

ABSTRACT
Manipulation of host cellular pathways is a strategy employed by gammaherpesviruses, including mouse gammaherpesvirus 68 (MHV68), in order to negotiate a chronic infection. Ataxia-telangiectasia mutated (ATM) plays a unique yet incompletely understood role in gammaherpesvirus infection, as it has both proviral and antiviral effects. Chronic gammaherpesvirus infection is poorly controlled in a host with global ATM insufficiency, whether the host is a mouse or a human. In contrast, ATM facilitates replication, reactivation, and latency establishment of several gammaherpesviruses in vitro, suggesting that ATM is proviral in the context of infected cell cultures. The proviral role of ATM is also evident in vivo, as myeloid-specific ATM expression facilitates MHV68 reactivation during the establishment of viral latency. In order to better understand the complex relationship between host ATM and gammaherpesvirus infection, we depleted ATM specifically in B cells, a cell type critical for chronic gammaherpesvirus infection. B cell-specific ATM deficiency attenuated the establishment of viral latency due to compromised differentiation of ATM-deficient B cells. Further, we found that during long-term infection, peritoneal B-1b, but not related B-1a, B cells display the highest frequency of gammaherpesvirus infection. While ATM expression did not affect gammaherpesvirus tropism for B-1 B cells, B cell-specific ATM expression was necessary to support viral reactivation from peritoneal cells during long-term infection. Thus, our study reveals a role of ATM as a host factor that promotes chronic gammaherpesvirus infection of B cells.
IMPORTANCE Gammaherpesviruses infect a majority of the human population and are associated with cancer, including B cell lymphomas. ATM is a unique host kinase that has both proviral and antiviral roles in the context of gammaherpesvirus infection. Further, there is insufficient understanding of the interplay of these roles in vivo during chronic infection. In this study, we show that ATM expression by splenic B cells is required for efficient establishment of gammaherpesvirus latency. We also show that ATM expression by peritoneal B cells is required to facilitate viral reactivation during long-term infection. Thus, our study defines a proviral role of B cell-specific ATM expression during chronic gammaherpesvirus infection.
KEYWORDS: ATM, gammaherpesvirus, B cell, T cell response, reactivation, chronic infection
INTRODUCTION
Ataxia-telangiectasia (A-T) mutated (ATM) kinase is a multifunctional kinase, insufficiency of which in humans manifests as A-T (1, 2). The function of ATM was originally defined in the context of response to double-stranded DNA breaks. However, it is now clear that ATM functions within multiple host networks, including oxidative stress, inflammation, and mitophagy (3). ATM facilitates replication of diverse RNA and DNA viruses in cell culture. Although this proviral activity is traditionally ascribed to the role of ATM in DNA damage response (4, 5), it is likely that regulation of additional signaling networks by ATM contributes to viral replication (6).
Gammaherpesviruses, a focus of the current study, are ubiquitous pathogens that establish lifelong infection and are associated with a number of malignancies, including B cell lymphoproliferative diseases and lymphomas (7). Similar to other DNA viruses, both human (Epstein-Barr virus [EBV] and Kaposi's sarcoma-associated herpesvirus [KSHV]), and rodent (mouse gammaherpesvirus 68 [MHV68]) gammaherpesviruses benefit from ATM in vitro. Specifically, ATM activity and expression facilitate EBV reactivation from transformed cell lines (8) and the establishment of KSHV latency in primary endothelial cell cultures (9). Further, ATM expression enhances MHV68 replication in primary macrophages, but not mouse embryonic fibroblasts (MEF), in vitro (10), indicating that ATM functions as a proviral host factor in the context of many infected cell types.
Intriguingly, in contrast to the proviral activity of ATM manifested in tissue culture, A-T patients are selectively susceptible to severe herpesvirus infections, including frequent complications of primary varicella-zoster virus infection (11–17). Further, EBV (and human herpesvirus 6 [HHV-6], when present) loads are significantly elevated in A-T patients, and abnormal EBV-driven lymphoproliferation observed early in life has been suggested as a possible diagnostic trigger for A-T (12). Consistent with the inadequate control of EBV in A-T patients, we showed that MHV68 chronic infection is poorly controlled in ATM-deficient mice, an animal model of A-T (18). The mechanism(s) by which ATM deficiency confers increased susceptibility to deregulated chronic gammaherpesvirus infection remains unclear.
In order to reconcile proviral and antiviral roles of ATM, we proposed that ATM functions as a proviral molecule within gammaherpesvirus-infected cells, whether in culture or in vivo. Additionally, in an intact host, ATM expression outside infected cells likely supports a gammaherpesvirus-specific immune response that restricts viral replication and reactivation. Thus, in an ATM-competent host, these pro- and antiviral functions of ATM occur simultaneously, establishing a virus-host balance that both promotes and limits chronic gammaherpesvirus infection. In support of this model, we showed that ATM-deficient mice display an inadequate and skewed MHV68-specific CD8 T cell response (18). Further, myeloid cell-specific ATM deficiency attenuated establishment of MHV68 latency in vivo, likely by directly facilitating MHV68 reactivation from macrophages (19).
The current study defines the extent to which B cell-specific ATM expression regulates chronic gammaherpesvirus infection. Specifically, we show that selective deficiency of ATM within B cells attenuated the establishment of MHV68 splenic latency. Because the establishment of gammaherpesvirus latency is intimately tied to the B cell differentiation program (20–25), the altered differentiation of ATM-deficient B cells observed in our study was likely responsible for the attenuated establishment of MHV68 latency in the spleen. Surprisingly, we found that B cell-specific ATM deficiency had no effect on long-term MHV68 infection in the spleen. In contrast, gammaherpesvirus reactivation from the peritoneal cells was attenuated in long-term-infected mice with ATM-depleted B cells. Extending this observation, we show that peritoneal B-1b cells are a significant reservoir of latent gammaherpesvirus during long-term infection and that ATM expression within B cells is required to support long-term gammaherpesvirus reactivation in the peritoneum.
RESULTS
Generation of mice with B cell-specific ATM deficiency.
To define the extent to which B cell-specific ATM expression alters chronic gammaherpesvirus infection, we generated a mouse model of B cell-restricted ATM deficiency (designated B-Cre). In this mouse model (Fig. 1A), conditional ATM alleles (26) were combined with a Cre recombinase allele driven by the endogenous CD19 promoter (27). B-Cre mice were maintained as heterozygotes with respect to the Cre allele to retain expression of endogenous CD19.
FIG 1.
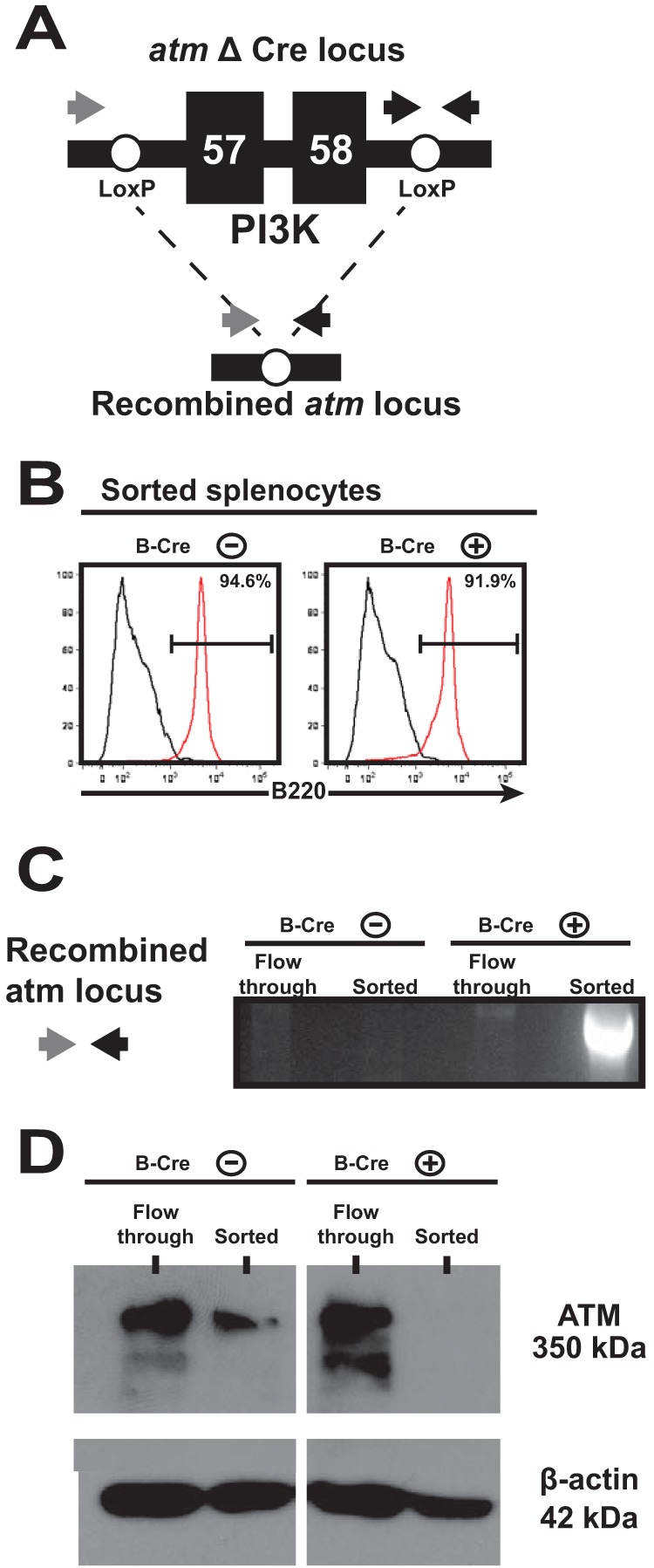
B cell-specific depletion of ATM. Recombination of the floxed ATM allele in B cells was achieved by placing expression of Cre recombinase under the control of the endogenous CD19 promoter. (A) Schematic representing the floxed ATM allele. Exons 57 and 58 encoding the ATM phosphatidylinositol 3-kinase (PI3K) domain are flanked by LoxP sites and were excised following Cre-mediated recombination. (B) Splenocytes were harvested from littermates positive or negative for the Cre transgene. CD19+ B cells were enriched by magnetic-column separation, and purity was assessed by expression of B220 (red lines, sorted B cells; black lines, flowthrough splenocytes). (C and D) ATM depletion was verified by PCR amplification of the recombined ATM locus (C) and detection of ATM protein in cell lysates (D) of the enriched B cells and flowthrough cells from B-Cre-positive and -negative littermates.
To evaluate ATM depletion, splenocytes from B-Cre-positive and -negative mice were sorted into B220+ B cells and non-B cells (flowthrough) (Fig. 1B). As expected, only B-Cre-positive sorted B cells contained the recombined ATM locus (Fig. 1C). Additionally, while comparable ATM protein levels were detected in non-B cells from B-Cre-positive and -negative splenocytes, ATM protein was below the level of detection in B-Cre-positive sorted B cells (Fig. 1D), indicating that ATM deficiency was B cell restricted.
The establishment of gammaherpesvirus splenic latency is attenuated under conditions of B cell-specific ATM depletion.
Following brief replication in the lungs, MHV68 transitions to the spleen, where the virus usurps the normal B cell differentiation program to establish lifelong infection. During the peak of MHV68 splenic latency, the viral genome is harbored by naive B cells, germinal center B cells, and plasma cells (20–25). Interestingly, while a majority of latently infected cells are found within the germinal center B cells, viral reactivation is restricted to the plasma cells (24). To determine the effect of B cell-specific ATM deficiency on the establishment of gammaherpesvirus latency, B-Cre-positive and -negative mice were intranasally infected with MHV68 and the parameters of viral latency were examined at 16 days postinfection.
The frequencies of MHV68 reactivation and viral-DNA-positive cells were decreased in the spleens of B-Cre-positive mice compared to their B-Cre-negative littermates (P < 0.05) (specific frequencies are indicated in Fig. 2A and B). This attenuation of MHV68 latency occurred in spite of similar frequencies and absolute numbers of splenic B220+ B cells in the B-Cre-positive and -negative groups (baseline or following infection) (Fig. 2C and D). No persistent virus was detected in either B-Cre-positive or -negative mice (data not shown). Thus, B cell-intrinsic expression of ATM facilitated the establishment of splenic MHV68 latency.
FIG 2.

B cell-specific depletion of ATM attenuates MHV68 latency at 16 days postinfection. (A and B) B-Cre-positive and -negative mice were infected intranasally with 104 PFU of MHV68. At 16 days postinfection, splenocytes were harvested and subjected to limiting-dilution analyses to determine the frequency of ex vivo reactivation (A) and the frequency of infected cells (B). The dashed lines in these and other frequency experiments are drawn at 62.5%, and the intercept of the line with the data curve indicates the frequency of a positive event in the analyzed population. The frequencies of positive cells are indicated next to each genotype. Data were pooled from three independent experiments with 3 to 5 mice per group. The error bars indicate standard error of measurement. (C and D) The frequency (C) and absolute number (D) of B220+ splenocytes were determined in B-Cre-positive and -negative mice at 16 days following either mock or intranasal infection with 104 PFU of MHV68. Each symbol represents an individual mouse; data were pooled from 3 independent experiments.
B cell-specific ATM deficiency attenuates class switching, germinal center response, and the generation of IgG-positive plasma cells.
The MHV68 splenic reservoir was decreased in B-Cre-positive mice in spite of similar total B cell numbers. Thus, we next tested the hypothesis that altered B cell differentiation in B-Cre-positive mice might be responsible for the attenuated viral latency in the spleen. ATM facilitates class switching and somatic hypermutation in splenic B cells (28–30), and both of these processes are CD4 T cell dependent, occur in the context of germinal center reaction, and lead to generation of plasma or memory B cells. While MHV68 latency is highest in germinal center B cells, MHV68 reactivation from B cells is uniquely limited to plasma cells (24).
To test for altered B cell differentiation in B-Cre-positive mice, the abundances of class-switched B cells, germinal center B cells, and plasma cells were assessed. The frequency and number of class-switched B cells (B220+ IgM/IgD−) were decreased in MHV68-infected B-Cre-positive compared to B-Cre-negative mice (Fig. 3A to C). Because class-switched B cells are found within both germinal center and plasma cell compartments, we next assessed germinal center B cells. Similar to those observed for class-switched B cells, frequencies and absolute numbers of germinal center B cells (B220+ GL7+ CD95+) were decreased in B-Cre-positive compared to B-Cre-negative splenocytes harvested from infected mice (Fig. 3D to F). Moreover, both the frequency and number of class-switched plasma cells (B220low CD138+ IgM− IgG− intracellular IgG+) were significantly decreased in the MHV68-infected B-Cre-positive group compared to the B-Cre-negative group (Fig. 3G to I).
FIG 3.

ATM deficiency in B cells attenuates MHV68-driven B cell differentiation. B-Cre-positive and -negative mice were either mock treated or infected intranasally with 104 PFU of MHV68. At 16 days postinfection (dpi), splenocytes were harvested and analyzed using flow cytometry. Each data point represents an individual mouse; data from 2 to 4 independent experiments were pooled. (A) Class-switched B cells were pregated on B220+ and identified as IgM− IgD− (a representative flow diagram is shown). Boxed areas identify immune populations of interest. (B and C) The frequencies (B) and the absolute numbers (C) of B220+ IgM− IgD− splenocytes were quantified. (D) Germinal center B cells were pregated on B220+ and further identified as CD95+ GL7+ (a representative flow diagram is shown). (E and F) Frequencies (E) and absolute numbers (F) of B220+ CD95+ GL7+ splenocytes. (G) Plasma cells were pregated on B220+, further gated as IgM− IgD−, and identified for surface expression of CD138+ and for intracellular IgG+ (representative flow diagrams are shown). (H and I) Frequencies (H) and absolute numbers (I) of B220+ IgM− IgD− CD138+ IgG+ plasma cells. (J to M) Serum total immunoglobulin (J) and MHV68-specific antibodies (total [K], IgM [L], and IgG [M]) were measured by ELISA at 16 days post-mock treatment or -MHV68 infection; pooled data are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard deviations.
To assess the functional outcomes of changes in B cell populations, total and virus-specific immunoglobulin levels were measured. Total serum immunoglobulin was decreased in B-Cre-positive compared to B-Cre-negative mice, both at baseline and following MHV68 infection (Fig. 3J). Similarly, the generation of MHV68-specific antibodies was reduced in B-Cre-positive mice, primarily due to decreased levels of MHV68-specific IgG (Fig. 3K to M). Thus, B cell-specific ATM deficiency attenuated germinal center responses and subsequent B cell differentiation induced by MHV68 infection.
ATM deficiency restricted to B cells fails to alter MHV68-specific CD8 T cell response.
We had previously found that MHV68-specific T cell responses are attenuated and skewed in viral epitope specificity in mice with global ATM deficiency (18). Given the known function of B cells as antigen-presenting cells in the context of gammaherpesvirus infection (31) and decreased splenic MHV68 latency observed in B-Cre-positive mice, we wanted to test the hypothesis that both global and MHV68-specific T cell responses are altered under conditions where ATM deficiency is limited to B cells. Similar numbers of CD8 and CD4 T cells were observed in the spleen and peritoneum in B-Cre-positive and -negative mice at baseline and following MHV68 infection (16 days) (Fig. 4A), indicating that B cell-specific ATM deficiency had no effect on the global T cell response to MHV68 infection. Further, similar frequencies and absolute numbers of MHV68-specific CD8 T cells (ORF6487/Db and ORF61524/Kb) were found in spleens of B-Cre-positive and -negative mice (Fig. 4B and C). Thus, in contrast to what was observed in mice with global ATM deficiency, neither polyclonal nor MHV68-specific T cell responses were affected by the B cell-specific ATM deficiency.
FIG 4.

ATM deficiency restricted to B cells fails to alter MHV68-specific CD8 T cell response. B-Cre-positive and -negative mice were mock infected or intranasally inoculated with 104 PFU of MHV68. (A) Numbers of T cells and CD4 and CD8 T cells were determined by flow cytometry in the spleen and peritoneum at 16 days postinfection. The error bars indicate standard deviations. (B and C) Proportions and numbers of orf6 or orf61 tetramer-positive CD8 T cells were determined by flow cytometry in the spleen and peritoneum. Data were pooled from 2 or 3 independent experiments. Each symbol represents an individual mouse.
B cell-specific ATM deficiency attenuates MHV68 reactivation in the peritoneum in long-term-infected mice.
Between 16 and 42 days postinfection, MHV68 transitions from early latency to stable long-term maintenance concurrent with a decrease and subsequent stabilization of the latent reservoir and attenuation of ex vivo reactivation. Having observed decreased establishment of splenic MHV68 latency in B-Cre-positive mice (Fig. 2), we assessed the relevance of B cell-specific ATM expression in long-term infection. In contrast to those observed at 16 days postinfection (Fig. 2A and B), the frequencies of MHV68 genome-positive splenocytes were similar in B-Cre-positive and -negative mice at 42 days postinfection, with little if any spontaneous reactivation from splenocytes in either group (Fig. 5A and B). Intriguingly, MHV68 reactivation from the peritoneal cells was significantly decreased in the long-term-infected B-Cre-positive cohort (Fig. 5C) (P = 0.007) in spite of similar frequencies of MHV68 DNA-positive peritoneal cells in the two experimental groups (Fig. 5D). Of note, persistent replication was not detected in any of the experimental groups (data not shown). Thus, B cell-specific ATM deficiency attenuated MHV68 reactivation from the peritoneal cells of long-term-infected mice.
FIG 5.

B cell-specific ATM deficiency attenuates MHV68 reactivation in the peritoneum in long-term-infected mice. B-Cre-negative and -positive littermates were mock treated or intranasally infected with 104 PFU of MHV68. At 42 dpi, splenocytes and PECs were harvested and subjected to limiting-dilution analysis to determine the frequency of ex vivo reactivation (A and C) and the frequency of infected cells (B and D). Data were pooled from 2 to 4 independent experiments. The error bars indicate standard deviations.
Peritoneal B-1b cells preferentially support long-term MHV68 latency following intranasal infection.
We have recently shown that peritoneal B-1 and B-2 B cells support MHV68 latency in BL6 mice, with the former cell type showing a greater frequency of infected cells (32). Our observation of decreased MHV68 reactivation from peritoneal cells of B-Cre-positive mice suggested that ATM may facilitate MHV68 latency in peritoneal B cells.
Peritoneal B cells are comprised of several distinct populations that have minimal overlap with the B cell populations found in the spleen (33). The peritoneal cavity hosts a distinct population of B-1 B cells (as opposed to splenic B-2 B cells) that have intermediate expression of B220 along with high surface levels of CD19 (B220Int CD19Hi) and are further differentiated into B-1a and B-1b subpopulations based on CD5 (expressed by B-1a but not B-1b cells) (Fig. 6A). These B-1 B cells have a limited B cell receptor (BCR) repertoire, are self-renewing, and produce “innate” antibodies in a T cell-independent manner. In addition to B-1 B cells, B220Hi CD19Hi B-2 B cells are also present in the peritoneum. Peritoneal B-2 B cells may not simply represent recirculating splenic B cells, as surface markers of the B-2 B cells in the peritoneum represent a phenotype that is intermediate between splenic B-2 B cells and peritoneal B-1 B cells (34). Functionally, peritoneal B-2 B cells are also closer to B-1b cells than to splenic B-2 B cells (34). In spite of showing tropism of MHV68 for peritoneal B-1 B cells in our previous study (32), the tropism of MHV68 for B-1 B subpopulations (B-1a and B-1b) remains unknown.
FIG 6.

Peritoneal B-1b cells preferentially support long-term MHV68 latency following intranasal infection. Mice of the indicated genotypes were infected as for Fig. 5. (A) Peritoneal cells of each genotype were pooled at 42 days postinfection and sorted into the indicated populations using cell surface markers. (B and C) Sorted cell populations were subjected to limiting-dilution PCR assay to determine the frequency of MHV68 DNA-positive cells. Data were pooled from 2 to 4 independent experiments. (D) Long-term-infected mice were intraperitoneally treated with clodronate or control liposomes at 39 days postinfection, and the frequency of viral reactivation was measured in peritoneal cells pooled from 5 mice of each genotype at 42 days postinfection (3 days following chlodronate treatment). The error bars indicate standard deviations.
To determine whether ATM regulated MHV68 infection of distinct peritoneal B cell populations, cohorts of B-Cre-positive and -negative mice were intranasally infected, peritoneal cells were harvested at 42 days postinfection, and the presence of the distinct peritoneal B cell populations was assessed by flow cytometry. The relative abundances of peritoneal B-1a, B-1b, and B-2 B cell populations defined by the markers outlined in Fig. 6A were similar between B-Cre-positive and -negative MHV68-infected groups (data not shown).
Sorted B and non-B cell populations (as defined in Fig. 6A) were next assessed for the frequency of MHV68 DNA-positive cells. MHV68-positive cells were enriched in most peritoneal B cell populations compared to peritoneal non-B cells (Fig. 6B and C), consistent with our observation in BL6 mice (32), suggesting that the genetic background does not have a profound influence on the peritoneal cell types hosting latent MHV68. Interestingly, while the highest frequency of MHV68 DNA-positive cells was found in the B-1b B cells of either B-Cre genotype (Fig. 6B), the frequency of MHV68 infection in B-1a B cells was very low to undetectable (data not shown). Additionally, CD5+ B-2 B cells harbored a lower frequency of MHV68-infected cells than CD5-negative B-2 B cells (Fig. 6B). Thus, following intranasal infection, MHV68 preferentially established long-term latency in CD5-negative peritoneal B cells, with B-1b B cells harboring the highest frequency of infected cells. Importantly, ATM expression by B cells did not alter MHV68 tropism for the peritoneal B cell populations examined in this study.
To determine the extent to which macrophages contributed to MHV68 reactivation from the peritoneum in long-term-infected mice, B-Cre-positive and -negative mice were intranasally infected with MHV68 and treated with clodronate at day 39 postinfection. In spite of >99% efficient depletion of CD11b+ F4/80+ peritoneal cells, clodronate treatment failed to significantly alter the frequency of viral reactivation in B-Cre-negative peritoneal cells harvested at 42 days postinfection (Fig. 6D). Clodronate treatment also had no effect on the very low levels of MHV68 reactivation observed in B-Cre-positive peritoneal cells (Fig. 6D). Thus, ATM expression by peritoneal B cells promoted MHV68 reactivation in long-term-infected mice.
DISCUSSION
The results of this study establish the importance of B cell-specific ATM expression during both the establishment of MHV68 latency and long-term reactivation. Based on the evidence presented, we propose the following model (Fig. 7). ATM regulates the establishment of viral splenic latency indirectly by promoting B cell differentiation, including MHV68-driven germinal center reaction, class switching, and efficient generation of plasma B cells. These plasma cells, when derived from MHV68-infected germinal center B cells, mediate MHV68 reactivation, producing infectious virus that reseeds the B cell compartment (Fig. 7A). In contrast, ATM is likely to directly facilitate MHV68 reactivation from peritoneal B cells during long-term infection (Fig. 7B).
FIG 7.
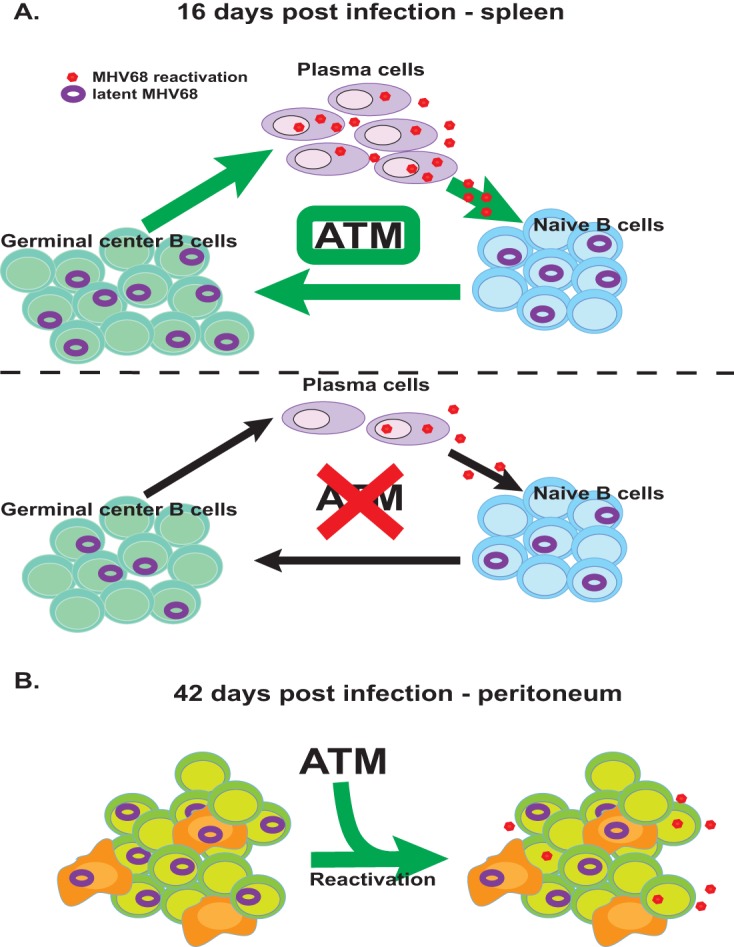
Working model. (A) ATM regulates the establishment of viral latency indirectly by promoting B cell differentiation, including MHV68-driven germinal center reaction, class switching, and efficient generation of plasma B cells. These plasma cells, when derived from MHV68-infected germinal center B cells, mediate MHV68 reactivation, producing infectious virus that reseeds the B cell compartment. Suboptimal differentiation of ATM-deficient B cells indirectly attenuates the establishment of peak MHV68 latent reservoir and reactivation. (B) ATM expression by peritoneal B cells likely directly facilitates MHV68 reactivation during long-term infection.
Control of chronic gammaherpesvirus infection in an ATM-deficient host.
ATM-deficient hosts, whether humans or mice, are selectively susceptible to severe chronic, but not acute, viral infections. Specifically, A-T patients display elevated EBV and HHV-6 loads, have prolonged and exacerbated primary varicella-zoster virus infection, and display more robust and persistent skin lesions associated with human papillomavirus (HPV) infection (11–17). These observations contradict the well-established proviral phenotypes in tissue culture systems, where ATM facilitates infection and replication of diverse herpesviruses and HPV. These opposing observations suggest a balancing nature of ATM, where it promotes viral replication/reactivation within the infected cell while at the same time supporting the generation of immune responses that are necessary to control chronic virus infections.
To support the hypothesis that ATM facilitates immune responses necessary to control chronic virus infections, we have previously demonstrated that MHV68-specific CD8 T cell responses are attenuated and skewed in MHV68-infected mice with global ATM deficiency (18). We have since developed and are evaluating additional mouse models, including a model of T cell-specific ATM deficiency that will facilitate identification of ATM-dependent immune mechanisms that promote control of chronic virus infections.
In support of the proviral role of ATM within infected cells in vivo, we have previously shown that myeloid-specific ATM deficiency attenuates chronic MHV68 infection, likely by directly promoting viral reactivation within infected myeloid cells (19). Our current study shows that ATM expression by B cells indirectly facilitates the establishment of MHV68 latency through optimizing B cell differentiation and subsequently supports MHV68 reactivation from peritoneal B cells during long-term infection. In the myeloid model of ATM deficiency, we have shown that activation of ATM directly promotes MHV68 reactivation from latently infected macrophages ex vivo. Further, data in our current study indicate a similar direct role of ATM in facilitating viral reactivation from peritoneal B cells during long-term infection. However, the mechanism remains unclear. We have recently found that ATM attenuates type I interferon (IFN) responses during MHV68 replication in primary macrophages, and it is this attenuation that fully accounts for the proviral activity of ATM during gammaherpesvirus replication in vitro (44). It is tempting to speculate that the infected-cell-intrinsic expression of ATM and subsequent attenuation of the type I IFN response also promote virus reactivation during chronic infection, a hypothesis that is being tested in ongoing studies.
ATM and B cell infection in the peritoneum.
A majority of MHV68 studies, similar to those of human gammaherpesviruses, focused on the interaction of the viruses with splenic B cells, with very little known about the infection of other B cell types. A study published by the Virgin group showed that peritoneal macrophages maintain the majority of latent MHV68 at 16 days postinfection following the intraperitoneal route of inoculation (35). Subsequently, the Tibbetts group demonstrated a higher frequency of MHV68 DNA-positive cells in peritoneal B cells than in macrophages at 42 days post-intraperitoneal infection (36). However, the type of B cells harboring MHV68 in the peritoneum was not identified. We have since shown that B-1 B cells predominantly support peritoneal MHV68 latency in BL6 mice, regardless of the route of inoculation (32). In the current study, we show that MHV68 tropism for peritoneal B-1 B cells is also observed in mice on a mixed genetic background, suggesting that infection of B-1 B cells is not unique to the BL6 mouse strain.
Interestingly, latent MHV68 was significantly enriched in B-1b B cells (Fig. 6B) but was at or below the level of detection in B-1a B cells (data not shown). In spite of several similarities between these two B cell populations (location in body cavities, T cell independence, and self-renewal), they can have distinct functions during immune response and, potentially, distinct origins of development (33). Further, CD5 is expressed by B-1a, but not B-1b, B cells. In B cells, CD5 recruits phosphatase Shp1 to the B cell receptor to attenuate B cell receptor signaling and B cell activation (37). B cell activation promotes the establishment of MHV68 latency, at least in splenic B cells (38); thus, decreased activation capacity of B-1a B cells may make the cells more restrictive for MHV68 latency.
Due to the distinct features of B-1 and B-2 B cells, it is likely that distinct mechanisms regulate MHV68 infection in these B cell populations. In this study, B cell-specific ATM expression no longer regulated splenic latency during long-term infection. As recently demonstrated by the Tibbetts group, MHV68 infection of developing B cells in the bone marrow is important to support long-term, but not early, viral splenic latency (39). ATM is not depleted in the developing B cells of CD19-Cre-positive ATMc/c mice (40), and this is likely the reason why attenuation of splenic MHV68 latency is limited to early times in this model. However, in the same long-term-infected mice, B cell-specific expression of ATM facilitated MHV68 reactivation in the peritoneum. Short-term depletion of peritoneal macrophages in long-term-infected mice did not affect MHV68 reactivation (Fig. 6D). Further, we have previously shown that myeloid-specific deletion of ATM has no effect on MHV68 reactivation from peritoneal cells of long-term-infected mice (19). Thus, the current study defines a novel direct role of ATM in maintaining long-term MHV68 reactivation from peritoneal B cells. Understanding the molecular mechanism by which ATM supports such reactivation will offer important insights into the biology and pathogenesis of long-term gammaherpesvirus infection.
MATERIALS AND METHODS
Mice.
All mice were housed and bred in a specific-pathogen-free barrier facility in accordance with federal and institutional guidelines. All experimental manipulations of mice were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (AUA971). C57BL/6J B-Cre (C.129P2-Cd19tm1(cre)Cgn/J) mice were originally obtained from Jackson Laboratories (Bar Harbor, ME). ATM flox (ATMc/c) mice were a kind gift from Fred Alt (Howard Hughes Medical Institute, Harvard Medical School). A combination of ATMgF86723 (5′ ATC AAA TGT AAA GGC GGC TTC 3′), BAC13 (5′ CAT CCT TTA ATG TGC CTC CCT TCG CC 3′), and BAC 7 (5′ GCC CAT CCC GTC CAC AAT ATC TCT GC 3′) primers was used to detect either the floxed AtmΔcre (420-bp floxed; 350-bp wild type) or the recombined AtmDel (903 bp) (26) allele in ATMc/c mice using a strategy outlined in Fig. 1. The Cre transgene was detected in the parental B-Cre (C.129P2-Cd19tm1(cre)Cgn/J) mice using primers 5′ ACG TAC TGA CGG TGG GAG AA 3′ and 5′ CAA AAA TCC CTT CCA GGG CG 3′.
Virus infections in vivo.
Wild-type MHV68 virus stock was prepared and the titer was determined on NIH 3T12 cells as previously described (41). Due to the mixed genetic background of the B-Cre mouse strain, Cre-positive and -negative littermates were directly compared. In each experiment, 3 to 5 mice/genotype were infected. Infections were performed by intranasal inoculation at 6 to 7 weeks of age with 104 PFU of MHV68 or sterile carrier in an inoculum volume of 15 μl per mouse. Virus was diluted in sterile serum-free Dulbecco's modified Eagle's medium (Corning, Tewksbury, MA). Upon termination of the experiment, splenocytes, peritoneal cells, and serum were collected from each mouse. In viral-latency experiments, splenocytes were pooled from mice of the same genotype. These pooled cell suspensions were subjected to limiting-dilution assays (42). In contrast, splenocytes and peritoneal cells harvested from individual mice were analyzed as individual samples by flow cytometry and enzyme-linked immunosorbent assay (ELISA).
Cell sorting.
In experiments validating B cell-specific depletion of ATM, homogenized spleens from B-Cre-negative and B-Cre-positive mice were incubated with αCD19 magnetic beads, and B cells were positively enriched by filtration through a magnetized column (Miltenyi Biotec, San Diego, CA) according to the manufacturer's protocol. To prepare peritoneal exudate cells (PECs) for cell sorting by FACS Aria (BD Biosciences, San Jose, CA), single-cell suspensions of PECs from 5 mice per group were pooled. The PECs were resuspended in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline [PBS] plus 2% fetal calf serum [FCS] and 0.05% sodium azide) at 1 × 107 cells/ml. The cells were stained with antibodies (in parentheses) purchased from eBioscience (San Diego, CA) against the following surface molecules at optimal concentrations: CD5 (53-7.3), B220/CD45R (RA3-6B2), CD19 (6D5), and CD11b (M1/70). Prior to sorting, 2 × 106 cells were removed from each group for limiting-dilution PCR (LD-PCR) analysis to represent the bulk PEC population.
Clodronate treatment.
Clodronate and control PBS liposomes were obtained from ClodronateLiposomes (Amsterdam, Netherlands) and administered intraperitoneally according to the manufacturer's instructions. Each mouse was treated with 200 μl (5 mg/ml) 3 days prior to testing for virus reactivation. Macrophage depletion was determined by surface expression of CD11b and F4/80 via flow cytometry. Depletion efficiency was >99% in clodronate-treated mice compared to PBS controls.
Limiting-dilution analysis.
Limiting-dilution ex vivo reactivation and nested-PCR analyses were performed as previously described to measure the frequency of cells reactivating MHV68 or harboring the MHV68 genome, respectively (43). Briefly, to determine the frequency of cells reactivating virus ex vivo, serial 2-fold dilutions of splenocytes or peritoneal cell suspensions were plated onto monolayers of MEF immediately following harvest at 24 replicates per dilution. In order to control for any preformed infectious virus, 2-fold serial dilutions of mechanically disrupted splenocytes or peritoneal cells were plated as described above. MHV68 was allowed to reactivate from explanted cells, and virus was further amplified within the same well via subsequent replication in MEF. The cytopathic effect was scored at 21 days postplating in all replicates and dilutions. Because primary MEF were used to amplify the virus, the sensitivity of the limiting-dilution reactivation assay was a single PFU of MHV68. Because the endpoint of viral amplification in MEF was measured, the limiting-dilution reactivation assay was not susceptible to variability of titers released from primary cells upon viral reactivation ex vivo.
Tissue culture.
NIH 3T12 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine. Mouse embryonic fibroblasts were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 25 U/ml penicillin, 25 mg/ml streptomycin, 2.6 mM l-glutamine, and nonessential amino acids. Bone marrow-derived macrophages were generated as previously described (10).
Flow cytometry.
Single-cell suspensions were resuspended in FACS buffer (PBS plus 2% FCS and 0.05% sodium azide) at 1 × 107 cells/ml. A total of 1 × 106 cells were prestained with Fc block (monoclonal antibody blocking Fc receptor, clone 24G2) and then incubated with an optimal amount of antibody conjugate (eFluor450, fluorescein isothiocyanate, r-phycoerythrin [PE], PE-Cy7, or allophycocyanin). Antibodies (in parentheses) to the following molecules were purchased from eBioscience (San Diego, CA): B220/CD45R (RA3-6B2), CD8a/Ly-2 (53-6.7), CD4 (RM4-5), CD11b (M1/70), CD11c (N418), CD95 (GL-7), IgM (RMM-1), IgD (11-26c), CD138 (281-2), CD19 (6D5), and CD5 (53-7.3). For plasma cell analysis, cell surface antigens were stained as described above. Following surface staining, cells were fixed and permeabilized using a BD Cytofix/Cytoperm kit (BD Biosciences, San Diego, CA). Intracellular IgG was detected with anti-IgG purchased from Jackson ImmunoResearch. Data acquisition was performed on an LSR II flow cytometer (BD Biosciences, Sparks, MD), and the data were analyzed using FlowJo software (Tree Star, Ashland, OR). MHV68 tetramers were provided by the NIH Tetramer Core Facility.
ELISA.
Sera were collected from uninfected or infected mice of both genotypes at 16 days postinfection. MHV68-specific antibody titers were determined as described previously (19). Serum levels of total IgM and IgG and total Ig levels were detected by coating Nunc MaxiSorp immunoplates (Fisher Scientific, Pittsburgh, PA) with goat anti-mouse IgM, goat anti-mouse IgG (Fcγ fragment specific), or goat anti-mouse IgG (heavy plus light chain) (all from Jackson ImmunoResearch, West Grove, PA) at 10 μg/ml in PBS. The plates were washed with PBS-Tween (0.05%), blocked for 1 h with PBS-Tween (0.05%)-bovine serum albumin (BSA) (3%), incubated with 3-fold dilutions of serum in PBS-Tween (0.05%)-BSA (0.5%) for 1 h, and washed with PBS-Tween (0.05%). Bound antibody was detected with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgM, goat anti-mouse IgG (heavy plus light chain), or goat anti-mouse IgG (Fcγ fragment specific) using 3,3′,5,5′-tetramethylbenzidine substrate (Life Technologies, Gaithersburg, MD). HRP enzymatic activity was stopped by the addition of 1 N HCl, and absorbance was read at 450 nm on a model 1420 Victor3V multilabel plate reader (PerkinElmer, Waltham, MA).
Western blot analysis.
Samples were collected in Laemmli buffer (0.1 M Tris, 4% SDS, 4 mM EDTA, 100 mM beta-mercaptoethanol, 3.2 M glycerol with 0.05% bromophenol blue), boiled for 10 min, and subjected to SDS-PAGE. The protein was transferred to a polyvinylidene difluoride membrane (Immunobilon; Millipore, Billerica, MA) using a Transblot transfer cell semidry system (Bio-Rad, Hercules, CA). The membranes were blocked overnight with 5% ovalbumin in TBST buffer (150 mM NaCl, 10 mM Tris, and 0.2% Tween) at 4°C. Proteins were visualized with a rabbit anti-ATM monoclonal antibody (1:500; clone D2E2; Cell Signaling Technology, Boston, MA) or an anti-β-actin antibody (1:15,000; clone AC-74; Novus Biologicals, Littleton, CO) followed by secondary HRP-conjugated antibodies (1:20,000; Jackson ImmunoResearch, West Grove, PA).
Statistical analyses.
All statistical analyses were performed using GraphPad (San Diego, CA) Prism software. Student's t test was used to measure statistical significance, with an α-value of 0.05.
ACKNOWLEDGMENTS
We are indebted to Linda van Dyk for her insightful comments, which greatly facilitated this study. We thank John Corbett, William Jackson, and their research teams for helpful discussions. Cell-sorting procedures were executed by Tamara N. Nelson, Children's Research Institute Shared Flow Cytometry Resource at the Medical College of Wisconsin.
This study was supported by the AHA (15PRE22640005; W.P.M.), ACS Research Scholar Grant RSG-12–174-01-MPC, and R01CA183593 (V.L.T.).
REFERENCES
- 1.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 2.Lavin MF. 2008. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 4.Weitzman MD, Weitzman JB. 2014. What's the damage? The impact of pathogens on pathways that maintain host genome integrity. Cell Host Microbe 15:283–294. doi: 10.1016/j.chom.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turnell AS, Grand RJ. 2012. DNA viruses and the cellular DNA-damage response. J Gen Virol 93:2076–2097. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- 6.Shah GA, O'Shea CC. 2015. Viral and cellular genomes activate distinct DNA damage responses. Cell 162:987–1002. doi: 10.1016/j.cell.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cesarman E. 2011. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagemeier SR, Barlow EA, Meng Q, Kenney SC. 2012. The cellular ataxia telangiectasia-mutated kinase promotes Epstein-Barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J. Virol 86:13360–13370. doi: 10.1128/JVI.01850-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh VV, Dutta D, Ansari MA, Dutta S, Chandran B. 2014. Kaposi's sarcoma-associated herpesvirus induces ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells which play roles in latency establishment. J. Virol 88:2821–2834. doi: 10.1128/JVI.03126-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarakanova VL, Leung-Pineda V, Hwang S, Yang C-W, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folgori L, Scarselli A, Angelino G, Ferrari F, Antoccia A, Chessa L, Finocchi A. 2010. Cutaneous granulomatosis and combined immunodeficiency revealing ataxia-telangiectasia: a case report. Ital J Pediatr 36:29. doi: 10.1186/1824-7288-36-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lankisch P, Adler H, Borkhardt A. 2013. Testing for herpesvirus infection is essential in children with chromosomal-instability syndromes. J Virol 87:3616–3617. doi: 10.1128/JVI.03279-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masucci G, Berkel I, Masucci MG, Ernberg I, Szigeti R, Ersoy F, Sanal O, Yegin O, Henle G, Henle W, and 1984. Epstein-Barr virus (EBV)-specific cell-mediated and humoral immune responses in ataxia-telangectasia patients. J Clin Immunol 4:369–382. doi: 10.1007/BF00917140. [DOI] [PubMed] [Google Scholar]
- 14.Morio T, Takahashi N, Watanabe F, Honda F, Sato M, Takagi M, Imadome KI, Miyawaki T, Delia D, Nakamura K, Gatti RA, Mizutani S. 2009. Phenotypic variations between affected siblings with ataxia-telangiectasia: ataxia-telangiectasia in Japan. Int J Hematol 90:455–462. doi: 10.1007/s12185-009-0408-0. [DOI] [PubMed] [Google Scholar]
- 15.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. 2004. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr 144:505–511. doi: 10.1016/j.jpeds.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 16.Saemundsen AK, Berkel AI, Henle W, Henle G, Anvret M, Sanal O, Ersoy F, Caglar M, Klein G. 1981. Epstein-Barr-virus-carrying lymphoma in a patient with ataxia-telangiectasia. Br Med J 282:425–427. doi: 10.1136/bmj.282.6262.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Zvi A, Soffer D, Yatziv S. 1978. Disseminated herpes simplex virus infection in ataxia-telangiectasia. Acta Paediatr Scand 67:667–670. doi: 10.1111/j.1651-2227.1978.tb17821.x. [DOI] [PubMed] [Google Scholar]
- 18.Kulinski JM, Leonardo SM, Mounce BC, Malherbe LP, Gauld SB, Tarakanova VL. 2012. Ataxia-telangiectasia mutated kinase controls chronic gammaherpesvirus infection. J Virol 86:12826–12837. doi: 10.1128/JVI.00917-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulinski JM, Darrah EJ, Broniowska KA, Mboko WP, Mounce BC, Malherbe LP, Corbett JA, Gauld SB, Tarakanova VL. 2015. ATM facilitates mouse gammaherpesvirus reactivation from myeloid cells during chronic infection. Virology 483:264–274. doi: 10.1016/j.virol.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman CB, Nealy MS, Tibbetts SA. 2010. Immature and transitional B cells are latency reservoirs for a gammaherpesvirus. J Virol 84:13045–13052. doi: 10.1128/JVI.01455-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins CM, Boss JM, Speck SH. 2009. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J Virol 83:6484–6493. doi: 10.1128/JVI.00297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol 165:1074–1081. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- 23.Flano E, Kim IJ, Woodland DL, Blackman MA. 2002. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J Exp Med 196:1363–1372. doi: 10.1084/jem.20020890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog 5:e1000677. doi: 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willer DO, Speck SH. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J Virol 77:8310–8321. doi: 10.1128/JVI.77.15.8310-8321.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zha S, Sekiguchi J, Brush JW, Bassing CH, Alt FW. 2008. Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc Natl Acad Sci U S A 105:9302–9306. doi: 10.1073/pnas.0803520105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rickert RC, Roes J, Rajewsky K. 1997. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res 25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, Morse HC III, Gearhart PJ, Wynshaw-Boris A, Max EE, Hodes RJ. 2004. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med 200:1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. 2004. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med 200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson A, Trip M, Warris A, van der Flier M, Wulffraat N, Verhagen MM, Taylor MA, van Zelm MC, van Dongen JJ, van Deuren M, van der Burg M. 2013. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. J. Allergy Clin Immunol 131:1367–1375. doi: 10.1016/j.jaci.2013.01.053. [DOI] [PubMed] [Google Scholar]
- 31.McClellan KB, Gangappa S, Speck SH, Virgin HW. 2006. Antibody-independent control of gamma-herpesvirus latency via B cell induction of anti-viral T cell responses. PLoS Pathog 2:e58. doi: 10.1371/journal.ppat.0020058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rekow MM, Darrah EJ, Mboko WP, Lange PT, Tarakanova VL. 2016. Gammaherpesvirus targets peritoneal B-1 B cells for long-term latency. Virology 492:140–144. doi: 10.1016/j.virol.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sindhava VJ, Bondada S. 2012. Multiple regulatory mechanisms control B-1 B cell activation. Front Immunol 3:372. doi: 10.3389/fimmu.2012.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hastings WD, Tumang JR, Behrens TW, Rothstein TL. 2006. Peritoneal B-2 cells comprise a distinct B-2 cell population with B-1b-like characteristics. Eur J Immunol 36:1114–1123. doi: 10.1002/eji.200535142. [DOI] [PubMed] [Google Scholar]
- 35.Weck KE, Kim SS, Virgin HW, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Ikuta K, Sixbey JW, Tibbetts SA. 2008. A replication-defective {gamma}-herpesvirus efficiently establishes long-term latency in macrophages but not B cells in vivo. J Virol 82:8500–8508. doi: 10.1128/JVI.00186-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sen G, Bikah G, Venkataraman C, Bondada S. 1999. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur J Immunol 29:3319–3328. [DOI] [PubMed] [Google Scholar]
- 38.Krug LT, Moser JM, Dickerson SM, Speck SH. 2007. Inhibition of NF-kB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog 3:97–118. doi: 10.1371/journal.ppat.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coleman CB, McGraw JE, Feldman ER, Roth AN, Keyes LR, Grau KR, Cochran SL, Waldschmidt TJ, Liang C, Forrest JC, Tibbetts SA. 2014. A gammaherpesvirus Bcl-2 ortholog blocks B cell receptor-mediated apoptosis and promotes the survival of developing B cells in vivo. PLoS Pathog 10:e1003916. doi: 10.1371/journal.ppat.1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamoto K, Lee BJ, Li C, Dubois RL, Hobeika E, Bhagat G, Zha S. 2015. Early B-cell-specific inactivation of ATM synergizes with ectopic CyclinD1 expression to promote pre-germinal center B-cell lymphomas in mice. Leukemia 29:1414–1424. doi: 10.1038/leu.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mounce BC, Tsan FC, Kohler S, Cirillo LA, Tarakanova VL. 2011. Dynamic association of gammaherpesvirus DNA with core histone during de novo lytic infection of primary cells. Virology 421:167–172. doi: 10.1016/j.virol.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mboko WP, Olteanu H, Ray A, Xin G, Darrah EJ, Kumar SN, Kulinski JM, Cui W, Dittel BN, Gauld SB, Tarakanova VL. 2015. Tumor suppressor IRF-1 counteracts germinal center reaction driven by a cancer-associated gammaherpesvirus. J Virol 90:2818–2829. doi: 10.1128/JVI.02774-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarakanova VL, Stanitsa E, Leonardo SM, Bigley TM, Gauld SB. 2010. Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo. Virology 405:50–61. doi: 10.1016/j.virol.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 44.Darrah EJ, Stoltz KP, Ledwith M, Tarakanova VL. 2017. ATM supports gammaherpesvirus replication by attenuating type I interferon pathway. Virology 510:137–146. doi: 10.1016/j.virol.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]