme was also observed in IGS region from Oryza sativa where two nucleotide motifs were defined: O1 (173 bp in length) repeated four times and O2 (57 bp) present in five copies. In IGS from Setaria italica two motifs (Si1 = 77 bp, Si2 = 25 bp) present in five tandem repeats were found. In Brachypodium distachyon the analysis revealed the presence of three different inter-repeats ranging from 39 to 78 bp. The IR region in Sorghum bicolor could be distinguished into two parts. In a part located near the 25 S nrRNA gene motif Sb2 and Sb4 were alternated. Downstream there was located a second part of IR region comprising Sb1, Sb3 and Sb5 (Fig. 6). In all the species tested, excluding S. italica and Z. mays IRs were located both within NTS and 5′ETS. In nine of 11 examined species, repeated motifs included putative TIS, what entailed its multiplication from two in Stipa spp. to four in B. distachyon (Table 2).
Figure 6.

Structure of the IGS of nrRNA in Poaceae. Blue line – plot of GC content, green line – plot of AT content; green bar – best global reconstruction of CpG islands. Colored bars – repeated regions.
GC/AT-content and CpG islands
Within the rDNA, the highest share of GC-content reaching up to 76.3% (ITS2 of Stipa magnifica) was noted in ITS1 and ITS2 regions. The analyzed IGS were characterized by a lower contribution of GC pairs (Table 3), ranging from 53.6% in Sorghum bicolor to 70.1% in Oryza sativa. The distribution of GC pairs within the IGS region was variable between the analyzed species as well. In Oryza sativa and Brachypodium distachyon their even arrangement and relatively high share resulted in lack of AT–rich regions. In S. bicolor and Zea mays IGS contained four regions where the rate of AT pairs exceeded 50%. In the analyzed representatives of the genus Stipa a short AT-rich region was present within the NTS, near its 5′ end (Fig. 5). IGS from Setaria italica also contained a short region with a high rate of AT pairs within the 5′ part of NTS.
Table 3.
Variability in length and GC-content. Comparison of selected functional elements and domains within the rDNA repeat unit between the analyzed species. GC – content of GC pairs in an analyzed nucleotide sequence. In the case of species where more than one putative TIS was found, the variant with the shortest 5′ETS was considered.
| Species | rDNA | ITS1 | ITS2 | IGS | 3′ETS + NTS | 5′ETS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC, % | Length, bp | GC, % | Length, bp | GC, % | Length, bp | GC, % | Length, bp | GC, % | Length, bp | GC, % | Length, bp | |
| Stipa caucasica | 58.5 | 7991 | 73.3 | 221 | 76.2 | 206 | 61.2 | 2193 | 59.5 | 1513 | 65.0 | 680 |
| S. lipskyi | 58.7 | 8451 | 73.8 | 221 | 75.7 | 206 | 61.1 | 2653 | 59.4 | 1969 | 65.9 | 684 |
| S. magnifica | 58.8 | 8897 | 72.9 | 221 | 207 | 61.2 | 3098 | 60.0 | 2414 | 65.6 | 684 | |
| S. narynica | 58.7 | 8844 | 74.9 | 221 | 73.8 | 207 | 60.9 | 3045 | 59.5 | 2361 | 65.7 | 684 |
| S. orientalis | 58.4 | 8038 | 71.5 | 221 | 75.2 | 206 | 61.0 | 2239 | 59.5 | 1558 | 64.8 | 681 |
| S. pennata | 58.6 | 8031 | 71.0 | 221 | 75.1 | 205 | 61.7 | 2233 | 60.5 | 1556 | 64.4 | 677 |
| Brachypodium distachyon | 58.3 | 8078 | 63.5 | 222 | 69.0 | 216 | 62.9 | 2271 | 64.0 | 1392 | 61.1 | 879 |
| Oryza sativa | 55.0 | 7923 | 72.2 | 198 | — | — | 70.1 | 2357 | 69.6 | 1254 | 70.5 | 1103 |
| Setaria italica | 56.1 | 7779 | 57.4 | 209 | 61.5 | 213 | 57.3 | 1991 | 53.9 | 1212 | 62.6 | 779 |
| Sorghum bicolor | 54.3 | 8689 | 56.1 | 212 | 68.1 | 116 | 53.6 | 2952 | 53.5 | 2307 | 54.2 | 645 |
| Zea mays | 59.7 | 8795 | 69.9 | 219 | 74.1 | 216 | 62.5 | 3421 | 60.4 | 2591 | 69.2 | 830 |
The analysis of CpG sites location revealed they comprised the whole IGS from B. distachyon, O. sativa and Z. mays. In the IGS from S. bicolor there were two CpG-rich sites within the NTS (Fig. 6). In Stipa spp. the region characterized by a high concentration of CpG sites covered most of the IGS including 3′ETS, 5′ETS and a substantial part of the NTS. Only in the AT-rich fragment and downstream (Fig. 5) the lack of CpG islands was noted. In Setaria italica there was one CpG-reach region covering a part of NTS and the whole 5′ETS.
Discussion
The parts of nuclear rDNA sequence data are widely used in phylogenetic inference at various taxonomic levels. The most prominent nuclear phylogenetic marker in plants is ITS10. The region was successfully applied for phylogeny reconstruction in Poaceae at the level of family18, subfamily19 and tribe20, 21. The ITS sequences were also shown to be useful for assessing evolutionary relationships among closely related grass species for example within the genera of Bromus 22, Miscanthus 23, Hordeum 24, Elymus 25, Setaria 26 and Festuca-Lolium complex27. However, the literature data have shown that phylogenetic trees derived from ITS sequences were unresolved within clades comprising Stipa 3, 28. The branch of ITS-based cladogram published by Hamasha et al.3 comprising Stipa spp. contained a lot politomies and only few clades with credibility value over 50%, which brought little information useful in reconstruction of phylogenetic relationships within the genus. Our study also revealed an insufficient level of ITS variability in the species from Stipa to address questions of their intrageneric relationships. Both the low-resolution phylogenetic tree derived from MP analysis and the profile of PI proved relatively low rate of ITS sequence evolution.
Much more information useful in phylogenetic assays concerning Stipa was carried by the IGS region. Numerous studies proved, that IGS was a valuable marker in phylogenetic analyses of angiosperms which could supplement nucleotide variation from generally shorter ITS9, 11, 29. The high rate of evolution of this spacer region makes it suitable even for detecting intraspecific polymorphism8, 30–32. The rate of evolution within IGS is not uniform, what is a rule for this region12, 32 and it presumably results from different functions of its components8. In general, more conservative region within IGS is the 5′ETS fragment11, 12. Literature data demonstrate that the variation of the fragment remains at level typical for ITS or slightly higher12. The same relation was demonstrated in Stipa, where the PI of IGS was greater than in ITSs, however the number of Pi sites did not exceed the number of Pi sites reported for ITS1.
A much higher level of informativeness identified in the IGS of Stipa was caused by a great variability of NTS containing repeated regions. Both the PI plot for 3′ETS + NTS and a cladogram, providing more resolution to phylogenetic reconstructions than the one obtained for ITS sequence, demonstrate that the phylogenetic signal carried by 3′ETS + NTS is strong enough to picture intrageneric relationships in Stipa. It was also confirmed by the MP and BI analysis conducted on a set of 36 Stipa species. 26 of 36 species included in our analysis were previously analyzed by Hamasha et al.3 and very low divergence of ITS sequence was reported for them. 3′ETS + NTS-based cladogram in comparison with ITS-based tree3 delivered much more information useful in phylogenetic inference in case of all the species common for the two studies. Considering only the species common to work by Hamasha et al.3 and our research, analysis based on ITS sequences resulted in only five clades at different levels of organization with bootstrap values over 50%, while the tree based on 3′ETS + NTS contained 19 clades with support >50%.
The part of intergenic spacer comprising 3′ETS + NTS is also more useful for phylogenetic implications in Stipa than cpDNA markers applied in the studies on this genus so far. For example, the phylogenetic analysis based on nine plastid DNA regions4 (Romaschenko et al. 2012) grouped Stipa capillata, S. caucasica and S. pennata, which represent three different sections within the genus, into one highly supported and unresolved clade. In turn, the analysis derived from the IGS variability placed these species into three different clades indicating their more distant relationship.
Unfortunately, the rapid evolution of NTS entails the presence of return mutations, homoplasies and substitution saturation, which make up a so-called informative noise33. Its presence is indicated by the PI profile with a peak placed near the right site of a diagram (Fig. 2). While the part of the curve right to the peak illustrates a high informativity of the analyzed region for the earliest evolutionary events, the part left to the peak, covering a large part of the analyzed timeline, indicates the accumulation of uninformative mutations33, 34. Therefore, the 3′ETS + NTS region is a suitable marker in shallower phylogenetic scale. Its application would not be appropriate in suprageneric level studies, as it would not reflect evolutionary history of higher taxonomic groups and could lead to erroneous conclusions. Another disadvantage resulting from applying in phylogenetic study a nucleotide fragment highly variable in length and sequence is a problem with alignment construction. However, identification of repeated motifs in NTS and the conservative TIS sites greatly facilitates aligning this extremely variable fragment, and in our opinion indeed is essential for the correct data analysis.
Arrangement of repeats distinguished in the NTS should best illustrate the evolutionary history of this region formed by multiplication or deletions of entire sequence fragments. Point mutations in type of indels or nucleotide substitutions happening simultaneously with motif multiplication obliterate their picture to a large extent. Therefore, assignment of best possible scheme of repeats is challenging and largely depending on available set of data. Having at disposal only one nucleotide sequence one would distinguish another set of repeats than while having set of sequences for several closely related species. Also, depending on the algorithm used for the designation of motifs, different set of repeats might be obtained. In this study methodology assumed the constant length of particular repeats, differing only in SNPs. In other studies31, 35, 36, authors decided to take into account the differences in the length of motifs. Due to different methodology, sometimes including searching for sequence motifs bye eye31, 35, often the results obtained in different studies are inconsistent with each other. Only in case of clear and simple patterns of repeats we can get similar results using various methods. That is a case of Zea mays, where ten IRs 192 bp in length distinguished in our study correspond with ten repeats between 165 and 232 nt in length described by McMullen et al.35. Also, the results of analysis of IGS from Brachypodium distachyon has a common ground with the results obtained by Borowska-Zuchowska et al.37 as described by them repeat REP corresponds with our B1 inter-repeat. The literature data on Oryza sativa demonstrated variation in IGS structure depending on the analyzed genotype, although the general scheme of alternating long and short repeat38 is similar to the pattern of repeats in IGS from rice analyzed in our study. In turn, the organization of IR region described by us for Setaria italica as an arrangement of two IRs repeated tandemly in five copies, doesn’t correspond to three regions of short subrepeats and one long repeat distinguished in IGS from S. italica by Fukunaga et al.31 who analyzed set of 40 accessions of landraces within two subspecies.
Our analysis of IGS structure within the genus of Stipa demonstrated its great applicability in phylogenetic studies at the generic level. However, the comparison of IGSs within the family of Poaceae leads to the conclusion that this region is highly divergent between the genera. Each of the analyzed genera was characterized by an individual pattern of inter-repeats and finding a common sequence motif or evolution scheme between the genera was impossible. Therefore, the IGS region is not an appropriate phylogenetic marker for studies at the supra-generic level in Poaceae.
The species included in the study also differed significantly regarding the presence of regions rich in AT sites. These regions are of special concern because they putatively contain protein binding sites involved in regulation of transcription and are associated with a proximity of gene promoter8, 39, 40. The presence of AT-rich sequence adjacent to TIS sites was reported, for example for Arabidopsis thaliana 41, Olea europea 42 Haplopappus gracilis 39, Fagus sylvatica, Quercus suber 43 and Brassica oleracea 32. Within the analyzed species representing Poaceae, the presence of AT-rich region upstream TIS was not a rule. Only in Sorghum bicolor three out of four short AT-rich regions, overlapping with fragments of SB1 repeat, preceded putative TIS sites. In Stipa spp. and Setaria italica a fragment of IGS representing a high rate of AT-sites was found, but closer to 25 S RNA gene, near the 5′ end of the NTS. In turn, the IGS from Zea mays contained four short AT-rich regions localized within the copies of IR Z1 but none of them was adjacent to TIS. The lack of IGS regions with a high share of AT pairs in Oryza sativa and Brachypodium distachyon completes the picture of sequence diversity within IGS.
The varied pattern of AT-rich sites distribution within the IGS may correspond with assumptions of some authors which discuss the significance of repeated elements in a control of nrRNA transcription. On the example of wheat, it was demonstrated that nrRNA genes located on the chromosomes with less subrepeats within the IGSs appeared relatively inactive44. Molecular studies showed that the subrepeat sequences in IGS attract similar proteins as sequences surrounding TIS and presumably play a role of promoter enhancers8, 45, 46. It is supposed that higher order structure formed by repeated motifs rather than a defined sequence plays a key role in these interactions8, 32, 46. In general, the IGS sequence is GC-rich32, however in the family of Poaceae the content of GC pairs is varied, and unless in Oryza sativa indeed it is very high and exceeds 70%, in Sorghum bicolor is less than 54%. Also in Fagus sylvatica and Quercus suber GC-content was at moderate level reaching 52% and 57% respectively. Characteristic for the IGS is the presence of CpG, CpCpG or CpNpG motifs, which are prone to methylation8, 32, 47. This one of the most important epigenetic modifications leads to gene silencing and the level of sequence methylation in the promoter region is strongly associated with the regulation of transcription of nuclear nrRNA genes48. It was shown that in species from Cucurbitaceae, equipped with a large number of ribosomal DNA, at least 70% of the repeats were completely methylated and therefore excluded from transcription8. The distribution of methylable sites along the IGS sequence is another feature of this spacer greatly diverse between the species40. For example, in Punica granatum over 100 methylable sites were detected, mainly in the subrepeat region and fewer within unique regions40. In turn, the IGS from F. sylvatica and Q. suber contained CpG island exclusively within the 5′ETS region32. Within the analyzed representatives of Poaceae there was no specific pattern of CpG sites distribution within the IGS in relation to its functional elements. In all the examined species, they were abundant and rather evenly distributed.
In conclusion, the present study shows that the IGS region, and especially its part adjacent to 26 S nrDNA, is a molecular marker giving a real chance for a phylogeny reconstruction of Stipa. The region seems to be the most phylogenetically informative for Stipa from all the chloroplast and nuclear markers tested so far. Moreover, as a nuclear spacer it enables the study of hybridization and introgression phenomena occurring in Stipa. Comparative analysis of nrDNA repeat units from Stipa to other representatives of Poaceae showed that their structure does not deviate from the general scheme. However, the rate of evolution within the inter-repeats region is extremely high and therefore it predestines the region for phylogenetic analyses of Stipa at the interspecific level in the genus.
Materials and Methods
Plant material and DNA extraction
All the examined Stipa spp. specimens were collected during field research in the years 2011–2014 (see Supplementary Table S3). Total genomic DNA was extracted from desiccated leaf tissue using ZR Plant/Seed DNA MiniPrepTM kit (Zymo Research Corp., USA) and Genomic Mini AX Plant Spin (A&A Biotechnology, Poland) following the manufacturers recommendations. DNA quality was assessed by the 1% agarose gel electrophoresis and quantity was estimated with the use of the Qubit fluorometer system and the Quant-IT ds-DNA BR Assay kit (Invitrogen, USA).4.
DNA library preparation and sequencing
A genomic library for MiSeq sequencing (for S. lipskyi, S. narynica & S. orientalis) was developed with the use of the Nextera XT Kit (Illumina, San Diego, CA, USA). DNA in the amount of 1 ng was used in the procedure described in the Nextera XT protocol. Constructed libraries were sequenced using the Miseq. 600v3 sequencing kit (Illumina, San Diego, CA, USA) that enable to obtain of 2 × 300-bp pair-end reads.
Three other species (S. caucasica, S. magnifica, S. pennata) were sequenced using the Illumina HiSeq. 2000 platform (Illumina, San Diego, CA, USA). A 350-bp paired-end library was constructed using Truseq DNA Nano kit and sequenced at Macrogene (Korea).
NGS sequencing
All the obtained reads were trimmed and parts with low quality (Q below 5) or containing N’s were excluded. Cleaned reads were assembled de novo using Velvet plugin in Geneious 7.0 (Biomatters, New Zealand). The contigs contained nrRNA genes were identified and were further expanded by mapping cleaned reads with 25 iterations using Geneious 7.0 with custom settings (70 bp overlap with 99% identity). The sequences obtained after iterations were assembled de novo using Geneious built-in assembler with high sensitivity settings.
Annotations were performed using Geneious software based on BLAST tool and NCBI resources49. Complete IGS sequences of six Stipa species were explored to discover the degree of conservation between different functional units. Sequences were aligned with Muscle genome alignment within the Geneious environment and corrected manually. The nrDNA sequences of Brachypodium distachyon, Oryza sativa, Setaria italica, Sorghum bicolor and Zea mays downloaded from the NCBI database (Supplementary Table S4) were not included into the alignment due to the vast variability in IGS region which made an accurate sequence alignment impossible. Six complete nrDNA (ribosomal DNA) sequences were deposited in the NCBI (Supplementary Table S3).
Sanger sequencing
PCR fragments for sequencing with Sanger method were amplified using TFL Epicentre polymerase and the primer combination: igsF: 5′-AGC CCC ACG TCG CAC GGA TTC GTC C-3′ with igsR: 5′-CCT CMC TTC AAC MGT TTC CRT GGG-3′. PCR experiments included an initial 95 °C denaturation followed by 35 cycles of 45 s at 95 °C, 50 s at 54-57 °C, 1 min 20 s at 72 °C, with a final 7-min extension at 72 °C. The amplification products were visualized on 2% agarose gel with GelView (Invitrogen™, Carlsbad, CA, USA) staining. Purified PCR products were sequenced in both directions using ABI BigDye 3.1 Terminator Cycle Kit (Applied Biosystems®, Foster City, CA, USA) with the same primers and then visualized using an ABI Prism 3130 Automated DNA Sequencer (Applied Biosystems®, Foster City, CA, USA). PCR recipe: 20 mM (NH4)SO4, 50 mM Tris-HCl (pH 9.0 at 25 °C), 1.5 mM MgCl2, 10 μg BSA, 0.2 mM of each dNTPs, 1.0 μM of each primer, 1U Taq polymerase (TFL Epicentre), 10 ng of the DNA, to 20 μl with dH20.
The analysis of IGS structure
For the discovery and comparative analysis of repetitive motifs in nucleotide sequences complete IGS regions were extracted from nrDNA units and submitted to the MEME Suite 4.11.250. Discovery of sequence inter-repeats was done with the MEME tool51 and verified with MAST52. Six IGS sequences of Stipa spp. were submitted to the MEME environment as a set while each of the other sequences were analyzed individually. The limit for the minimum and maximum width of the repetitive motif was 20-200 bp for Stipa spp. and Zea mays, 15-250 bp for Brachypodium distachyon, and 25-250 bp for Setaria italica, Sorghum bicolor and Oryza sativa. The most suitable length of predicted inter-repeats was established basing on their E-values.
The prediction of putative transcription initiation sites (TIS) and putative transcription termination sites (TTS) was based on the comparative analysis of sequences with literature data concerning different plant species8, 29, 32, 37, 43, 46. AT-rich regions and CpG islands were identified using the Geneious software. The Geneious environment was also used to perform dot plot analysis.
Phylogenetic informativeness
Phylogenetic informativeness (PI) was estimated using PhyDesign server53, 54. PI profiles were plotted with reference to an uncalibrated tree. The tree used to overlay the historic changes in substitution rates was obtained with Maximum Likelihood method using MEGA v.655 and ultrametrisized using PATHd856. To obtain relative ages for the clades, the root of the tree was set at an evolutionary time of 1.0 and tips at time of 0. The HyPhy program57 which is using empirical base frequencies and a time-reversible model of substitution was used to calculate PI of nucleotide data sets.
The analysis of ITS and 3′ETS + NTS resolving power
MP analysis was done using MEGA v.655 The MP tree was obtained using the Subtree-Pruning-Regrafting (SPR) algorithm with search level 1 and the number of initial trees equal to 10. The tree was tested with bootstrap method with the number of bootstrap replications at the level of 500.
BI analysis was done using MrBayes plugin in Geneious 7.0 (Biomatters, New Zealand) with the priors set according to the output of DNA model testing in MEGA v.655. The parameters of the likelihood model applied for 3′ETS + NTS were adequate for general time reversible model with a gamma-shaped distribution of rates across sites (GTR + Γ), (n st = 5). BI was estimated running four incrementally heated chains (MCMC algorithm) for 1,000,000 generations, sampling one out of every 200 generations of rando trees. The first 200,000 generations were discarded as “burn-in”. The remaining generations were used to construct The Bayesian consensus tree.
Electronic supplementary material
Acknowledgements
Financial support for this study came from the National Science Center, Poland (DEC-2013/09/B/NZ8/03287).
Author Contributions
Conception and design: All authors. Collection and determination of plant material: M.N., A.N. Analysis of NGS data: J.S., M.Sz. Sanger sequencing and interpretations of results: K.K. Drafting of manuscript: K.K., J.S., M.N. Final approval of manuscript: All authors.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-11804-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Barkworth ME, et al. Molecules and morphology in South American Stipeae (Poaceae) Syst. Bot. 2008;33(4):719–731. doi: 10.1600/036364408786500235. [DOI] [Google Scholar]
- 2.Barkworth, M. E. & Everett, J. Evolution in the Stipeae: identification and relationships of its monophyletic taxa in Grass systematics and evolution: an International Symposium held at the Smithsonian Institution (ed. Soderstrom, T. R. et al.) 251–264 (Smithsonian Institution Press 1987).
- 3.Hamasha HR, von Hagen KB, Röser M. Stipa (Poaceae) and allies in the Old World: molecular phylogenetics realigns genus circumscription and gives evidence on the origin of American and Australian lineages. Plant Syst. Evol. 2012;298(2):351–367. doi: 10.1007/s00606-011-0549-5. [DOI] [Google Scholar]
- 4.Romaschenko K, et al. Systematics and evolution of the needle grasses (Poaceae: Pooideae: Stipeae) based on analysis of multiple chloroplast loci, ITS, and lemma micromorphology. Taxon. 2012;61(1):18–44. [Google Scholar]
- 5.Cialdella AM, et al. Phylogeny of Nassella (Stipeae, Pooideae, Poaceae) based on analyses of chloroplast and nuclear ribosomal DNA and morphology. Syst. Bot. 2014;39(3):814–828. doi: 10.1600/036364414X681419. [DOI] [Google Scholar]
- 6.Romaschenko K, et al. Molecular phylogenetic analysis of the American Stipeae (Poaceae) resolves Jarava sensu lato polyphyletic: evidence for a new genus. Pappostipa. J. Bot. Res. Inst. Texas. 2008;2(1):165–192. [Google Scholar]
- 7.Romaschenko, K., Peterson, P. M., Soreng, R. J., Garcia-Jacas, N. & Susanna, A. Phylogenetics of Stipeae (Poaceae: Pooideae) based on plastid and nuclear DNA sequences. Div. Phyl. Evol. Monocotyledons 511–537 (2010).
- 8.Hemleben, V. & Zentgraf, U. Structural organization and regulation of transcription by RNA polymerase I of plant nuclear ribosomal RNA genes in Plant Promoters andTranscription Factors (ed. Nover, L.) 3–24 (Springer, 1994). [DOI] [PubMed]
- 9.Baldwin BG, Markos S. Phylogenetic utility of the external transcribed spacer (ETS) of 18S-26S rDNA: Congruence of ETS and ITS Trees of Calycadenia (Compositae) Mol. Phyl. Evol. 1998;10(3):449–463. doi: 10.1006/mpev.1998.0545. [DOI] [PubMed] [Google Scholar]
- 10.Álvarez I, Wendel JF. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phyl. Evol. 2003;29(3):417–434. doi: 10.1016/S1055-7903(03)00208-2. [DOI] [PubMed] [Google Scholar]
- 11.Poczai P, Hyvönen J. Nuclear ribosomal spacer regions in plant phylogenetics: problems and prospects. Mol. Biol. Rep. 2010;37(4):1897–1912. doi: 10.1007/s11033-009-9630-3. [DOI] [PubMed] [Google Scholar]
- 12.Calonje M, et al. Non-coding nuclear DNA markers in phylogenetic reconstruction. Plant Syst. Evol. 2009;282:257–280. doi: 10.1007/s00606-008-0031-1. [DOI] [Google Scholar]
- 13.Volkov RA, Komarova NY, Hemleben V. Ribosomal DNA in plant hybrids: inheritance, rearrangement, expression. Syst. Biodivers. 2007;5(3):261–276. doi: 10.1017/S1477200007002447. [DOI] [Google Scholar]
- 14.Smirnov PA. Stiparum Armeniae minus cognitarum descriptiones. Byulleten Moskovskogo Obshchestva Ispytatelej Prirody, Otdel Biologicheskii. 1970;75:113–115. [Google Scholar]
- 15.Tzvelev, N. N. Z SSSR [Grasses of the Soviet Union] 1–778 (Nauka, 1976).
- 16.Nobis M. Taxonomic revision of the Stipa lipskyi group (Poaceae: Stipa section Smirnovia) in the Pamir alai and Tian-Shan Mountains. Plant Syst. Evol. 2013;299(7):1307–1354. doi: 10.1007/s00606-013-0799-5. [DOI] [Google Scholar]
- 17.Nobis M, Gudkova PD. Taxonomic notes on feather grasses (Poaceae: Stipa) from eastern Kazakhstan with typification of seven names and one new combination. Phytotaxa. 2016;245(1):31–42. doi: 10.11646/phytotaxa.245.1.3. [DOI] [Google Scholar]
- 18.Hsiao C, Jacobs SWL, Chatterton NJ, Asay KH. A molecular phylogeny of the grass family (Poaceae) based on the sequences of nuclear ribosomal DNA (ITS) Aust. Syst. Bot. 1999;11(6):667–688. doi: 10.1071/SB97012. [DOI] [Google Scholar]
- 19.Hsiao C, Chatterton NJ, Asay KH, Jensen KB. Molecular phylogeny of the Pooideae (Poaceae) based on nuclear rDNA (ITS) sequences. Theor. Appl. Genet. 1995;90:389–398. doi: 10.1007/BF00221981. [DOI] [PubMed] [Google Scholar]
- 20.Grebenstein B, Röser M, Sauer W, Hemleben V. Molecular phylogenetic relationships in Aveneae (Poaceae) species and other grasses as inferred from ITS1 and ITS2 rDNA sequences. Plant Syst. Evol. 1998;213:233–250. doi: 10.1007/BF00985203. [DOI] [Google Scholar]
- 21.Hodkinson TR, et al. The use of DNA sequencing (ITS and trnL-F), AFLP, and fluorescent in situ hybridization to study allopolyploid Miscanthus (Poaceae) Am. J. Bot. 2002;89:279–286. doi: 10.3732/ajb.89.2.279. [DOI] [PubMed] [Google Scholar]
- 22.Ainouche ML, Bayer RJ. On the origins of the tetraploid Bromus species (section Bromus, Poaceae): insights from internal transcribed spacer sequences of nuclear ribosomal DNA. Genome. 1997;40(5):730–743. doi: 10.1139/g97-796. [DOI] [PubMed] [Google Scholar]
- 23.Hodkinson TR, Chase MW, Lledó DM, Salamin N, Renvoize SA. Phylogenetics of Miscanthus, Saccharum and related genera (Saccharinae, Andropogoneae, Poaceae) based on DNA sequences from ITS nuclear ribosomal DNA and plastid trnL intron and trnL-F intergenic spacers. J. Plant Res. 2002;115(5):381–392. doi: 10.1007/s10265-002-0049-3. [DOI] [PubMed] [Google Scholar]
- 24.Blattner FR. Phylogenetic analysis of Hordeum (Poaceae) as inferred by nuclear rDNA ITS sequences. Mol. Phyl. Evol. 2004;33(2):289–299. doi: 10.1016/j.ympev.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Gao G, et al. Phylogenetic relationships among Elymus and related diploid genera (Triticeae: Poaceae) based on nuclear rDNA ITS sequences. Biologia. 2015;70(2):183–189. doi: 10.1515/biolog-2015-0019. [DOI] [Google Scholar]
- 26.Kim S, et al. Phylogenetic relationships among species of Setaria (Paniceae; Panicoideae; Poaceae) in Korea: insights from nuclear (ITS and kn1) and chloroplast DNA sequence data. Plant Syst. Evol. 2015;301(2):725–736. doi: 10.1007/s00606-014-1111-z. [DOI] [Google Scholar]
- 27.Gaut BS, Tredway LP, Kubik C, Gaut RL, Meyer W. Phylogenetic relationships and genetic diversity among members of the Festuca-Lolium complex (Poaceae) based on ITS sequence data. Plant Syst. Evol. 2000;224:33–53. doi: 10.1007/BF00985265. [DOI] [Google Scholar]
- 28.Sclovich SE, Giussani LM, Cialdella AM, Sede SM. Phylogenetic analysis of Jarava (Poaceae, Pooideae, Stipeae) and related genera: testing the value of the awn indumentum in the circumscription of Jarava. Plant. Syst. Evol. 2015;301:1625–1641. doi: 10.1007/s00606-014-1175-9. [DOI] [Google Scholar]
- 29.Markos S, Baldwin BG. Structure, molecular evolution, and phylogenetic utility of the 5′ region of the external transcribed spacer of 18S-26S rDNA in Lessingia (Compositae, Astereae) Mol. Phyl. Evol. 2002;23(2):214–228. doi: 10.1016/S1055-7903(02)00004-0. [DOI] [PubMed] [Google Scholar]
- 30.Sano Y, Sano R. Variation of the intergenic spacer region of ribosomal DNA in cultivated and wild rice species. Genome. 1990;33(2):209–218. [Google Scholar]
- 31.Fukunaga K, Ichitani K, Kawase M. Phylogenetic analysis of the rDNA intergenic spacer subrepeats and its implication for the domestication history of foxtail millet, Setaria italica. Theor. Appl. Genet. 2006;113(2):261–269. doi: 10.1007/s00122-006-0291-5. [DOI] [PubMed] [Google Scholar]
- 32.Yang K, et al. Diversity and inheritance of intergenic spacer sequences of 45S ribosomal DNA among accessions of Brassica oleracea L. var. capitata. Int. J. Mol. Sci. 2015;16(12):28783–28799. doi: 10.3390/ijms161226125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend JP. Profiling phylogenetic informativeness. Syst. Biol. 2007;56:222–231. doi: 10.1080/10635150701311362. [DOI] [PubMed] [Google Scholar]
- 34.Townsend JP, Leuenberger C. Taxon sampling and the optimal rates of evolution for phylogenetic inference. Syst. Biol. 2011;60:358–365. doi: 10.1093/sysbio/syq097. [DOI] [PubMed] [Google Scholar]
- 35.McMullen MD, Hunter B, Phillips RL, Rubenstein I. The structure of the maize ribosomal DNA spacer region. Nucleic Acids Res. 1986;14(12):4953–4968. doi: 10.1093/nar/14.12.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang KD, Fang SA, Chang FC, Chung MC. Chromosomal conservation and sequence diversity of ribosomal RNA genes of two distant Oryza species. Genomics. 2010;96(3):181–190. doi: 10.1016/j.ygeno.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 37.Borowska-Zuchowska, N., Kwaśniewski, M. & Hasterok, R. Cytomolecular analysis of ribosomal DNA evolution in a natural allotetraploid Brachypodium hybridum and its putative ancestors - dissecting complex repetitive structure of intergenic spacers. Fron. Plant Sci. 7, doi:10.3389/fpls.2016.01499 (2016). [DOI] [PMC free article] [PubMed]
- 38.Cordesse F, Grellet F, Reddy AS, Delseny M. Genome specificity of rDNA spacer fragments from Oryza sativa L. Theor. Appl. Genet. 1992;83:864–870. doi: 10.1007/BF00226709. [DOI] [PubMed] [Google Scholar]
- 39.Castiglione MR, Gelati MT, Cremonini R, Frediani M. The intergenic spacer region of the rDNA in Haplopappus gracilis (Nutt.) Gray. Protoplasma. 2013;250:683–689. doi: 10.1007/s00709-012-0441-3. [DOI] [PubMed] [Google Scholar]
- 40.Parvaresh M, Talebi M, Sayed-Tabatabaei BE. Molecular characterization of ribosomal DNA intergenic spacer (IGS) region in pomegranate (Punica granatum L.) Plant Syst. Evol. 2014;300(5):899–908. doi: 10.1007/s00606-013-0928-1. [DOI] [Google Scholar]
- 41.Doelling JH, Gaudino RJ, Pikaard CS. Functional analysis of Arabidopsis thaliana rRNA gene and spacer promoters in vivo and by transient expression. Proc. Nat. Acad. Sci. 1993;90(16):7528–7532. doi: 10.1073/pnas.90.16.7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maggini F, Gelati MT, Spolverini M, Frediani M. The intergenic spacer region of the rDNA in Olea europaea L. Tree Genet. Genomes. 2008;4(2):293–298. doi: 10.1007/s11295-007-0109-x. [DOI] [Google Scholar]
- 43.Inácio V, Rocheta M, Morais-Cecílio L. Molecular organization of the 25S–18S rDNA IGS of Fagus sylvatica and Quercus suber: A comparative analysis. PloS one. 2014;9(6):e98678. doi: 10.1371/journal.pone.0098678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flavell, R. B., O’Dell, M. & Thompson, W. F. Cytosine methylation of ribosomal RNA genes and nucleolus organizer activity in wheat in Kew Chromosome Conference II (ed. Brandham, P. E. & Bennett, M.D.) 11–17 (George Allen and Unwin, 1983).
- 45.Kramerov DA, Vassetzky NS. Short retroposons in eukaryotic genomes. Int. Rev. Cytol. 2005;247:165–221. doi: 10.1016/S0074-7696(05)47004-7. [DOI] [PubMed] [Google Scholar]
- 46.Rogers SO, Bendich AJ. Ribosomal RNA genes in plants: variability in copy number and in the intergenic spacer. Plant Mol. Biol. 1987;9(5):509–520. doi: 10.1007/BF00015882. [DOI] [PubMed] [Google Scholar]
- 47.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 48.Grummt I, Pikaard CS. Epigenetic silencing of RNA polymerase I transcription. Nat. Rev. Mol. Cell. Biol. 2003;4(8):641–649. doi: 10.1038/nrm1171. [DOI] [PubMed] [Google Scholar]
- 49.Geer LY, et al. The NCBI BioSystems database. Nucleic Acids Res. 2010;38:D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucl. Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers in Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology (ed. Altman, R.) 28–36 (CAAAI Press, 1994). [PubMed]
- 52.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14(1):48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 53.Christelová P, Valárik M, Hřibová E, De Langhe E, Doležel J. A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol. Biol. 2011;11(1):103. doi: 10.1186/1471-2148-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.López-Giráldez F, Townsend JP. PhyDesign: an online application for profiling phylogenetic informativeness. BMC Evol. Biol. 2011;11(1):152. doi: 10.1186/1471-2148-11-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Britton T, Anderson CL, Jacquet D, Lundqvist S, Bremer K. Estimating divergence times in large phylogenetic trees. Syst. Biol. 2007;56:741–752. doi: 10.1080/10635150701613783. [DOI] [PubMed] [Google Scholar]
- 57.Pond SLK, Muse SV. HyPhy: Hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.