

ABSTRACT
The recent approval of oncolytic virus for therapy of melanoma patients has increased the need for precise evaluation of the mechanisms by which oncolytic viruses affect tumor growth. Here we show that the human NK cell-activating receptor NKp46 and the orthologous mouse protein NCR1 recognize the reovirus sigma1 protein in a sialic-acid-dependent manner. We identify sites of NKp46/NCR1 binding to sigma1 and show that sigma1 binding by NKp46/NCR1 leads to NK cell activation in vitro. Finally, we demonstrate that NCR1 activation is essential for reovirus-based therapy in vivo. Collectively, we have identified sigma1 as a novel ligand for NKp46/NCR1 and demonstrated that NKp46/NCR1 is needed both for clearance of reovirus infection and for reovirus-based tumor therapy.
IMPORTANCE Reovirus infects much of the population during childhood, causing mild disease, and hence is considered to be efficiently controlled by the immune system. Reovirus also specifically infects tumor cells, leading to tumor death, and is currently being tested in human clinical trials for cancer therapy. The mechanisms by which our immune system controls reovirus infection and tumor killing are not well understood. We report here that natural killer (NK) cells recognize a viral protein named sigma1 through the NK cell-activating receptor NKp46. Using several mouse tumor models, we demonstrate the importance of NK cells in protection from reovirus infection and in reovirus killing of tumors in vivo. Collectively, we identify a new ligand for the NKp46 receptor and provide evidence for the importance of NKp46 in the control of reovirus infections and in reovirus-based cancer therapy.
KEYWORDS: NCR1, NK, NKp46, reovirus
INTRODUCTION
Reovirus is a nonenveloped double-stranded RNA virus. In humans, reovirus infection is usually associated with mild respiratory or gastrointestinal disease (1–3). To establish infection, the virus must first attach to its target cell. This is mediated by the binding of the reovirus capsid protein, sigma1, to different cellular targets, such as the tight junction protein JAM-A and sialic acids, on the target cell surface (4–11). Reovirus is an oncolytic virus that has been tested in experimental animal models and in human clinical trials for the treatment of head and neck cancers, melanoma, lung cancer, ovarian cancer, and colorectal cancer (1, 7, 8, 12, 13). Reovirus was shown to affect tumor growth by direct killing of tumor cells and by activation of the immune system against tumor cells (12–18). Despite the relative success of the current reovirus-based therapy, attempts are being made to make this treatment even more efficient (19). For example, coadministration of reovirus and cyclophosphamide was used to improve tumor cell sensitivity to reovirus (20). In addition, genetically engineered reovirus with greater infectivity was used to increase reovirus-mediated oncolysis (21).
Improved reovirus-based therapy could also be potentially achieved by boosting the antitumor immune response, especially by boosting NK cell activity, as it has been shown that reovirus can induce NK cell activation against tumor cells (14, 22, 23). However, the molecular mechanisms by which reovirus activates NK cell cytotoxicity are not fully understood (14, 22, 23).
The human NKp46 receptor and the orthologous mouse protein NCR1 are members of the natural cytotoxicity receptor (NCR) family. They are expressed on all NK cells and on some innate lymphoid cells and are essential for NK cell-mediated elimination of virus-infected cells and tumor cells (24–30). The identity of the membrane-bound tumor/cellular ligand of NKp46/NCR1 is still obscure; however, several viral ligands that trigger NKp46/NCR1 have been identified (28, 31–33). We have previously demonstrated that NK cells can recognize and eliminate influenza virus-infected cells through NKp46/NCR1 receptor binding to the viral hemagglutinin (HA) protein (28). We also established that the binding of NKp46 to viral HA is mediated by α2,6-linked sialic acid residues expressed on specific threonine residues of NKp46 and NCR1 (24, 34, 35).
The importance of NCR1 in viral recognition in vivo was demonstrated by the generation of Ncr1 knockout (KO) mice in which exons 5 to 7, which encode the cytoplasmic tail and transmembrane portion of NCR1, were replaced with green fluorescent protein (GFP) (Ncr1gfp/gfp mice) (27). In these mice, NK cells are labeled with GFP and NCR1 is absent. The NK cells in the heterozygous (Het) mice (Ncr1+/gfp mice) function normally and express GFP, while Ncr1gfp/gfp mice are significantly more susceptible to viral infection (27).
Here, we identify sigma1 as a novel ligand for NKp46/NCR1 and show that NKp46/NCR1 is important for the control of reovirus infection and for successful reovirus-based therapy of tumors.
RESULTS
The NKp46 receptor recognizes reovirus.
NKp46 is a receptor particularly important in the recognition of viruses (24, 32, 33). To test if NKp46 recognizes reovirus, we initially incubated Vero cells with reovirus type 3 (Dearing) and determined that the virus adheres to the cells by staining them with an anti-sigma1 monoclonal antibody (MAb) (Fig. 1A). Next, we prepared fusion proteins containing the extracellular portion of NKp46 fused to human IgG1 and stained Vero cells in the presence or absence of reovirus. NKp46-Ig recognized uninfected Vero cells (Fig. 1B), suggesting that Vero cells express an unknown ligand for NKp46/NCR1. Importantly, following incubation with reovirus, increased NKp46-Ig binding was seen (Fig. 1B). The binding was specific, since little or no increase in the binding of D1-Ig (prepared in a manner similar to that used for NKp46-Ig) was noticed (Fig. 1B, left histogram; the binding of all fusion proteins is summarized in panel C). D1-Ig is the membrane-distal Ig-like domain of NKp46 that is not involved in the binding of NKp46 to its ligands (24). The integrity of the fusion protein was analyzed by Coomassie-stained gels under nonreducing conditions. As expected, NKp46-Ig appears as a single band slightly larger than 250 kDa (Fig. 1D).
FIG 1.

NKp46 is activated by reovirus. (A) Vero cells were incubated with reovirus for 14 h and stained with anti-sigma1 antibody (open gray histogram). The filled gray histogram depicts the background staining of Vero cells with the secondary MAb in the absence of reovirus. The background staining of Vero cells in the presence of reovirus was similar and is not shown. The empty black histogram depicts the staining of uninfected Vero cells with anti-sigma1 antibody. (B) FACS staining of Vero cells incubated for 14 h in the presence or absence of reovirus. Staining was performed with D1-Ig and NKp46-Ig, as indicated on the x axis. The filled gray histograms depict the background staining of Vero cells with the secondary MAb in the absence of reovirus. The background staining of Vero cells in the presence of reovirus was similar and is not shown. The empty black histograms depict the staining of uninfected Vero cells with the fusion proteins indicated. The empty gray histograms depict the staining of Vero cells preincubated with reovirus and stained with the fusion proteins indicated. Shown are the results of one representative experiment out of three performed. (C) The median fluorescence intensity (MFI) of anti-sigma1 antibody, D1-Ig, and NKp46-Ig staining of uninfected and reovirus-infected cells in three different experiments. Each error bar represents the standard deviation (SD). Statistically significant differences are indicated. *, P < 0.05; ns, not significant. (D) Coomassie staining of the NKp46-Ig fusion protein used in panel B after gel electrophoresis under nonreducing conditions. The image was cropped and the background was adjusted for better clarity. (E) FACS staining of BW cells expressing NKp30-zeta (BW NKp30) and NKp46-zeta (BW NKp46). The empty black histograms depict staining with the MAb indicated, and the filled gray histograms depict background staining with the secondary MAb only. (F) The various BW cells expressing the chimeric proteins shown in panel E were cocultured with Vero cells preincubated in the presence or absence of reovirus for 14 h. IL-2 secretion was determined by ELISA. Relative IL-2 secretion, determined as described in Materials and Methods, is shown. Mean values and SD of three independent experiments are shown. Statistically significant differences are indicated. *, P < 0.05; ns, not significant. (G) Vero cells were incubated in the absence (designated uninfected) or presence of reovirus for 14 h and then cocultured with human NK cells. The human NK cells were preblocked with anti-NKp46 antibodies (designated anti-NKp46) or without antibodies (designated reovirus). Killing was performed for 5 h. The effector-to-target cell ratios ranged from 2:1 to 10:1. The mean values and SD of three independent experiments are shown. Statistically significant differences are indicated. **, P < 0.01. In all flow cytometry experiments, antibodies and fusion proteins were incubated with target cells on ice. Fusion proteins were incubated for 2 h, and antibodies were incubated for 1 h.
NKp46 binding to reovirus leads to increased NKp46-mediated cytotoxicity.
To test whether the NKp46 interaction with reovirus is functional, we initially used a cell-based reporter system. We generated chimeric proteins composed of the extracellular portion of NKp46 or NKp30 fused to the mouse zeta chain and expressed each one separately in mouse BW cells (BW NKp46 and BW NKp30, respectively) (Fig. 1E). NKp30 is a member of the NCR family, to which NKp46 also belongs, and was used as a negative control. In this cell-based reporter assay, BW cells secrete interleukin-2 (IL-2) if the chimeric protein is bound and triggered by a ligand (36). We incubated the parental BW cells and the various BW transfectants with reovirus-infected Vero cells or uninfected Vero cells. When BW NKp46 cells were cocultured with Vero cells infected with reovirus, increased IL-2 secretion was seen (Fig. 1F). No significant increase in IL-2 secretion was seen when BW NKp30 cells were cocultured with Vero cells infected with reovirus (Fig. 1F). To test whether NKp46 binding to reovirus is functional and leads to increased NK cell cytotoxicity, we assayed NK cytotoxicity against 35S-methionine-labeled uninfected and infected Vero cells. Primary bulk-activated human NK cell cultures were added to the cells, and radioactivity levels in the medium were measured. A significant increase in the NK-mediated killing of reovirus-infected cells was noticed (Fig. 1G). This increased killing was mediated by NKp46, as blocking with anti-NKp46 polyclonal antibodies reduced the killing back to basal levels (Fig. 1G).
NCR1, the murine NKp46 orthologue, binds reovirus and induces NK cell cytotoxicity.
Next, we prepared a fusion protein made of the extracellular portion of the mouse ortholog of NKp46, NCR1, fused to human IgG1 and used it to stain Vero cells in the presence or absence of reovirus. Like NKp46, NCR1 also recognizes an unknown ligand expressed by Vero cells, and upon incubation with reovirus, increased binding of NCR1-Ig was seen (Fig. 2A; a summary of three experiments is shown in panel B). The integrity of the fusion protein used for staining was analyzed by Coomassie-stained gels under nonreducing conditions. As expected, NCR1-Ig presented a band similar in size to its human analog NKp46-Ig (compare Fig. 2C and 1D). Next, we fused the extracellular portion of NCR1 to mouse zeta chain and expressed this construct in mouse BW cells (BW NCR1, Fig. 2D). The parental BW and BW NCR1 cells were incubated with uninfected or reovirus-infected Vero cells. IL-2 secretion was observed when BW NCR1 cells were cocultured with Vero cells, even in the absence of reovirus, because of the presence of an unknown NCR1 ligand on these cells (Fig. 2A). Importantly, in the presence of reovirus, a significant increase in IL-2 secretion was observed (Fig. 2E).
FIG 2.

NCR1 is activated by reovirus. (A) FACS staining of Vero cells incubated for 14 h in the presence or absence of reovirus. The filled gray histogram depicts the background staining of Vero cells with the secondary MAb in the absence of reovirus. The background staining of Vero cells in the presence of reovirus was similar and is not shown. The background used here is identical to the background used in Fig. 1B, as the experiments were performed at the same time. The empty black histogram depicts the staining of Vero cells with NCR1-Ig. The empty gray histogram depicts the staining of Vero cells preincubated with reovirus and stained with NCR1-Ig. Shown are the results of one representative experiment out of three performed. (B) The median fluorescence intensity (MFI) of NCR1-Ig staining of uninfected and infected cells obtained from three different experiments. Each error bar represents the standard deviation (SD). Statistically significant differences are indicated. *, P < 0.05. (C) Coomassie staining of the NCR-1-Ig used in in panel A after gel electrophoresis under nonreducing conditions. The image was cropped and the background was adjusted for better clarity. (D) FACS staining of BW cells expressing NCR1-zeta (BW NCR1). The empty black histogram depicts staining with the anti-NCR1 MAb. The filled gray histogram depicts staining with the secondary MAb only. (E) BW cells or BW cells expressing NCR1-zeta (BW NCR1) were cocultured for 48 h with Vero cells preincubated for 14 h in the presence (reovirus) or absence (uninfected) of reovirus. IL-2 secretion was determined 24 h later by ELISA. Relative IL-2 secretion, determined as described in Materials and Methods, is shown. The mean values and SD of three independent experiments are shown. Statistically significant differences are indicated. *, P < 0.05. (F) Vero cells incubated with reovirus for 14 h were tested in a killing assay against mouse Ncr1+/gfp (Ncr1 Het) and Ncr1gfp/gfp (Ncr1 KO) NK cells. The effector-to-target cell ratio was 1:1. The mean values and SD of three independent experiments are shown. Statistically significant differences are indicated. *, P < 0.05; **, P < 0.01. In all flow cytometry experiments, antibodies and fusion proteins were incubated with target cells on ice. Fusion proteins were incubated for 2 h, and antibodies were incubated for 1 h.
To test whether NCR1 activation by reovirus can induce mouse NK cell cytotoxicity, we isolated mouse NK cells from immunocompetent C57BL/6 Ncr1+/gfp (Ncr1 Het) mice (the NK cells in these mice are fully functional but express GFP, enabling their efficient isolation [27, 34, 37]) and from C57BL/6 Ncr1gfp/gfp (Ncr1 KO) mice. We then incubated the NK cells with 35S-methionine-labeled uninfected and reovirus-infected Vero cells. When NK cells obtained from immunocompetent C57BL/6 Ncr1+/gfp mice were used, increased NK-mediated killing was seen (Fig. 2F). In contrast, no increase in NK-mediated killing was observed with NCR1-deficient C57BL/6 Ncr1gfp/gfp NK cells (Fig. 2F).
NKp46 and NCR1 directly bind sigma1 in a sialic-acid-dependent manner.
Since NKp46 and NCR1 were previously shown to interact with influenza virus HA via their sialylated residues (5, 24, 26, 38), we hypothesize that both proteins might recognize reovirus by interacting with the reovirus sialic-acid-binding protein sigma1. This is because both influenza virus HA and reovirus sigma1 are expressed as trimers on virions and both are sialic-acid-binding proteins. We have previously shown that HA-Ig fusion protein dimers have binding properties similar to those of trimeric HA (26, 38). Therefore, to test whether sigma1 is indeed recognized by the NK cell receptors, we cloned the glycan-binding portion of the sigma1 protein and fused it to the Fc domain of human IgG1. The purity of the sigma1-Ig fusion protein was validated by using nonreducing SDS-PAGE (Fig. 3A). Two protein bands are shown, a 150-kDa band corresponding to the sigma1-Ig dimer (most likely due to dimerization through the Ig domain) and a second band slightly larger than 250 kDa corresponding to a higher-level complex. These results suggest that similarly to the influenza virus HA-Ig fusion protein, the sigma1-Ig fusion protein is present as either dimers or higher-order complexes and is likely to still possess the same binding capabilities as the native sigma1 protein found on reovirus virions (26, 38). Next, we tested whether sigma1-Ig is directly recognized by NKp46 and NCR1. For this, D1-Ig, NKp46-Ig, or NCR1-Ig was bound to enzyme-linked immunosorbent assay (ELISA) plates and tested for direct binding of sigma1-Ig by ELISA. Direct sigma1-Ig binding to NKp46-Ig and NCR1-Ig was seen, while no detectable binding to D1-Ig was observed (Fig. 3B).
FIG 3.

Sialylated residues of NKp46 and NCR1 are essential for reovirus recognition. (A) Coomassie staining of the fusion proteins used in panels C to H after gel electrophoresis under nonreducing conditions. The background was adjusted for better clarity. (B) Binding of biotinylated sigma1-Ig to D1-Ig, NKp46-Ig, and NCR1-Ig. Binding was detected by ELISA. The mean values and SD of three independent experiments are shown. Statistically significant differences are indicated. *, P < 0.05; **, P < 0.01. (C) FACS staining of Vero cells in the presence (empty gray histograms) or absence (empty black histograms) of reovirus. The cells were incubated with reovirus for 14 h, and then staining was performed with NKp46-Ig (left histogram) or T225A NKp46-Ig (right histogram). The filled gray histograms depict the background staining of Vero cells with the secondary MAb only. The background staining of Vero cells in the presence or absence of reovirus was similar and is not shown. Shown are the results of one representative experiment out of three performed. (D) FACS staining of Vero cells in the presence (empty gray histograms) or absence (empty black histograms) of reovirus. Cells were incubated with reovirus for 14 h, and then staining was performed with NCR1-Ig (left histogram) or T222A T225A NCR-Ig (right histogram). The empty black histograms depict staining of Vero cells with the fusion proteins indicated. The filled gray histograms depict the background staining of Vero cells with the secondary MAb only. The background staining of Vero cells in the presence or absence of reovirus was similar and is not shown. Shown are the results of one representative experiment out of three performed. (E) Median fluorescence intensity (MFI) of the results shown in panels C and D. Averages and SD are derived from three independent repeats. ns, not significant. (F to G) SPR sensograms representing binding of sigma1-Ig to NKp46-Ig (F) and T225A NKp46-Ig (G) fusion proteins immobilized on a CM5 sensor chip via amine coupling. The sigma1-Ig analyte was injected in increasing concentrations of 10.5, 21.1, 42.2, 84.4, and 169 nM. R.U., response units. (H) Summary of SPR analysis. (I) Vero cells were incubated in the absence (designated uninfected) or presence of reovirus for 14 h and then cocultured with human NK cells for 5 h. The infected cells were preincubated with SLL or lactose for 30 min at 37°C after the addition of reovirus but before the addition of NK cells. Presented are averages of four independent experiments. Error bars represent the standard error of the mean. Statistically significant differences are indicated. *, P < 0.05. (J) Plates coated with UV-inactivated reovirus particles or RPMI medium were incubated with either lactose (black columns) or SLL (white columns) for 30 min at 37°C and then cocultured with BW cells expressing NKp46-zeta (BW Np46) or NCR1-zeta (BW NCR1). IL-2 secretion was determined 48 h later by ELISA. Relative IL-2 secretion, determined as described in Materials and Methods, is shown. Shown are the average and standard error of the mean of three independent experiments. Statistically significant differences are indicated. *, P < 0.05. In all flow cytometry experiments, antibodies and fusion proteins were incubated with target cells on ice. Fusion proteins were incubated for 2 h, and antibodies were incubated for 1 h.
The reovirus sigma1 protein recognizes α2,6- and α2,3-linked sialic acid residues (5). These types of sialic acid linkages are present on NKp46/NCR1 (30). We have previously shown that the sialylated threonine residue located at position 225 (T225) of NKp46 is essential for its binding to influenza virus HA (24). To test whether T225 also facilitates sigma1 recognition, we mutated this residue, replaced it with alanine, and generated a fusion protein containing the mutation (T225A NKp46-Ig). The purity of the fusion protein was determined (Fig. 3A), and remarkably, the mutated T225A NKp46-Ig fusion protein failed to bind reovirus-infected cells (Fig. 3C).
We recently identified two novel O-glycosylated residues located at positions 222 and 225 of NCR1 that are essential for influenza virus HA recognition (35). To test whether these two residues are essential for NCR1 recognition of sigma1, we generated a mutated fusion protein in which the two sialylated threonine residues were replaced with alanine (T222A T225A NCR1-Ig) and checked its purity by SDS-PAGE (Fig. 3A). The mutated NCR1-Ig fusion protein T222A T225A NCR1-Ig failed to bind reovirus (Fig. 3D). A summary of all of our experiments with mutated NKp46 and NCR1 fusion proteins is presented in Fig. 3E.
We also tested the binding of NKp46-Ig, T225A NKp46-Ig, and D1-Ig to sigma1-Ig by using surface plasmon resonance (SPR) analysis. Consistent with the results presented in Fig. 3B to E, NKp46-Ig bound sigma1-Ig (Fig. 3F) and this binding was reduced when the T225A NKp46-Ig protein was used (Fig. 3G). No binding of D1-Ig to sigma1-Ig was observed. The NKp46-Ig binding to sigma1 was of high affinity (KD [equilibrium dissociation constant] = 8.48 × 10−10 M), and approximately 10-fold lower affinity was seen with T225A NKp46-Ig (KD = 9.24 × 10−9 M) (Fig. 3H).
To test directly if the sialic-acid-binding properties of sigma1 are essential for its NKp46/NCR1 recognition, we used 3′-sialyllactose (SLL), a soluble sugar containing a sialic acid residue attached to a lactose backbone, to block this interaction. This carbohydrate, found in the milk of several mammals, has been shown to interfere with the interaction between reovirus sigma1 and its ligands and also to interfere with the hemagglutination ability of sigma1. These effects were shown to be dose dependent and affect only sialic-acid-binding strains of reovirus (10, 39–41).
We repeated the NK cytotoxicity experiments in the presence of SLL and in the presence of the lactose backbone only (used as a control). While lactose did not have any observable effect on the killing of reovirus-infected cells, the presence of SLL significantly inhibited NK cell cytotoxicity (Fig. 3I). In addition, we coated plates with UV-inactivated reovirus particles, incubated them with either SLL or lactose, and then added the BW reporter cells expressing either NKp46 or NCR1 fused to the mouse zeta chain. In the presence of lactose, cell-free reovirus virions were able to activate the NKp46 and NCR1 reporter cells, while this activation was significantly reduced in the presence of SLL (Fig. 3J).
NCR1 is essential for the control of reovirus infection in vivo.
To test whether NCR1 controls reovirus infection in vivo, we infected C57BL/6 Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice with 2.5 × 106 PFU of reovirus each. Lungs were harvested 5 days postinfection, and viral loads were determined (Fig. 4A). Significantly greater viral loads were observed in the lungs of Ncr1 KO mice than in those of WT mice (Fig. 4B). C57BL/6 mice were relatively resistant to reovirus infection; the viral loads in their lungs were quite light (Fig. 4B), and the mice showed no symptoms of illness such as reduced weight (data not shown).
FIG 4.
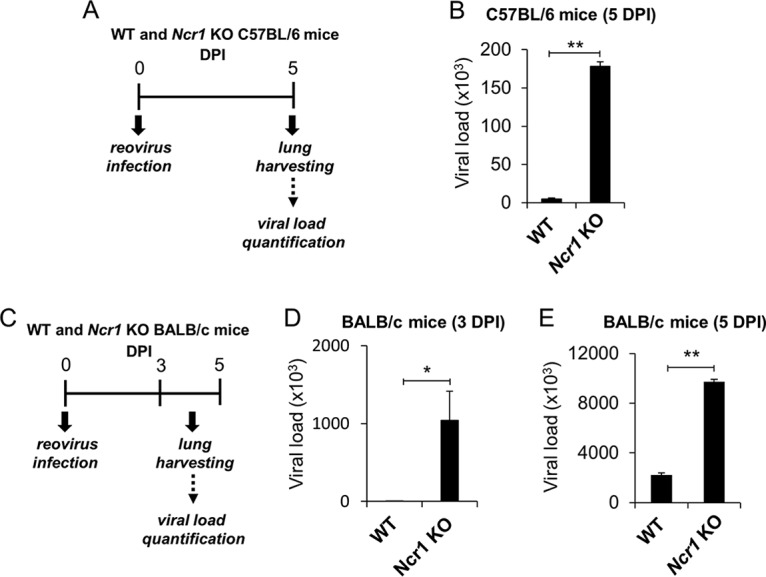
NCR1 control reovirus infection in vivo. (A) Schematic representation of the experimental procedure used. Four- to 6-week-old C57BL/6 and BALB/c Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice were each infected intranasally with 2.5 × 106 PFU of reovirus type 3 (Dearing). Lungs were harvested 5 days postinfection, and viral loads in the lungs were determined by qRT-PCR. All results were normalized to mouse GAPDH and ActB expression. (B) Reovirus loads in the lungs of C57BL/6 Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice (five per group). (C) Schematic representation of the experimental procedure used. Four- to 6-week-old BALB/c Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice were each infected intranasally with 2.5 × 106 PFU of reovirus type 3 (Dearing). Lungs were harvested 3 and 5 days postinfection, and viral loads in the lungs were determined by qRT-PCR. All results were normalized to mouse GAPDH and ActB expression. (D, E) Viral loads were determined 3 (D) and 5 (E) days postinfection (DPI). Shown are mean values derived from a pool of five mice per group. The y axes depict viral load measurements. Mean values and standard errors (bars) are from triplicate experiments. Statistically significant differences are indicated. *, P < 0.05; **, P < 0.01.
BALB/c mice are usually more sensitive to viral infections (42, 43). Therefore, we also infected BALB/c Ncr1+/+ (WT) mice and Ncr1gfp/gfp (Ncr1 KO) mice with reovirus at 2.5 × 106 PFU/mouse. Lungs were harvested 3 and 5 days postinfection, and viral loads were determined (Fig. 4C). Although we still did not observe any symptoms of illness, the BALB/c mice were indeed more sensitive to reovirus infection, as greater viral loads were observed in their lungs than in those of C57BL/6 mice (compare Fig. 4B and E). Significantly greater reovirus loads were detected in the lungs of Ncr1 KO mice than in those of WT mice at 3 (Fig. 4D) and 5 (Fig. 4E) days postinfection. Collectively, we demonstrated that NCR1 plays an important role in the control of reovirus infection in vivo.
NCR1 is essential for reovirus-based cancer therapy.
Reovirus is currently being used in phase III clinical trials for the treatment of various cancers (7, 8, 12, 44). To test whether NKp46/NCR1 is important for the antitumor activity of reovirus, we first injected increasing doses (104, 105, or 106) of the melanoma cell line B16F10.9 (B16) subcutaneously (s.c.) into C57BL/6 Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice and monitored tumor progression. In accordance with our previous reports (44), when 104 B16 cells were injected into WT and Ncr1 KO mice, the tumor volume was significantly greater in Ncr1 KO mice. However, when larger amounts of B16 cells were injected (105 and 106), no significant difference in tumor volume was seen (Fig. 5A). Consistent with these results, when 5 × 105 B16 cells were injected, no significant difference in survival between Ncr1 KO and WT mice was seen (Fig. 5B). Therefore, we concluded that to assess the effect of NKp46/NCR1 during the course of reovirus treatment, at least 105 B16 cells should be injected, because with that amount, the tumors are equally controlled by WT and Ncr1 KO mice. We injected 5 × 105 B16 cells into Ncr1 KO and WT mice and monitored tumor progression daily. Once tumors were visible, reovirus was injected directly into the tumors (Fig. 5C). Importantly, reovirus treatment significantly delayed tumor progression, leading to delayed death, but only in mice having functional NCR1 alleles (Fig. 5D).
FIG 5.

NCR1 plays an important role in reovirus-based treatment of B16 cells. (A) Tumor volumes of C57BL/6 Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice 20 days after s.c. injection of 104, 105, or 106 B16 cells. Statistically significant differences are indicated. *, P < 0.05; ns, not significant. (B) Percent survival of C57BL/6 Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice following the injection of 5 × 105 B16 cells. (C) Schematic representation of the experimental procedure used. Each reovirus injection consisted of 5 × 109 PFU/mouse. I.T., intratumor. (D) Percent survival of the mouse groups (five mice per group) indicated in two independent experiments. Mice were euthanized when tumors reached a size of >1 cm3.
To investigate the specific contribution of NCR1 to reovirus therapy of tumors independently of the oncolytic effect of the virus, we used a UV-inactivated reovirus. UV treatment completely inactivated the virus, as no visible plaques were seen when the UV-inactivated virus was used in a plaque assay (Fig. 6A). To test whether the UV-inactivated virus could still bind the cells and be recognized by NCR1, we incubated B16 cells with the UV-inactivated virus and, using flow cytometry, observed that the virus still adhered to the cells (Fig. 6B) and was recognized by NCR1-Ig (Fig. 6C).
FIG 6.

Specific evaluation of the role of NCR1 in a B16 melanoma model by reovirus-based treatment. (A) Viral titer quantification in reovirus stocks left untreated or inactivated by UV irradiation. The values on the y axis are the numbers of PFU per milliliter of viral stock. Presented are averages derived from two independent experiments. Each error bar represents the standard deviation (SD). (B to D) FACS staining of B16 cells incubated for 14 h in the presence (empty gray histograms) or absence (empty black histograms) of UV-inactivated reovirus. Staining was performed with anti-sigma1 antibody (B), NCR1-Ig (C), and NKG2D-Ig (D), as indicated on the x axes. The filled gray histograms depict the background staining of the parental Vero cells with the secondary MAb only. The background staining of Vero cells in the presence of UV-inactivated reovirus was similar and is not shown. Shown are the results of one representative experiment out of three performed. (E) Coomassie staining of NKG2D-Ig used in panel D after gel electrophoresis under nonreducing conditions. The image was cropped and the background was adjusted for better clarity. (F) Median fluorescence intensity (MFI) of the results presented in panels C and D. Presented are averages derived from three independent experiments. Each error bar represents the standard deviation (SD). Statistically significant differences are indicated. *, P < 0.05; ns, not significant. (G) B16 cells incubated with reovirus for 14 h were tested in a 5-h killing assay against Ncr1+/gfp (Ncr1 Het, “UV-inactivated reovirus”) and Ncr1gfp/gfp (Ncr1 KO) NK cells. The effector-to-target cell ratio was 1:1. The mean values and SD of three independent experiments are shown. Statistically significant differences are indicated. **, P < 0.01. (H) Percent survival of C57BL/6 Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice treated with UV-inactivated reovirus or left untreated. Each UV-inactivated reovirus injection consisted of 2.5 × 1010 PFU per mouse. The empty circles represent untreated WT mice. The filled circles represent UV-inactivated-reovirus-treated WT mice. The empty squares represent untreated KO mice. The filled squares represent UV-inactivated-reovirus-treated KO mice. Shown are the results of one representative experiment out of two performed. Statistically significant differences in survival between treated and untreated WT mice are indicated in the graph. In all flow cytometry experiments, antibodies and fusion proteins were incubated with target cells on ice. Fusion proteins were incubated for 2 h, and antibodies were incubated for 1 h.
In addition to NKp46/NCR1, NKG2D is the other main mouse NK cell-activating receptor. NKG2D recognizes so-called stress-induced ligands (45). To further demonstrate the specificity of NKp46/NCR1, we also stained UV-inactivated-reovirus-coated cells with a soluble murine NKG2D-Ig fusion protein. Incubation with UV-inactivated reovirus did not seem to upregulate the NKG2D ligands, as no increase in NKG2D-Ig staining was seen in the presence of UV-inactivated reovirus (Fig. 6D). The integrity and purity of this fusion protein were validated by gel electrophoresis (Fig. 6E), and three independent repeats of the flow cytometry analysis are quantified in Fig. 6F.
We also verified that UV-inactivated-virus-bound cells were better recognized by NK cells in vitro and that this recognition was NCR1 dependent by using NK cell cytotoxicity experiments with NK cells extracted from Ncr1+/gfp (Het) or Ncr1gfp/gfp (KO) mice. We used Het mice in these experiments because they are immunocompetent and serve as a better littermate control than WT mice. While the killing of B16 cells by Het NK cells was increased in the presence of the inactivated reovirus, Ncr1 KO NK cells showed no effect (Fig. 6G). Next, we injected 5 × 106 B16 cells s.c. into Ncr1 KO and WT mice. Once tumors were visible, the UV-inactivated reovirus was injected into the tumors (5 × 109 PFU per mouse per injection). Remarkably, UV-inactivated reovirus treatment significantly improved the survival of WT mice but not that of Ncr1 KO mice (Fig. 6H), indicating that the reovirus oncolytic effect is at least partially mediated by NCR1-dependent immune activity.
To demonstrate that the role of NCR1 in reovirus-based cancer therapy is not restricted to a particular tumor model, we used the Lewis lung carcinoma cell line D122. We first tested the binding of NKp46-Ig and NCR1-Ig and found that both fusion proteins recognized an unknown tumor ligand expressed by D122 cells (Fig. 7A). Following incubation with UV-inactivated reovirus, increased NKp46-Ig (Fig. 7A, quantified in panel B) and NCR1-Ig (Fig. 7C, quantified in panel D) binding was seen. We also validated that the UV-inactivated virus did not lead to upregulation of ligands for NKG2D, and similar to our observation regarding B16 melanoma cells, no change in the expression of murine NKG2D ligands was observed (Fig. 7E, quantified in panel F). We next injected 106 D122 cells into the abdomens of C57BL/6 Ncr1+/gfp (Ncr1 Het) and C57BL/6 Ncr1gfp/gfp (Ncr1 KO) mice. Once tumors were visible, UV-inactivated reovirus (5 × 109 PFU per mouse per injection) was directly injected into the tumors. In accordance with our previous report (44), when 106 D122 cells were injected, no significant difference in survival between the two mouse genotypes was seen (Fig. 7G). Importantly, UV-inactivated reovirus treatment significantly improved Ncr1 Het but not Ncr1 KO mouse survival (Fig. 7G). Taken together, these combined results highlight the pivotal role played by NKp46/NCR1 in reovirus-based cancer therapy.
FIG 7.

NCR1 is essential for reovirus treatment in a model of lung carcinoma. (A) FACS staining of D122 cells incubated for 14 h in the presence (empty gray histogram) or absence (empty black histogram) of reovirus. Staining was performed with NKp46-Ig (A, B), NCR1-Ig (C, D), and NKG2D-Ig (E, F), as indicated on the x axes. The filled gray histograms depict the background staining of D122 cells with the secondary MAb only in the absence of reovirus. The background staining of D122 cells in the presence of reovirus was similar and is not shown. Shown are the results of one representative experiment out of three (NKp46-Ig) or five (NCR1-Ig and NKG2D-Ig) performed. The median fluorescence intensity (MFI) of the different repeats of NKp46-Ig (B), NCR1-Ig (D), and NKG2D (F) staining is shown. Error bars represent the standard error of the mean. Statistically significant differences are indicated. *, P < 0.05; ns, not significant. (G) Percent survival of C57BL/6 Ncr1+/gfp (Ncr1 Het) and Ncr1gfp/gfp (Ncr1 KO) mice treated with UV-inactivated reovirus or left untreated. Each UV-inactivated reovirus injection consisted of 2.5 × 1010 PFU per mouse. Empty circles represent untreated Het mice. Filled circles represent UV-inactivated-reovirus-treated Het mice. Empty squares represent untreated KO mice. Statistically significant differences are indicated in the graph. Shown are the results of one representative experiment out of two performed. In all flow cytometry experiments, antibodies and fusion proteins were incubated with target cells on ice. Fusion proteins were incubated for 2 h, and antibodies were incubated for 1 h.
DISCUSSION
Reovirus infections are frequent, and most adults develop antireovirus antibodies (46). Reovirus infects the respiratory and gastrointestinal tracts, causing influenza-like symptoms and diarrhea. The mechanisms by which reovirus infections are controlled by immune cells are not fully understood. Here we demonstrate direct interaction between the NK cell-activating receptor NKp46/NCR1 and the sigma1 protein of reovirus and further show that this recognition is important in the elimination of reovirus-infected cells. Further in-depth analysis of NK cell activation by reovirus should be performed to analyze the impact of other factors that might contribute to the antiviral effect of NK cells. These might include secreted cytokines, improved NK cell migration to the tumor site upon reovirus infection, or induction of further unrecognized ligands for additional NK cell-activating receptors.
The reovirus sigma1 protein is a fiber-like molecule that can bind both the tight junction protein JAM-A and sialic acid residues on the cell surface and enables viral attachment to target cells (9–11, 47, 48). The ability to use sialic acids when binding target cells is conserved in different pathogens (5). Influenza virus, for example, uses its viral HA to bind sialic acids on the surface of respiratory epithelial cells (49). NK cells utilize HA's ability to recognize sialic acids and eliminate the influenza virus-infected cells via NKp46/NCR1 in a sialic-acid-dependent manner (24, 28, 38).
We show here that NKp46/NCR1 also directly recognizes the sigma1 protein of reovirus in a sialic-acid-dependent manner. We demonstrate that following the incubation of viruses with SLL, a soluble sugar containing sialic acid, recognition and activation by NKp46 and NCR1 are lost. We show that the sialylated residue T225 of NKp46 and T222 and T225 of NCR1 are essential for NKp46 and NCR1 binding to sigma1. However, additional factors might also be important for sigma1 recognition by NKp46, as mutating the T225 residue of NKp46 significantly reduced its binding to sigma1, as observed by SPR analysis, but did not completely abolish it.
Reovirus is currently being used in human clinical trials for the treatment of various tumors. It was suggested before that the antitumor effect of reoviruses is mediated both by the direct lysis of tumor cells and by activation of the immune system and that NK cells play an important role in the reovirus attack on tumors (12–18, 22, 23). However, the molecular mechanisms through which NK cells recognize reovirus-treated tumors are unknown. Importantly, we show that NKp46/NCR1 is an essential component of reovirus-based cancer treatment, as reovirus injection significantly improved mouse survival only in the presence of NCR1. By using UV-inactivated reovirus, we were able to efficiently distinguish between the oncolytic effect of the virus and the NKp46/NCR1-dependent effect. Strikingly, UV-inactivated reovirus was able to significantly prolong mouse survival in two tumor models in an NCR1-dependent manner. Together with previous evidence that UV-inactivated reovirus is able to induce an immune response in vitro and in vivo (23, 50), this indicates a central role for the immune system, and specifically NKp46/NCR1, during reovirus-based therapy. One study did not observe an NK cell effect when using inactivated virus (51). The observed discrepancies could be due to differences between the reagents or protocols used. In the study referred to (51), NK cells were incubated with virions and only then were target cells added. Thus, the free virions might block NKp46 (and perhaps additional activating receptors) and interfere with the interaction between NK cells and their target cells. Here, the target cells were initially incubated with reovirus and only then were NK cells added.
Our findings are of special interest in light of the recent success of immunotherapy and the approval of the first oncolytic virus therapy for melanoma (52, 53). It would be interesting to test if the recent advances in gene delivery into NK cells and in genome editing (54) could be used to enhance the NK cell antitumor activity we describe here. We propose that boosting NKp46 activity during reovirus treatment could improve the current reovirus-based therapy.
MATERIALS AND METHODS
Cells, viruses, and viral infection.
The cell lines used in this study were the Cercopithecus aethiops kidney epithelial Vero cell line, the murine thymoma BW cell line, the mouse fibroblast CLL-1 cell line (ATCC CCL-1.2), Lewis lung carcinoma line D122, and the B16F10.9 mouse melanoma cell line. Vero, D122, and B16F10.9 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich D5796), BW cells were grown in RPMI 1640 medium (Sigma-Aldrich R8758), and CLL-1 cells were grown in Eagle minimum essential medium (EMEM; Sigma-Aldrich M4655). All media were supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich F7524) and with 1% each penicillin-streptomycin, l-glutamine, sodium pyruvate, and EMEM nonessential amino acid solutions (Biological Industries 03-031-1B, 03-020-1B, 03-042-1B, and 01-340-1B, respectively).
Mammalian reovirus type 3 (Dearing) isolated from a child with diarrhea was purchased from ATCC (VR-824) and kept in its purified form diluted in EMEM (ATCC 30-2003TM) plus 2% FBS (ATCC 30-2020TM) at −80°C until used. The virus was propagated on cultures of CLL-1 cells in serum-free EMEM as previously described (55). Viral loads were determined by using plaque assays as previously described (56). Briefly, plaque assays were performed with confluent cultures of CCL-1 cells. After initial infection, cells were grown in a 1:1 solution of DMEM and 2% Bacto agar (diluted in 1× phosphate-buffered saline [PBS]) at 37°C for 4 days. Plaques were visualized with Neutral Red (Sigma-Aldrich N4638). Unless otherwise indicated, in all infections, cells were incubated with the virus at a multiplicity of infection (MOI) of 100 in a minimal volume of medium for 1 h at 37°C and 5% CO2, and then the medium was added. The infected cells were grown for 14 h at 37°C and 5% CO2 and analyzed.
For UV inactivation, reovirus was exposed to UV light (365 nm) for 45 min before the virus was incubated with the target cells at an MOI of 100. Unless otherwise indicated, viral infection or incubation of UV-inactivated virus with the relevant cell culture was performed at 37°C and 5% CO2 for 14 h.
Antibodies, fusion proteins, and reagents.
The MAbs used in this study included phycoerythrin (PE)-conjugated anti-human NKp46 (Beckman Coulter IM3711), PE-conjugated anti-mouse NCR1 (BioLegend 137604), PE-conjugated anti-NKp30 (BioLegend 325208), and anti-sigma1 (Merck Millipore MAB994) MAb. The secondary antibodies used were Alexa Fluor 647-conjugated goat anti-mouse IgG (Jackson ImmunoResearch 115-606-062) and allophycocyanin-conjugated donkey anti-human IgG (Jackson ImmunoResearch709-136-098) antibodies. For ELISAs, the antibodies used were anti-mouse IL-2 (BioLegend BLG-503702), biotinylated anti-mouse IL-2 (BioLegend BLG-503804), and streptavidin-horseradish peroxidase (HRP) (Jackson ImmunoResearch 016-030-084) antibodies. Blocking antibodies for NKp46 were generated by injecting 50 μg of NKp46-Ig s.c. into C57BL/6 mice. Seven days later, the mice were boosted with an additional 50 μg of NKp46-Ig. Blood serum was collected 30 days following the second injection. NKp46-Ig, T225A NKp46-Ig (in which the threonine at position 225 was replaced with an alanine), NCR1-Ig, T222A T225A NCR1-Ig (in which the threonines at positions 222 and 225 were replaced with alanines), and murine NKG2D-Ig were produced as previously described (24, 28, 31, 35, 57–59). Briefly, the extracellular fragment of the relevant genes from cDNA prepared from RNA extracted from human or murine NK cells was amplified by PCR. The DNA was cloned into a mammalian expression vector containing a mutated Fc portion of human IgG with impaired Fc receptor binding, expressed in human embryonic kidney 293T cells, and purified on a HiTrap protein G HP column (GE Healthcare Life Sciences 17-0405-01) as previously described (57). For cloning of sigma1-Ig, viral RNA was isolated from reovirus type 3 Dearing-infected Vero cells and used as a template to generate cDNA. The glycan-binding portion of the sigma1 protein (positions 101 to 455) (5) was cloned with forward primer GACTAGTAGTGTTACCCAGTTGGGTGCT and reverse primer TCCCGCGTAGTTTCACGTGAACCGGTG. The PCR product was digested with restriction enzymes SpeI and AgeI (New England BioLabs) and cloned downstream of a mutated Fc portion of human IgG with impaired Fc receptor binding (36). The integrity of all fusion proteins was analyzed by Coomassie-stained gels under nonreducing conditions.
Biotinylation of sigma1-Ig was performed with the EZ-link Sulfo-NHS-SS Biotin kit (Thermo Scientific 21331). For sigma1 sialic-acid-binding domain blocking experiments, SLL sodium salt (Santa Cruz Biotechnology sc-216626) and d-lactose (Santa Cruz Biotechnology sc-285369) were used, both at a final concentration of 10 mM (for NK cell cytotoxicity assays) or 50 mM (for BW assays). All of the fusion proteins used in this study contain a mutation in the Fc domain that impairs binding to Fc receptors (36), and D1-Ig was used to detect background staining.
Flow cytometry.
For fluorescence-activated cell sorter (FACS) staining, 0.5 to 5 μg/well fusion protein or 0.3ul/well anti-sigma1 antibody was used and incubated with the target cells (50 × 103 to 150 × 103/well) for 2 h (Ig fusion proteins) or 1 h (antibodies). Cells were then washed with FACS medium (1× PBS, 0.05% bovine serum albumin [BSA], 0.05% NaN3) and stained with a secondary antibody (0.25 to 0.75 μg/well) for 1 h. Cells were then washed twice with FACS medium and analyzed with a FACSCalibur machine (BD Biosciences) and the CellQuest software. All procedures were performed on ice. In all experiments, cells stained with the secondary antibody alone were used to measure background staining. In all flow cytometry experiments, the cells were not permeabilized.
ELISA.
For experiments measuring direct binding of sigma1-Ig and NKp46/NCR1-Ig, ELISA plates were coated with D1-Ig, NKp46-Ig, and NCR1-Ig at 0.5 μg/well for 14 h at 4°C. The ELISA plates were then incubated with 5% BSA diluted in PBS-Tween for 2 h at room temperature, washed, and then incubated on ice for 2 h with biotinylated sigma1-Ig at 5 to 10 ng/well. Direct binding was measured with streptavidin-HRP and 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (SouthernBiotech 0410-01).
For experiments measuring IL-2 secretion from BW cells, a similar protocol was used, but the ELISA plates were coated with an anti-mouse IL-2 antibody (0.2 μg/well). Detection of binding was performed by using biotinylated anti-mouse IL-2 antibodies (0.1 μg/well) and then detection with streptavidin-HRP and TMB substrate.
BW reporter assay.
BW transfected cells were prepared as previously described (28, 36). Fifty thousand BW or BW transfected cells were cocultured with 25,000 Vero cells (either incubated with reovirus or not incubated with reovirus) for 48 h at 37°C and 5% CO2 or in wells coated with cell-free virions (1 h at 37°C prior to the addition of BW cells; MOI of 0.2 to 20). Vero cells were irradiated just prior to their addition to the BW cells (3,000 rads), and cell-free reovirus virions were UV inactivated as described above. In the relevant experiments, SLL sodium salt or d-lactose was added to the wells containing cell-free virions for incubation (37°C, 30 min) before the addition of BW cells. Incubation was performed in flat-bottom 96-well plates. After 48 h of incubation, supernatants were collected and the level of IL-2 was quantified by ELISA and analyzed with the Gen5 software at a wavelength of 650 nm. Background IL-2 secretion from parental BW cells was normalized to 1, and normalized results are presented as increased IL-2 secretion observed in wells containing infected cells/cell-free viruses relative to the no-target wells from the same BW cell line.
Cytotoxicity assay.
The cytotoxic activity of human NK cells was assessed by 5-h 35S-methionine release assays as previously described (60). Briefly, NK cells were incubated with 35S-methionine-labeled cells for 5 h at 37°C and 5% CO2 at different effector-to-target cell ratios in U-bottom 96-well plates. The assay was terminated by centrifugation (1,600 rpm, 5 min, 4°C), and 100-μl volumes of supernatants containing 35S-methionine were taken for analysis. Reovirus infection of 35S-methionine-labeled cells or incubation of 35S-methionine-labeled cells with UV-inactivated reovirus was performed for 14 h prior to the addition of NK cells. For NKp46 blocking, NK cells were incubated with NKp46-blocking polyclonal antibodies on ice for 1 h. For sigma1 sialic-acid-binding domain blocking experiments, target cells were incubated for 30 min at 37°C and 5% CO2 with 10 to 50 mM SLL sodium salt or d-lactose prior to the addition of NK cells. NK cells were isolated from Ncr1gfp/+ (Ncr1 Het) and Ncr1gfp/gfp (Ncr1 KO) mice at 18 h following the intraperitoneal injection of 200 μg of poly(I·C) (P1530; Sigma). The effector-to-target cell ratio was 1:1.
SPR analysis.
SPR affinity measurements were performed with a Biacore T100 biosensor (GE Healthcare, Uppsala, Sweden). NKp46-Ig, T225A NKp46-Ig (in which the threonine at position 225 is replaced with an alanine), and D1-Ig were dissolved in 10 mM sodium acetate (pH 4.0) to 50 mM and coupled to a CM5 sensor chip (GE Healthcare, Uppsala, Sweden) by using the manufacturer-recommended amine coupling protocol to an immobilization level of 500 response units. Sigma1-Ig was dissolved in HBS-EP+ buffer (10 mM HEPES [pH 7.3], 150 mM NaCl, 3 mM EDTA [pH 8], 0.05% Tween 20) and injected as an analyte in a series of injections at concentrations of 10.5, 21.1, 42.2, 84.4, and 169 nM at a 50-μl/min flow rate with a contact time of 120 s. Dissociation was measured for 120 s. The surface was regenerated by a single pulse (30 s, 20 μl/min) of 4 M MgCl2 at the end of each cycle. Kinetic parameters and the affinity constant were derived by using the Kinetic 1:1 Binding model in BIAevaluation software v. 3.0.1 (GE Healthcare, Uppsala, Sweden.). Dilutions and pretreatment of the fusion proteins were performed on ice.
Mouse infection.
Four- to 6-week-old C57BL/6 and BALB/c Ncr1+/+ (WT) and Ncr1gfp/gfp (Ncr1 KO) mice (five per group) were infected intranasally with reovirus type 3 (Dearing). Lungs were harvested 3 and 5 days postinfection, and viral loads in the lungs were determined by quantitative RT-PCR (qRT-PCR) as previously described (26, 61). All results were normalized to mouse GAPDH and ActB expression. The following primer sequences were used: sigma1, GTATAGGGTTGTCGTCGGG (forward) and CCCCTCAACACGTAACCGAA (reverse); GAPDH, GAGTCAACGGATTTGGTCGT (forward) and GATCTCGCTCCTGGAAGATG (reverse), ActB, CCCTGAACCCTAAGGCCA (forward) and GGTACGACCAGAGGCATACAG (reverse). For melanoma model experiments, 7- to 9-week-old C57BL/6 WT and Ncr1 KO mice were injected s.c. in the flank region with 5 × 105 B16 cells in 200 μl of PBS. Tumor growth was evaluated daily. Once tumors were visible, mice were injected with 5 × 109 PFU of live reovirus or 2.5 × 1010 PFU of UV-inactivated virus diluted in 100 μl of PBS. Each mouse received three intratumoral injections at 2-day intervals. Mouse survival and tumor growth were monitored daily. Tumor size was evaluated with a digital caliper, and mice were sacrificed when the tumor size reached 1.0 by 1.0 cm in two perpendicular directions or >1.5 cm in any one of the directions (data not shown). For the lung cancer model, 106 D122 cells were injected s.c. into the abdomens of 10- to 14-week-old C57BL/6 and BALB/c Ncr1+/+ (WT) and Ncr1+/gfp (Ncr1 Het) mice (5 to 10 per group). All experiments were done in the specific-pathogen-free unit of the Hebrew University-Hadassah Medical School (Ein-Kerem, Jerusalem) in accordance with the guidelines of the Declaration of Helsinki and the local research ethics committee.
Statistical methods.
Student's t test was used to determine the statistical significance of differences. For survival curves, the log rank test was used to determine the statistical significance of differences.
ACKNOWLEDGMENTS
This study was supported by the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC (grant agreement number 320473-BacNK). Further support came from the Israel Science Foundation, the GIF Foundation, the Lewis Family Foundation, the ICRF professorship grant, the Helmholtz Israel grant, and the Rosetrees Trust (all to O.M.). Further support came from the I-CORE Program of the Planning and Budgeting Committee and the Israel Science Foundation and from the I-Core on Chromatin and RNA in Gene Regulation. O.M. is a Crown Professor of Molecular Immunology. The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Norman KL, Lee PW. 2000. Reovirus as a novel oncolytic agent. J Clin Invest 105:1035–1038. doi: 10.1172/JCI9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leary TP, Erker JC, Chalmers ML, Cruz AT, Wetzel JD, Desai SM, Mushahwar IK, Dermody TS. 2002. Detection of mammalian reovirus RNA by using reverse transcription-PCR: sequence diversity within the lambda3-encoding L1 gene. J Clin Microbiol 40:1368–1375. doi: 10.1128/JCM.40.4.1368-1375.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim M, Garant KA, zur Nieden NI, Alain T, Loken SD, Urbanski SJ, Forsyth PA, Rancourt DE, Lee PW, Johnston RN. 2011. Attenuated reovirus displays oncolysis with reduced host toxicity. Br J Cancer 104:290–299. doi: 10.1038/sj.bjc.6606053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danthi P, Holm GH, Stehle T, Dermody TS. 2013. Reovirus receptors, cell entry, and proapoptotic signaling. Adv Exp Med Biol 790:42–71. doi: 10.1007/978-1-4614-7651-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stencel-Baerenwald JE, Reiss K, Reiter DM, Stehle T, Dermody TS. 2014. The sweet spot: defining virus-sialic acid interactions. Nat Rev Microbiol 12:739–749. doi: 10.1038/nrmicro3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villalona-Calero MA, Lam E, Otterson GA, Zhao W, Timmons M, Subramaniam D, Hade EM, Gill GM, Coffey M, Selvaggi G, Bertino E, Chao B, Knopp MV. 2016. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer 122:875–883. doi: 10.1002/cncr.29856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolb EA, Sampson V, Stabley D, Walter A, Sol-Church K, Cripe T, Hingorani P, Ahern CH, Weigel BJ, Zwiebel J, Blaney SM. 2015. A phase I trial and viral clearance study of reovirus (Reolysin) in children with relapsed or refractory extra-cranial solid tumors: a Children's Oncology Group Phase I Consortium report. Pediatr Blood Cancer 62:751–758. doi: 10.1002/pbc.25464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowan K. 2010. Oncolytic viruses move forward in clinical trials. J Natl Cancer Inst 102:590–595. doi: 10.1093/jnci/djq165. [DOI] [PubMed] [Google Scholar]
- 9.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 11.Coyne CB. 2009. The distinct roles of JAM-A in reovirus pathogenesis. Cell Host Microbe 5:3–5. doi: 10.1016/j.chom.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Coffey MC, Strong JE, Forsyth PA, Lee PW. 1998. Reovirus therapy of tumors with activated Ras pathway. Science 282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 13.Gujar SA, Pan DA, Marcato P, Garant KA, Lee PW. 2011. Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther 19:797–804. doi: 10.1038/mt.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, de Bono J, Selby P, Coffey M, Vile R, Melcher A. 2008. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol 180:6018–6026. doi: 10.4049/jimmunol.180.9.6018. [DOI] [PubMed] [Google Scholar]
- 15.Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T, Thompson J, Galivo F, Harrington KJ, Pandha HS, Selby PJ, Vile RG, Melcher AA. 2009. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res 15:4374–4381. doi: 10.1158/1078-0432.CCR-09-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Errington F, White CL, Twigger KR, Rose A, Scott K, Steele L, Ilett LJ, Prestwich R, Pandha HS, Coffey M, Selby P, Vile R, Harrington KJ, Melcher AA. 2008. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther 15:1257–1270. doi: 10.1038/gt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirasawa K, Nishikawa SG, Norman KL, Coffey MC, Thompson BG, Yoon CS, Waisman DM, Lee PW. 2003. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res 63:348–353. [PubMed] [Google Scholar]
- 18.Rajani K, Parrish C, Kottke T, Thompson J, Zaidi S, Ilett L, Shim KG, Diaz RM, Pandha H, Harrington K, Coffey M, Melcher A, Vile R. 2016. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther 24:166–174. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinemann L, Simpson GR, Annels NE, Vile R, Melcher A, Prestwich R, Harrington KJ, Pandha HS. 2010. The effect of cell cycle synchronization on tumor sensitivity to reovirus oncolysis. Mol Ther 18:2085–2093. doi: 10.1038/mt.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiao J, Wang H, Kottke T, White C, Twigger K, Diaz RM, Thompson J, Selby P, de Bono J, Melcher A, Pandha H, Coffey M, Vile R, Harrington K. 2008. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res 14:259–269. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shmulevitz M, Gujar SA, Ahn DG, Mohamed A, Lee PW. 2012. Reovirus variants with mutations in genome segments S1 and L2 exhibit enhanced virion infectivity and superior oncolysis. J Virol 86:7403–7413. doi: 10.1128/JVI.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prestwich RJ, Errington F, Steele LP, Ilett EJ, Morgan RS, Harrington KJ, Pandha HS, Selby PJ, Vile RG, Melcher AA. 2009. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J Immunol 183:4312–4321. doi: 10.4049/jimmunol.0901074. [DOI] [PubMed] [Google Scholar]
- 23.Parrish C, Scott GB, Migneco G, Scott KJ, Steele LP, Ilett E, West EJ, Hall K, Selby PJ, Buchanan D, Varghese A, Cragg MS, Coffey M, Hillmen P, Melcher AA, Errington-Mais F. 2015. Oncolytic reovirus enhances rituximab-mediated antibody-dependent cellular cytotoxicity against chronic lymphocytic leukaemia. Leukemia 29:1799–1810. doi: 10.1038/leu.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnon TI, Achdout H, Lieberman N, Gazit R, Gonen-Gross T, Katz G, Bar-Ilan A, Bloushtain N, Lev M, Joseph A, Kedar E, Porgador A, Mandelboim O. 2004. The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood 103:664–672. doi: 10.1182/blood-2003-05-1716. [DOI] [PubMed] [Google Scholar]
- 25.Arnon TI, Markel G, Mandelboim O. 2006. Tumor and viral recognition by natural killer cells receptors. Semin Cancer Biol 16:348–358. doi: 10.1016/j.semcancer.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Bar-On Y, Seidel E, Tsukerman P, Mandelboim M, Mandelboim O. 2014. Influenza virus uses its neuraminidase protein to evade the recognition of two activating NK cell receptors. J Infect Dis 210:410–418. doi: 10.1093/infdis/jiu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O. 2006. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol 7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]
- 28.Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A. 2001. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409:1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 29.Jaron-Mendelson M, Yossef R, Appel MY, Zilka A, Hadad U, Afergan F, Rosental B, Engel S, Nedvetzki S, Braiman A, Porgador A. 2012. Dimerization of NKp46 receptor is essential for NKp46-mediated lysis: characterization of the dimerization site by epitope mapping. J Immunol 188:6165–6174. doi: 10.4049/jimmunol.1102496. [DOI] [PubMed] [Google Scholar]
- 30.Mendelson M, Tekoah Y, Zilka A, Gershoni-Yahalom O, Gazit R, Achdout H, Bovin NV, Meningher T, Mandelboim M, Mandelboim O, David A, Porgador A. 2010. NKp46 O-glycan sequences that are involved in the interaction with hemagglutinin type 1 of influenza virus. J Virol 84:3789–3797. doi: 10.1128/JVI.01815-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandelboim O, Porgador A. 2001. NKp46. Int J Biochem Cell Biol 33:1147–1150. doi: 10.1016/S1357-2725(01)00078-4. [DOI] [PubMed] [Google Scholar]
- 32.Jarahian M, Watzl C, Fournier P, Arnold A, Djandji D, Zahedi S, Cerwenka A, Paschen A, Schirrmacher V, Momburg F. 2009. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J Virol 83:8108–8121. doi: 10.1128/JVI.00211-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SH, Miyagi T, Biron CA. 2007. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol 28:252–259. doi: 10.1016/j.it.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Glasner A, Zurunic A, Meningher T, Lenac Rovis T, Tsukerman P, Bar-On Y, Yamin R, Meyers AF, Mandeboim M, Jonjic S, Mandelboim O. 2012. Elucidating the mechanisms of influenza virus recognition by Ncr1. PLoS One 7:e36837. doi: 10.1371/journal.pone.0036837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glasner A, Roth Z, Varvak A, Miletic A, Isaacson B, Bar-On Y, Jonjic S, Khalaila I, Mandelboim O. 2015. Identification of putative novel O-glycosylations in the NK killer receptor Ncr1 essential for its activity. Cell Discov 1:15036. doi: 10.1038/celldisc.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. 2009. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A 106:17858–17863. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, Achdout H, Ghadially H, Dor Y, Nir T, Doviner V, Hershkovitz O, Mendelson M, Naparstek Y, Mandelboim O. 2010. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol 11:121–128. doi: 10.1038/ni.1834. [DOI] [PubMed] [Google Scholar]
- 38.Bar-On Y, Glasner A, Meningher T, Achdout H, Gur C, Lankry D, Vitenshtein A, Meyers AF, Mandelboim M, Mandelboim O. 2013. Neuraminidase-mediated, NKp46-dependent immune-evasion mechanism of influenza viruses. Cell Rep 3:1044–1050. doi: 10.1016/j.celrep.2013.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 40.Iskarpatyoti JA, Morse EA, McClung RP, Ikizler M, Wetzel JD, Contractor N, Dermody TS. 2012. Serotype-specific differences in inhibition of reovirus infectivity by human-milk glycans are determined by viral attachment protein sigma1. Virology 433:489–497. doi: 10.1016/j.virol.2012.08.036. [DOI] [PubMed] [Google Scholar]
- 41.Connolly JL, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol 75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brisse E, Imbrechts M, Put K, Avau A, Mitera T, Berghmans N, Rutgeerts O, Waer M, Ninivaggi M, Kelchtermans H, Boon L, Snoeck R, Wouters CH, Andrei G, Matthys P. 2016. Mouse cytomegalovirus infection in BALB/c mice resembles virus-associated secondary hemophagocytic lymphohistiocytosis and shows a pathogenesis distinct from primary hemophagocytic lymphohistiocytosis. J Immunol 196:3124–3134. doi: 10.4049/jimmunol.1501035. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Z, Yang Y, Feng Y, Shi B, Chen L, Zheng Y, Tian D, Song Z, Xu C, Qin B, Zhang X, Guan W, Liu F, Yang T, Yang H, Zeng D, Zhou W, Hu Y, Zhou X. 2013. Infection of inbred BALB/c and C57BL/6 and outbred Institute of Cancer Research mice with the emerging H7N9 avian influenza virus. Emerg Microbes Infect 2:e50. doi: 10.1038/emi.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, Mandelboim O. 2012. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol 188:2509–2515. doi: 10.4049/jimmunol.1102461. [DOI] [PubMed] [Google Scholar]
- 45.Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, Goldman-Wohl D, Greenfield C, Yagel S, Hengel H, Altuvia Y, Margalit H, Mandelboim O. 2007. Host immune system gene targeting by a viral miRNA. Science 317:376–381. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ilett EJ, Prestwich RJ, Kottke T, Errington F, Thompson JM, Harrington KJ, Pandha HS, Coffey M, Selby PJ, Vile RG, Melcher AA. 2009. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther 16:689–699. doi: 10.1038/gt.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shmulevitz M, Marcato P, Lee PW. 2005. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene 24:7720–7728. doi: 10.1038/sj.onc.1209041. [DOI] [PubMed] [Google Scholar]
- 48.Ilett E, Kottke T, Donnelly O, Thompson J, Willmon C, Diaz R, Zaidi S, Coffey M, Selby P, Harrington K, Pandha H, Melcher A, Vile R. 2014. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol Ther 22:1851–1863. doi: 10.1038/mt.2014.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. 1988. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333:426–431. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- 50.Samson A, Bentham MJ, Scott K, Nuovo G, Bloy A, Appleton E, Adair RA, Dave R, Peckham-Cooper A, Toogood G, Nagamori S, Coffey M, Vile R, Harrington K, Selby P, Errington-Mais F, Melcher A, Griffin S. 15 November 2016. Oncolytic reovirus as a combined antiviral and anti-tumour agent for the treatment of liver cancer. Gut doi: 10.1136/gutjnl-2016-312009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao X, Rajasekaran N, Chester C, Yonezawa A, Dutt S, Coffey M, Kohrt HE. 2015. Reovirus activated NK cells show enhanced cetuximab mediated antibody-dependent cellular cytotoxicity against colorectal cancer cells, abstr B082. Abstracts: CRI-CIMT-EATI-AACR Inaugural International Cancer Immunotherapy Conference: Translating Science into Survival, 16–19 September 2015, New York, NY. [Google Scholar]
- 52.Ledford H. 2015. Cancer-fighting viruses win approval. Nature 526:622–623. doi: 10.1038/526622a. [DOI] [PubMed] [Google Scholar]
- 53.Topalian SL, Weiner GJ, Pardoll DM. 2011. Cancer immunotherapy comes of age. J Clin Oncol 29:4828–4836. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carlsten M, Childs RW. 2015. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol 6:266. doi: 10.3389/fimmu.2015.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Butler M, Burgener A, Patrick M, Berry M, Moffatt D, Huzel N, Barnabe N, Coombs K. 2000. Application of a serum-free medium for the growth of Vero cells and the production of reovirus. Biotechnol Prog 16:854–858. [DOI] [PubMed] [Google Scholar]
- 56.Gonzalez-Hernandez MB, Bragazzi Cunha J, Wobus CE. 2012. Plaque assay for murine norovirus. J Vis Exp 66:e4297. doi: 10.3791/4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mandelboim O, Malik P, Davis DM, Jo CH, Boyson JE, Strominger JL. 1999. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci U S A 96:5640–5644. doi: 10.1073/pnas.96.10.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glasner A, Simic H, Miklic K, Roth Z, Berhani O, Khalaila I, Jonjic S, Mandelboim O. 2015. Expression, function, and molecular properties of the killer receptor Ncr1-Noe. J Immunol 195:3959–3969. doi: 10.4049/jimmunol.1501234. [DOI] [PubMed] [Google Scholar]
- 59.Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. 2001. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol 31:2680–2689. doi:. [DOI] [PubMed] [Google Scholar]
- 60.Mandelboim O, Reyburn HT, Vales-Gomez M, Pazmany L, Colonna M, Borsellino G, Strominger JL. 1996. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J Exp Med 184:913–922. doi: 10.1084/jem.184.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hindiyeh M, Levy V, Azar R, Varsano N, Regev L, Shalev Y, Grossman Z, Mendelson E. 2005. Evaluation of a multiplex real-time reverse transcriptase PCR assay for detection and differentiation of influenza viruses A and B during the 2001 to 2002 influenza season in Israel. J Clin Microbiol 43:589–595. doi: 10.1128/JCM.43.2.589-595.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]