

ABSTRACT
Like other enteroviruses, enterovirus 71 (EV71) relies on phosphatidylinositol 4-kinase IIIβ (PI4KB) for genome RNA replication. However, how PI4KB is recruited to the genome replication sites of EV71 remains elusive. Recently, we reported that a host factor, ACBD3, is needed for EV71 replication by interacting with viral 3A protein. Here, we show that ACBD3 is required for the recruitment of PI4KB to RNA replication sites. Overexpression of viral 3A or EV71 infection stimulates the interaction of PI4KB and ACBD3. Consistently, EV71 infection induces the production of phosphatidylinositol-4-phosphate (PI4P). Furthermore, PI4KB, ACBD3, and 3A are all localized to the viral-RNA replication sites. Accordingly, PI4KB or ACBD3 depletion by small interfering RNA (siRNA) leads to a reduction in PI4P production after EV71 infection. I44A or H54Y substitution in 3A interrupts the stimulation of PI4KB and ACBD3. Further analysis suggests that stimulation of ACBD3-PI4KB interaction is also important for the replication of enterovirus 68 but disadvantageous to human rhinovirus 16. These results reveal a mechanism of enterovirus replication that involves a selective strategy for recruitment of PI4KB to the RNA replication sites.
IMPORTANCE Enterovirus 71, like other human enteroviruses, replicates its genome within host cells, where viral proteins efficiently utilize cellular machineries. While multiple factors are involved, it is largely unclear how viral replication is controlled. We show that the 3A protein of enterovirus 71 recruits an enzyme, phosphatidylinositol 4-kinase IIIβ, by interacting with ACBD3, which alters cellular membranes through the production of a lipid, PI4P. Consequently, the viral and host proteins form a large complex that is necessary for RNA synthesis at replication sites. Notably, PI4KB-ACBD3 interaction also differentially mediates the replication of enterovirus 68 and rhinovirus 16. These results provide new insight into the molecular network of enterovirus replication.
KEYWORDS: enterovirus 68, enterovirus 71, PI4KB-ACBD3, replication
INTRODUCTION
Previous work suggested that replication of enterovirus 71 (EV71) is dependent on phosphatidylinositol 4-kinase IIIβ (PI4KB), although the regulatory mechanism is unclear (1–3). PI4KB catalyzes the synthesis of phosphatidylinositol-4-phosphate (PI4P), a lipid involved in signal transduction and vesicle transport to the plasma membrane (1). PI4KB is normally recruited to the Golgi apparatus by ARF1, a small GTPase that cycles between cytosolic and membrane-associated forms (4, 5). Once activated by the large guanine nucleotide exchange factor GBF1, ARF1 induces the recruitment of effectors, such as PI4KB, to the Golgi membranes (6, 7). In cells infected with enteroviruses, PI4KB, as well as PI4P, is enriched in the replication organelles or sites. This microenvironment provides a docking site for the 3D polymerase to initiate genomic replication (1). Several lines of evidence suggest that GBF1, required for viral RNA replication, localizes to replication organelles in cells infected with poliovirus and coxsackievirus B3 (CVB3) (8, 9). Furthermore, the 3A proteins of several enteroviruses bind to GBF1 (6, 8, 10). A postulated model suggests that 3A recruits PI4KB through its interaction with ARF1 or GBF1 and thereby facilitates viral replication.
PI4KB also contributes to the replication of Aichi virus, a member of the genus Kobuvirus (2, 11–13). Upon infection with Aichi virus, PI4KB is recruited to the replication sites by nonstructural proteins via host acyl-coenzyme A (acyl-CoA)-binding protein domain 3 (ACBD3), a Golgi apparatus-resident protein (12). Knockdown of ACBD3 inhibits Aichi virus replication, suggesting a link of ACBD3 to viral infection. While incompletely characterized, ACBD3 participates in a variety of processes, from lipid transport to apoptosis (14–16). In addition to Aichi virus, the 3A proteins from bovine kobuvirus, human rhinovirus (HRV), poliovirus, and coxsackieviruses associate with ACBD3 and PI4KB (17–19). On the other hand, the 3A protein of EV71 is reported to be unable to bind ACBD3 (17). Based on these observations, ACBD3 is proposed to mediate the recruitment of PI4KB and to regulate enterovirus replication. However, depletion of ACBD3 does not inhibit the replication of rhinovirus or poliovirus (20). Further complicating matters, knockdown of ACBD3, ARF1, and GBF1 has no inhibitory effect on coxsackievirus B3 replication (21). The mechanisms of enterovirus replication remain unresolved.
Recently, we showed that ACBD3 is needed for EV71 replication by interacting with viral 3A protein (22). Here, we report that EV71 3A promotes the recruitment of PI4KB through ACBD3 to replication sites, forming a large complex that contains the 3D polymerase. We show that ACBD3 is also indispensable for the replication of EV68, but not human rhinovirus 16. Our results demonstrate that enterovirus 3A selectively utilizes ACBD3 to recruit PI4KB to the replication organelles, which facilitates PI4P production and subsequent viral RNA replication.
RESULTS
Inhibition of PI4KB impedes EV71 replication.
To investigate the link of PI4KB to EV71, we determined viral RNA replication in the presence or absence of PI4KB inhibitors, which include PIK93 (23), enviroxime (24), and GW5074 (2). The data in Fig. 1A show that EV71 RNA replication increased as infection progressed in control cells. Treatment with PIK93, enviroxime, or GW5074 clearly reduced viral RNA replication. Under these conditions, PI4KB inhibitors had no toxic effect (Fig. 1B). To specifically assess the role of PI4KB, we performed small interfering RNA (siRNA) knockdown assays. As shown in Fig. 1C, addition of siRNA-PI4KB resulted in a decrease of EV71 RNA compared to the control, indicating a requirement for PI4KB in EV71 replication. Western blot analysis showed that PI4KB expression was effectively reduced by PI4KB siRNA but not scrambled siRNA (Fig. 1D). These phenotypes were not due to a toxic effect (Fig. 1E). Consistent with this, siRNA knockdown of PI4KB reduced the efficiency of virus production (Fig. 1F). In addition, as shown in Fig. 1G, siRNA knockdown of PI4KB expression also inhibited RNA replication of other EV71 strains (22). These results suggest that PI4KB is critically required for EV71 replication.
FIG 1.
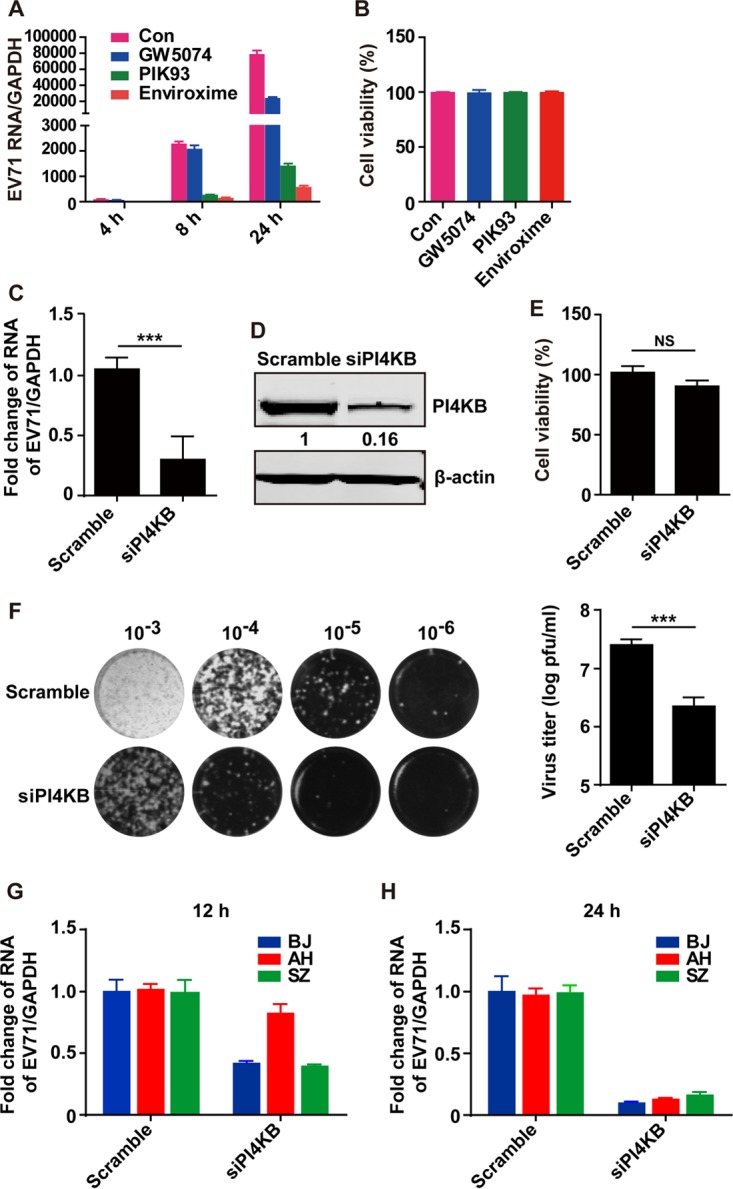
PI4KB plays a critical role in EV71 replication. (A) Inhibitory effects of GW5074, PIK93, and enviroxime on EV71 replication. RD cells were pretreated with GW5074 (3 μM), PIK93 (0.25 μM), or enviroxime (0.5 μg/ml) for 4 h. Then, the cells were infected with EV71 at an MOI of 1 PFU/cell. At 4, 8, and 24 h, cells were harvested and total RNA was extracted. The levels of EV71 RNA were evaluated by quantitative real-time PCR using SYBR green. The data are expressed as fold change of the EV71 RNA level relative to the control (Con) using the ΔΔCT method. The data are mean values and standard errors of the mean (SEM) (n = 3). (B) The effects of the inhibitors on cell viability were detected by using a Cytotoxicity Detection Kit Plus (LDH). (C) RD cells were transfected with siRNA against PI4KB. At 48 h after transfection, the cells were treated with EV71 at an MOI of 1 PFU/cell. Total RNA was extracted 24 h after infection, and the EV71 RNA levels were evaluated by quantitative real-time PCR using SYBR green. (D) Detection of PI4KB levels by Western blotting assay. Important blots were quantified with ImageJ software. (E) Effects of siPI4KB on cell viability. (F) RD cells were treated as for panel C. EV71 replication was detected by plaque assay as described in Materials and Methods. (G and H) RD cells were transfected with siRNA against PI4KB. At 48 h, the cells were infected with different EV71 strains as indicated. After 12 (G) or 24 (H) h, total RNA was extracted, and the EV71 RNA levels were evaluated by quantitative real-time PCR using SYBR green. ***, P < 0.001; NS, not significant.
EV71 3A enhances the binding of ACBD3 to PI4KB.
In cells infected with Aichi virus, ACBD3 forms a complex with PI4KB (11–13, 17). Our previous study showed that ACBD3 is also necessary for EV71 replication (22). To determine whether EV71 3A modulates the interaction between ACBD3 and PI4KB, 293T cells were transfected with plasmids expressing green fluorescent protein (GFP)-3A, Flag-ACBD3, and Myc-PI4KB. After 24 h, the cell lysates were immunoprecipitated with antibody against Flag tag. As shown in Fig. 2A, although expressed at comparable levels, ACBD3 alone bound to PI4KB weakly (lane 1). Coexpression of EV71 3A led to an increase in the binding of ACBD3 to PI4KB (lane 2), suggesting that EV71 3A facilitates the interaction of ACBD3 and PI4KB in the absence of other viral proteins. We further analyzed protein-protein interaction in virus-infected cells. As shown in Fig. 2B, in mock-infected human rhabdomyosarcoma (RD) cells, a smaller amount of ACBD3 precipitated with PI4KB when using anti-PI4KB antibody (lane 1). However, EV71 infection increased the amount of ACBD3 that precipitated with PI4KB (lane 2). This phenomenon was not cell line specific, as it was also detected in HeLa cells (Fig. 2C). Reverse coimmunoprecipitation assays using anti-ACBD3 antibody confirmed that EV71 infection stimulated the interaction between PI4KB and ACBD3 in RD (Fig. 2D) and HeLa (Fig. 2E) cells. The reason for a weaker signal associated with precipitated PI4KB is unknown, but it is possibly due to spatial hindrance of anti-ACBD3 antibody. When visualized by confocal microscopy, PI4KB only partially colocalized with ACBD3 in mock-infected cells (Fig. 2F, top). However, EV71 infection directed PI4KB and ACBD3 to discrete cytoplasmic structures, with a notable fraction colocalized (Fig. 2F, bottom). These results suggest that EV71 3A promotes the formation of a stable ACBD3-PI4KB complex.
FIG 2.

3A promotes recruitment of PI4KB by ACBD3. (A) EV71 3A increases the interaction between ACBD3 and PI4KB. 293T cells were transfected with plasmids expressing Myc-PI4KB, Flag-ACBD3 along with GFP, or GFP-3A as indicated. After 24 h, the cell lysates were immunoprecipitated (IP) using anti-Flag, followed by Western blotting using the indicated antibodies. IB, immunoblotting. WCL, whole-cell lysates. (B and C) RD (B) or HeLa (C) cells were mock infected or infected with EV71 at an MOI of 1 PFU/cell. At 24 h after infection, the lysates were immunoprecipitated with antibody against PI4KB. Samples were analyzed by Western blotting using the indicated antibodies. (D and E) Cells were treated as for panels B and C. The cell lysates were immunoprecipitated with antibody against ACBD3. (F) RD cells were mock infected or infected with EV71 at an MOI of 5 PFU/cell. After 8 h, the cells were fixed and stained with anti-ACBD3 (red) and anti-PI4KB (green) antibodies. The images were obtained using laser scanning confocal microscopy. The data shown are representative of the results of three independent experiments.
EV71 stimulates PI4P synthesis that is dependent on PI4KB and ACBD3.
Since EV71 replication relies on PI4KB, we reasoned that infection with EV71 might stimulate the synthesis of PI4P that remodels membranes for RNA replication. As such, we determined the effect of EV71 on PI4P production. RD cells were mock infected or infected for 8 h, and the levels of PI4P were determined by flow cytometry. As illustrated in Fig. 3A, although PI4P was detectable in mock-infected cells, its level was apparently upregulated in EV71-infected cells. To examine whether PI4P was localized to the replication sites upon infection, we carried out immunofluorescence analysis. As shown in Fig. 3B, in mock-infected cells, PI4P was weakly detectable where aggregates were seen. In EV71-infected cells, PI4P was enriched in the cytoplasm where 3A was present. These results suggest that EV71 induces the production of PI4P at the replication organelles. Interestingly, a large fraction of PI4P colocalized with EV71 3A. To define whether EV71 affected PI4P via PI4KB or ACBD3, we silenced the expression of PI4KB or ACBD3 in RD cells. As indicated by flow cytometry analysis (Fig. 3C), scrambled siRNA alone had no effect on PI4P, whereas EV71 stimulated its production. However, such upregulation of PI4P was precluded in cells treated with siRNA against PI4KB or ACBD3. Therefore, these results suggest EV71 upregulates PI4P production that is dependent on PI4KB and ACBD3.
FIG 3.

EV71 induces PI4P production. (A) RD cells were mock infected or infected with EV71. At 8 h after infection, the cells were fixed and stained with antibodies specific for PI4P. The cells were analyzed using flow cytometry. The data are mean values and SEM. (B) Localization of PI4P with 3A. RD cells were mock infected or infected with EV71 for 8 h. The cells were fixed and labeled with antibodies against PI4P (red) and 3A (green). (C) Effects of PI4KB and ACBD3 on PI4P production. RD cells were transfected with siACBD3 or siPI4KB. After 48 h, the cells were mock infected or infected with EV71 for 8 h. The cells were labeled with anti-PI4P antibody and anti-EV71 antibody and then analyzed by flow cytometry. PE, phycoerythrin. ***, P < 0.001.
PI4KB, ACBD3, and viral 3A are localized to RNA replication sites.
In cells infected with enteroviruses, viral and host proteins are thought to replicate the viral genome at discrete sites (25, 26). To examine the replication machinery, we visualized the localization of EV71 RNA and 3A, ACBD3, or PI4KB protein by confocal microscopy. Wild-type RD cells were mock infected or infected with EV71 at a multiplicity of infection (MOI) of 5 PFU/cell. At 8 h postinfection, the cells were processed and stained with antibodies against double-stranded RNA (dsRNA), 3A, ACBD3, and PI4KB. As shown in Fig. 4A, unlike mock-infected cells, both viral RNA and 3A were readily seen within the cytoplasm of EV71-infected cells, where viral RNA formed punctate patterns. A fraction of EV71 RNA colocalized with 3A, which suggests the formation of active replication sites. Although centered on the Golgi apparatus in mock-infected cells, ACBD3 was relocalized to the replication sites, with some colocalized with viral RNA (Fig. 4B). Not surprisingly, PI4KB colocalized with viral RNA in EV71-infected cells (Fig. 4C). Strikingly, when ACBD3 was deleted, a different phenotype was noted. In ACBD3−/− cells infected with EV71, PI4KB spread out in the cytoplasm (Fig. 5A), which resembled a pattern seen in uninfected cells (Fig. 4C). 3A in ACBD3−/−cells diffused as puncta that were no longer colocalized with PI4KB (Fig. 5B). Little viral RNA was detectable in ACBD3−/− cells, suggesting a block in the formation of RNA replication sites. In fact, ACBD3 knockout resulted in a diffused pattern of EV71 3A and PI4P (Fig. 5C), which differed from that seen in wild-type cells (Fig. 3B). Therefore, ACBD3 is essential for recruitment of PI4KB to the RNA replication sites and production of PI4P in EV71-infected cells.
FIG 4.
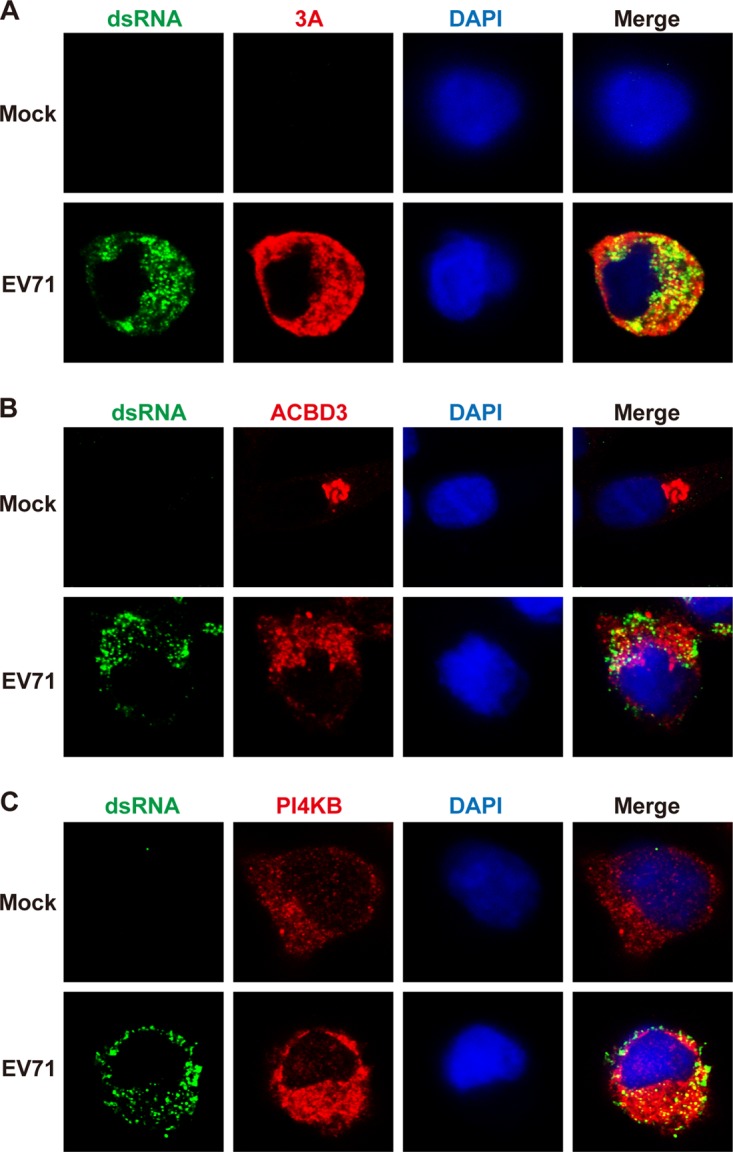
ACBD3 is necessary for PI4KB localization in replication centers. (A) RD cells were mock infected or infected with EV71 at an MOI of 5 PFU/cell. After 8 h, the cells were fixed and labeled with anti-dsRNA (MAb J2) (green) and anti-3A (red) antibodies. (B and C) RD cells were treated as for panel A. The cells were labeled with anti-dsRNA (MAb J2) (green) and anti-ACBD3 (red) (B) or anti-PI4KB (red) (C) antibodies.
FIG 5.
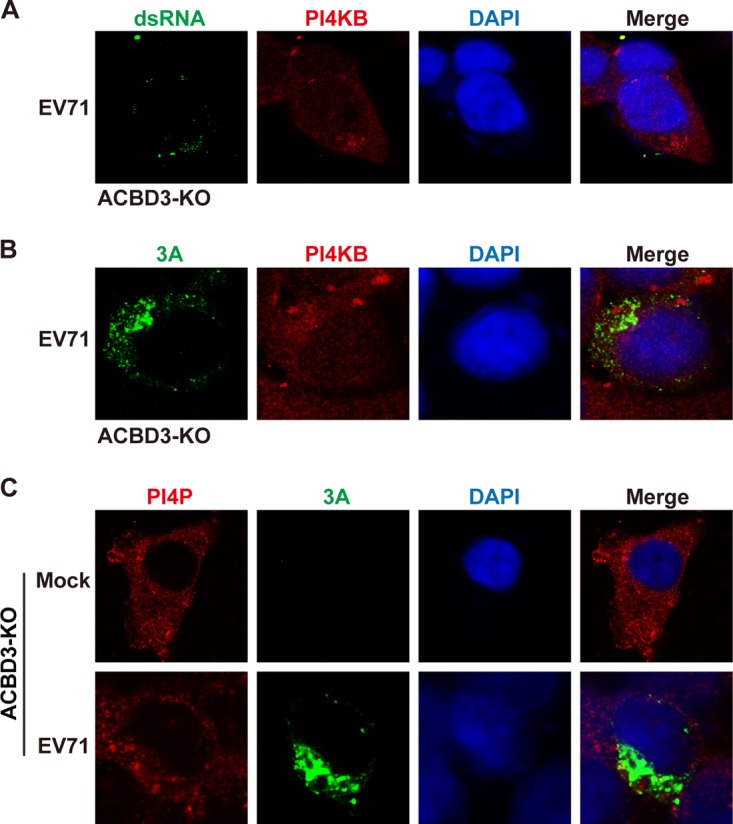
Effect of ACBD3 on localization of PI4KB. (A and B) ACBD3 knockout (KO) cells were infected with EV71 at an MOI of 5 PFU/cell. After 8 h, the cells were fixed and labeled with anti-PI4KB (red) and anti-dsRNA (green) (A) or anti-3A (red) (B) antibodies. (C) ACBD3-KO cells were treated as for panels A and B. At 8 h after infection, the cells were fixed and labeled with antibodies against PI4P (red) and 3A (green).
EV71 3A assembles a large protein complex via ACBD3.
To explore the way in which EV71 3A promotes viral replication, we examined whether ACBD3, PI4KB, and EV71 3A and 3D are localized in a complex by size exclusion chromatography (SEC) assay. RD cells were mock infected or infected with EV71 for 12 h. Cell lysates were fractionated on a Superdex 200 10/300 GL column and then analyzed by Western blotting. As illustrated in Fig. 6A, in mock-infected cells, PI4KB and ACBD3 coeluted, with the elution peaks centered around fractions 9 to 12, which corresponded to a size of approximately 240 kDa. Hence, ACBD3 and PI4KB formed a complex in the absence of EV71 infection. However, upon EV71 infection, a significant amount of ACBD3 migrated into higher-molecular-mass fractions (Fig. 6B), with a second peak corresponding to a size over 600 kDa. In parallel, PI4KB and EV71 3D, a polymerase, migrated to the higher-molecular-mass fractions. This correlated well with a distribution of EV71 3A or 3AB. These results strongly suggest that EV71 3A, 3D, ACBD3, and PI4KB form a higher-order protein complex in EV71-infected cells.
FIG 6.

EV71 3A, 3D, ACBD3, and PI4KB form a higher-order protein complex in EV71-infected cells. RD cells were mock infected (A) or infected with EV71 (B) for 12 h. Cell lysates were fractionated on a size exclusion column using Superdex 200 10/300 GL. Each fraction was analyzed by Western blotting using the indicated antibodies. The positions corresponding to the elution of the standard markers of molecular mass are indicated.
Isoleucine 44 and histidine 54 of 3A are functionally important.
Our previous results showed that isoleucine 44 and histidine 54 are important for the interaction of EV71 3A and ACBD3 (22). As ACBD3 directly binds to PI4KB (12), we asked whether 3A mutations affected the interaction of ACBD3 and PI4KB. As shown in Fig. 7, ACBD3 bound to PI4KB weakly (lane 1). Addition of wild-type 3A enhanced the binding of ACBD3 to PI4KB (lane 2). This effect was also observed in the presence of L12I (lane 3). In contrast, addition of I44A or H54Y did not have any effect compared to the control (lane 4 and 5). Hence, isoleucine 44 and histidine 54 are necessary to stabilize the interaction of ACBD3 and PI4KB. This is consistent with the efficiency of viral replication (22). These results suggested that the 3A-ACBD3-PI4KB complex is critically important for EV71 replication.
FIG 7.
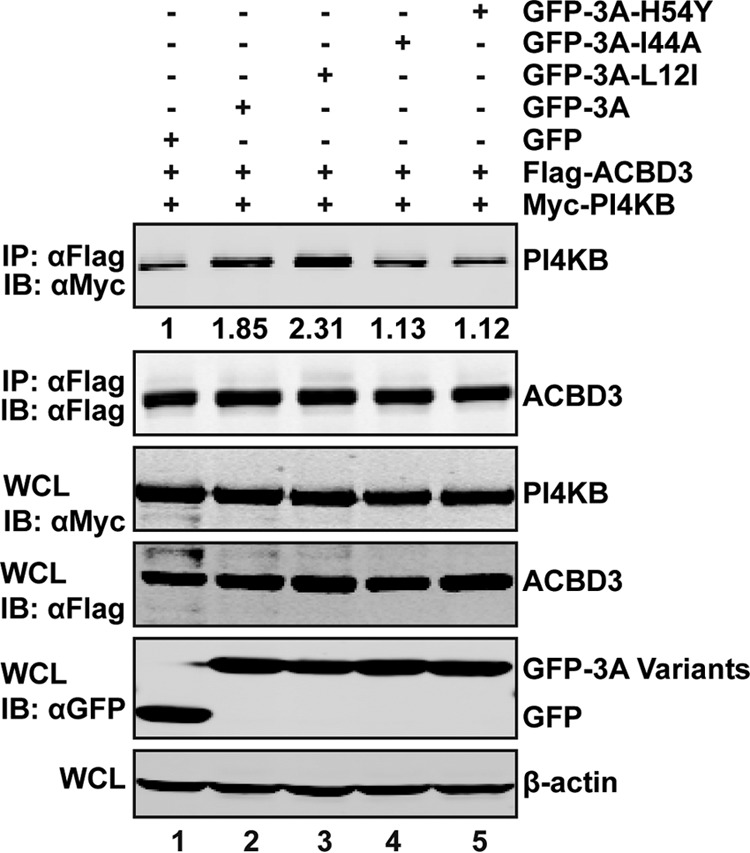
Role of 3A-ACBD3 interaction in forming the 3A-ACBD3-PI4KB complex. 293T cells were transfected with Flag-ACBD3 and Myc-PI4KB plasmids, along with 3A and its mutants as indicated. The immunoprecipitations were done by using anti-Flag antibody as described in the text.
ACBD3 is required for EV68 but not HRV16 replication.
To further explore PI4KB and ACBD3, we investigated their impacts on replication of other enteroviruses. We tested EV68, a member of the EV-D species, about whose replication little is known. As shown in Fig. 8A, siRNA knockdown of ACBD3 reduced viral RNA replication by 60% compared to control siRNA. Indeed, when ACBD3 was knocked out, viral RNA replication was almost abrogated (Fig. 8B). Similarly, viral protein production was barely detectable (Fig. 8C), indicating a functional role of ACBD3 in EV68 replication. We next assessed ACBD3 in HRV16 replication. Figure 8D shows that knockdown of ACBD3 by siRNA did not inhibit RNA replication of HRV16. Instead, viral replication was notably enhanced. This was further confirmed in ACBD3−/− cells infected with HRV16 (Fig. 8E and F), suggesting that ACBD3 is dispensable for HRV16 replication.
FIG 8.
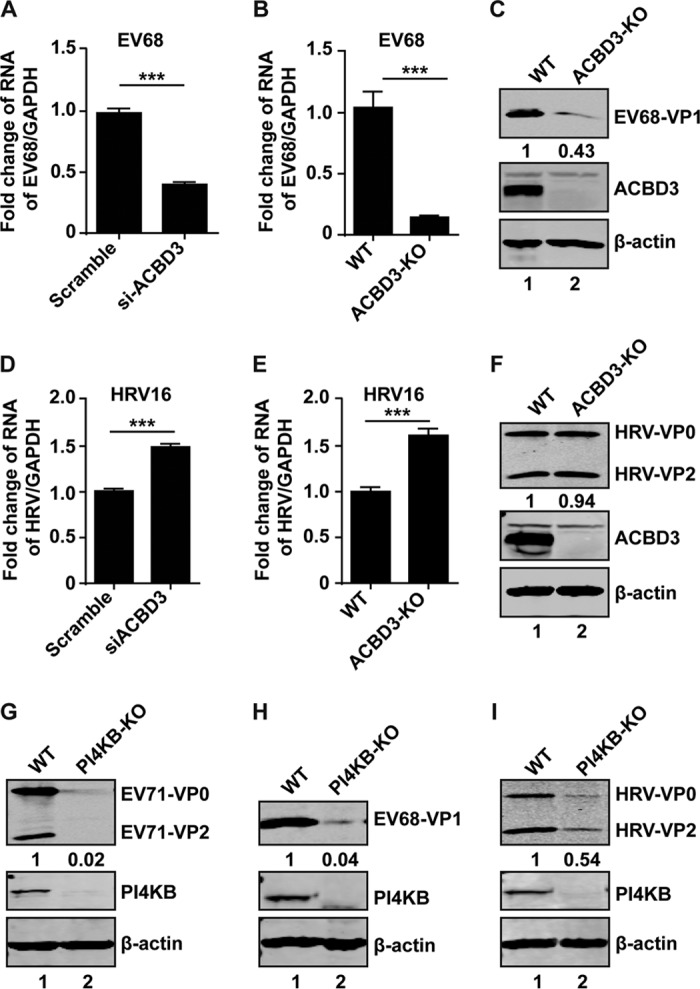
Effects of ACBD3 and PI4KB on replication of EV68 and HRV16. (A) RD cells were transfected with siRNA against ACBD3. After 48 h, the cells were infected with EV68 at an MOI of 1. At 24 h, EV68 replication was detected by reverse-transcription (RT)-PCR. (B) RD ACBD3-KO cells were infected with EV68 at an MOI of 1. At 24 h, EV68 replication was detected by RT-PCR. (C) RD ACBD3-KO cells were treated as described for panel B. At 24 h after infection, the cells were harvested and detected by Western blotting assays. WT, wild type. (D) HeLa cells were transfected with siRNA against ACBD3. After 48 h, the cells were infected with HRV16 at an MOI of 2. At 24 h, the replication of HRV16 was detected by RT-PCR assay. (E) HeLa ACBD3-KO cells were infected with HRV16 at an MOI of 2 PFU/cell. At 24 h, HRV16 replication was detected by RT-PCR. (F) HeLa ACBD3-KO cells were treated as described for panel E. At 24 h after infection, the cells were harvested and detected by Western blotting assay. (G to I) RD PI4KB-KO cells were infected with EV71 (G), EV68 (H), or HRV16 (I). At 24 h after infection, the cells were harvested and detected by Western blotting assay. ***, P < 0.001. The data are mean values and SEM.
To define the role of PI4KB in viral replication, we generated a PI4KB−/− cell line by clustered regularly interspaced short palindromic repeat (CRISPR)-mediated disruption. As illustrated in Fig. 8G, EV71 proteins were detected in wild-type cells. However, they were severely reduced in PI4KB−/− cells. This is consistent with the knockdown result in Fig. 1C. Similarly, depletion of PI4KB reduced protein expression of EV68 and HRV16 (Fig. 8H and I), suggesting PI4KB is important for both EV68 and HRV16 replication.
EV68 3A interacts with ACBD3 and stimulates PI4KB-ACBD3 interaction.
To clarify the different requirements for ACBD3, we analyzed 3A proteins from EV71, EV68, and HRV16. 293T cells were transfected with ACBD3, along with 3A from EV71, EV68, and HRV16. At 24 h postinfection, lysates of the cells were subjected to immunoprecipitation. As illustrated in Fig. 9A, EV71 3A, as well as EV68 3A, readily precipitated with ACBD3. However, 3A of HRV16 minimally precipitated with ACBD3. We next tested the impact of 3A on the binding of ACBD3 to PI4KB in transfected 293T cells (Fig. 9B). While ACBD3 formed a complex with PI4KB (lane 1), coexpression of EV71 3A or EV68 3A increased the interaction of ACBD3 and PI4KB (lanes 2 and 3). Therefore, the capacity of 3A to stabilize the ACBD3-PI4KB interaction was functionally linked to replication of EV71 and EV68. Interestingly, coexpression of HRV16 3A did not enhance the binding of ACBD3 to PI4KB, suggesting that HRV16 3A promotes viral replication independently of ACBD3. These results indicated that PI4KB-ACBD3 is necessary for EV71 and EV68 replication, but not that of HRV16.
FIG 9.

EV71 or EV68 3A interacts with ACBD3 and promotes its interaction with PI4KB. (A) Interaction of ACBD3 with 3A of EV71, EV68, or HRV16. 293T cells were transfected with Flag-ACBD3, along with 3A as indicated. After 24 h, the cell lysates were immunoprecipitated with antibody against Flag. Samples were then subjected to Western blot assay with antibodies against Flag, GFP, and β-actin. (B) EV71 3A and EV68 3A, but not HRV16 3A, promote recruitment of PI4KB by ACBD3. 293T cells were transfected with plasmids expressing Myc-PI4KB or Flag-ACBD3, along with GFP or GFP-3A as indicated. After 24 h, the cell lysates were immunoprecipitated using anti-Flag, followed by Western blotting using the indicated antibodies.
DISCUSSION
Recently, we reported that 3A protein of EV71 mediated viral replication by interacting with the host factor ACBD3 (22). Here, we demonstrate that the interaction between EV71 3A and ACBD3 facilitates recruitment of PI4KB and the 3D polymerase for viral RNA replication. In addition, EV68, but not HRV16, utilizes ACBD3 for genome replication. These results support the concept that PI4KB-ACBD3 differentially contributes to the replication of enteroviruses.
PI4KB-ACBD3 is crucial in Aichi virus replication, yet its relationship with enteroviruses is a puzzle (12, 17, 20, 21, 27, 28). ACBD3 is not involved in the replication of rhinovirus 14 and coxsackievirus (20, 21). On the other hand, ACBD3 inhibits viral replication upon poliovirus infection (18). Previous studies suggested that replication of enterovirus 71 relies on PI4KB, but the underlying mechanism is unknown (1, 2, 29, 30). In cells treated with PI4KB inhibitors, EV71 replication was reduced (29, 30). We found that specific depletion of PI4KB also sharply impaired viral RNA replication and subsequent production of EV71. Indeed, the interaction of EV71 3A and ACBD3 is key to the recruitment of PI4KB. When infected with EV71, PI4KB was recruited to the discrete regions where viral RNA interacted with viral 3A, ACBD3, and PI4KB. This suggests that EV71 3A, ACBD3, and PI4KB form a functional complex that mediates viral RNA replication. In line with this model, EV71 3A, 3D, ACBD3, and PI4KB were detectable in a large complex. We suspect that a higher-order protein complex likely represents the functional replication machinery and that this complex rests on the interaction of EV71 3A and ACBD3 because depletion of ACBD3 prevented PI4KB recruitment and 3A no longer interacted with PI4KB in infected cells.
Our data indicate that enterovirus 71 stimulates the synthesis of PI4P in an ACBD3-dependent manner. This is attributable to the interplay of EV71 and ACBD3. We noted that EV71 3A directed PI4KB to discrete areas in infected cells. Depletion of ACBD3 impaired PI4P production, which correlated well with a defect in viral replication. Although ACBD3 bound to PI4KB in uninfected cells, such interaction was weak. Expression of EV71 3A or virus infection strongly increased their interaction. Conversely, a disruptive mutation in EV71 3A reversed the phenotype. A plausible explanation is that EV71 3A may stabilize the interaction of ACBD3 and PI4KB. Alternatively, it may act as a chaperone that is required to remodel membranes. Because membrane recruitment of PI4KB by ACBD3 increases its enzymatic activity (13, 30–32), we believe that by binding to ACBD3, viral 3A may alter its conformation, leading to PI4KB activation and PI4P production.
In addition to EV71, EV68 modulates ACBD3 for its RNA replication. Depletion of ACBD3 severely reduced replication of EV68, which further highlights a crucial role of ACBD3. On the other hand, ACBD3 was dispensable for HRV16 replication, which is similar to coxsackievirus B3 and rhinovirus 3A (20, 21). These phenotypes paralleled the ability of 3A to interact with ACBD3. We favor the argument that 3A-mediated stabilization of the ACBD3-PI4KB complex is responsible. An increase in RNA replication of HRV16 with ACBD3 knockdown or knockout suggests that the presence of ACBD3 may be disadvantageous to HRV16. This seems consistent with the observation in poliovirus and CVB3 replication in ACBD3 knockdown cells (18), where viral replication is stimulated. In HRV16 infection, PI4KB is likely recruited into RNA replication sites by other host factors, but not ACBD3 and GBF1 (20). Collectively, these results suggest that the role of ACBD3 in PI4KB recruitment and viral replication may depend on virus species and strains. Whether it is related to differences in the enterovirus 3A structure warrants further investigation.
In summary, enteroviruses remodel cellular membranes into specialized structures where viral replication complexes are assembled coordinately. This study highlights an essential role of ACBD3 in the replication of EV71 and EV68. However, ACBD3 is dispensable in cells infected with rhinovirus 16. This is probably relevant to the biology of different picornaviruses. It is likely that the ACBD3 pathway is selectively exploited by additional RNA viruses.
MATERIALS AND METHODS
Cell lines and viruses.
Human embryonic kidney HEK-293T cells, RD cells, and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone, Logan, UT), 100 U/ml penicillin, and 100 μg/ml streptomycin. All the cells were cultured at 37°C in a 5% CO2 humidified atmosphere. Enterovirus 71 infection was carried out as described previously (21, 33–35). The EV71 strains SHZH98 (GenBank accession no. AF302996.1), Anhui Fuyang-0805 (GenBank accession no. FJ439769.1), and BJ/CHN/2008 (GenBank accession no. HQ615421.1) were used in this study. EV68 (GenBank accession no. KF726085.1) infection was carried out as described previously (36). HRV16 was purchased from ATCC (ATCC VR-283).
Plasmids.
The plasmid pEGFP-3A and its variants have been described previously (22, 35). EV68 and HRV16 3A coding sequences were amplified from cDNA that was reverse transcribed from EV68 or HRV16 mRNA by PCR using a specific primer pair (EV68-3A, 5′-ATCGTAATCAAGCTTCGGGACCTCCACAGTTTAAAGAGATCA-3′ and 5′-TGATTCGATGTCGACGTCTTATTGGATACCAGCAAAAAGTTTATAT-3′; HRV16-3A, 5′-ATCGTAATCAAGCTTCGGGGCCTATATCCATGGATAAACC-3′ and 5′-TGATTCGATGTCGACGTCTTACTGTAGAGAGCAAAAGAGCTTAT-3′). Then, EV68-3A or HRV16-3A was cloned into the pEGFP-C1 vector using HindIII and SalI restriction sites. The plasmid expressing human ACBD3 was purchased from Origene (Rockville MD). The plasmid expressing human PI4KB was a gift from Zhang Leiliang (Institute of Pathogen Biology, Chinese Academy of Medical Sciences, Beijing, China) (37). The ACBD3 mutants were constructed by site-directed mutagenesis using Pfu DNA polymerase (Strategene, La Jolla, CA) (22).
Antibodies and reagents.
Anti-ACBD3 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma-Aldrich (St. Louis, MO). Anti-PI4KB antibody was purchased from Chemicon (Billerica, MA) and BD Biosciences (Lexington, KY). Mouse anti-enterovirus 71 was purchased from Chemicon (Billerica, MA). The mouse anti-dsRNA (monoclonal antibody [MAb] J2) antibody was obtained from English and Scientific Consulting. The monoclonal antibody against PI4P was obtained from Echelon. Primary rabbit or mouse EV71 3A antibody was described previously (22). Antibodies against Flag, GFP, Myc, and β-actin were purchased from Sigma-Aldrich (St. Louis, MO). Goat anti-mouse or -rabbit secondary antibodies were purchased from Li-Cor Inc. (Lincoln, NE). Alexa Fluor 488-conjugated anti-mouse IgG and anti-rabbit IgG antibodies and Alexa Fluor 549-conjugated anti-mouse IgG, anti-mouse IgM, and anti-rabbit IgG were purchased from Molecular Probes (Eugene, OR, USA).
siRNA-mediated gene silencing in RD and HeLa cells.
RD or HeLa cells were transfected with 40 nM siRNA oligonucleotides for 48 h using DharmaFect1 (Dharmacon). The siRNA target sequences (forward) were ACBD3, 5′-GGAUGCAGAUUCCGUGAUU-3′, and PI4KB, 5′-CCUUUAAGCUGACCACAGA-3′.
Generation of a CRISPR-Cas9 knockout cell line.
The target sequence used to generate a CRISPR-Cas9 knockout cell line was CCTGCTAAGTGTCATCACGG for human PI4KB. The knockout cell lines were constructed as described in our recent publication (22).
Reverse transcription-PCR.
Total RNA was extracted from cells by using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA samples were treated with DNase I (Pierce, Rockford, IL), and reverse transcription was carried out using a Superscript cDNA synthesis kit (Invitrogen) according to the manufacturer's instructions. cDNA samples were subjected to real-time PCR using SYBR green. The primers used were as previously described (22). Levels of mRNA were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA. The results are reported as fold change using the ΔΔCT method.
Plaque assay.
RD cells (2 × 105) were seeded in 24-well plates with growth medium (DMEM-10% FBS). The next day, 200-μl serial dilutions of EV71 stocks were added to the wells. Then, the plates were incubated at 37°C for 2 h, and 1 ml 1.2% Avicel (R-591; FMC) (1 volume of 2.4% Avicel with the same volume of 2× DMEM) per well was added to the plate. After 72 h, the cells were fixed and stained with crystal violet (22, 38).
Western blot analysis.
Cells were washed and lysed in RIPA buffer containing 150 mM NaCl, 25 mM Tris, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA with protease inhibitor cocktail (Roche, Indianapolis, IN). Aliquots of the cell lysates were electrophoresed on SDS-PAGE gels and transferred to a nitrocellulose membrane (Pall, Port Washington, NY). The membranes were blocked with 5% nonfat dry milk and then probed with the indicated primary antibodies at 4°C overnight. This was followed by incubation with the corresponding IRD Fluor 800-labeled IgG or IRD Fluor 680-labeled IgG secondary antibody (Li-Cor Inc., Lincoln, NE) for 1 h at room temperature. After washing, the membranes were scanned with the Odyssey infrared imaging system (Li-Cor, Lincoln, NE) at a wavelength of 700 to 800 nm, and the molecular sizes of the developed proteins were determined by comparison with prestained protein markers (Fermentas, Rockville, MD).
Immunoprecipitation.
Transfected cells were lysed with radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN). Lysates of the cells were incubated with primary antibodies in 500 μl RIPA buffer at 4°C overnight on a rotator in the presence of protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Immunocomplexes captured on the protein A/G agarose beads were fractionated by 8 to 15% SDS-PAGE and transferred to nitrocellulose membranes for analysis.
Fluorescence microscopy.
Immunofluorescence assays were done as described previously (39). Briefly, cells were fixed with 4% paraformaldehyde, followed by washing with phosphate-buffered saline (PBS), and permeabilized with 0.5% Triton X-100. Then, the cells were blocked and stained with primary antibodies, followed by staining with Alexa Fluor 488/594 secondary antibodies. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, MO). Fluorescence images were obtained and analyzed using a laser scanning confocal microscope (Leica TCS SP5).
SEC using Superdex 200 10/300 GL.
RD cells (6 × 107) were mock infected or infected with EV71 for 12 h. The cells were harvested and lysed in buffer comprised of 14 mM CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 150 mM NaCl, and 20 mM Tris-HCl (pH 7.4). The lysed cells were centrifuged at 10,000 × g for 10 min. The supernatants were filtered with 0.2-μm membrane filters and loaded onto a Superdex 200 10/300 GL column (GE Healthcare, Life Sciences). Elution was performed at 4°C and monitored by absorbance at 280 and 260 nm (21, 40). Every 0.5-ml fraction was collected. Each fraction was analyzed by Western blotting assay. Molecular weight was determined using standard proteins (Fermentas, Rockville, MD).
Flow cytometric analysis.
PI4P synthesis was assayed by flow cytometry using a BD FACSCanto II flow cytometer (BD Biosciences). Cells were harvested after EV71 infection and fixed with 4% formalin, followed by washing with PBS. The cells were then incubated with digitonin (20 nM) in PBS for permeabilization. After staining with anti-PI4P and anti-EV71 antibodies, the cells were analyzed by flow cytometry.
Statistics.
The two-tailed Student t test was used for two-group comparisons. P values of <0.05, <0.01, and <0.001 were considered significant.
ACKNOWLEDGMENTS
We thank Leiliang Zhang for providing the PI4KB-expressing plasmid.
This work was supported by grants from the National Natural Science Foundation of China (81672032, 81225014, and 31270200), the Program for Changjiang Scholars and Innovative Research Team in Universities (IRT13007), the Changjiang Scholars Program (J.W.), the CAMS Innovation Fund for Medical Sciences (2016-I2M-1-014), the Ten Thousands Talent Program (J.W.), the PUMC Youth Fund, the Fundamental Research Funds for the Central Universities (3332015007), and the U.S. National Institute of Allergy and Infectious Diseases (AI112755).
REFERENCES
- 1.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2011. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol 85:2364–2372. doi: 10.1128/JVI.02249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin Y, Lin L, Chen Y, Wu S, Si X, Wu H, Zhai X, Wang Y, Tong L, Pan B, Zhong X, Wang T, Zhao W, Zhong Z. 2014. Curcumin inhibits the replication of enterovirus 71 in vitro. Acta Pharm Sin B 4:284–294. doi: 10.1016/j.apsb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, lurisci C, Luini A, Corda D, De Matteis MA. 1999. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns (4,5) P2 on the Golgi complex. Nat Cell Biol 1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- 5.Altan-Bonnet N, Sougrat R, Lippincott-Schwartz J. 2004. Molecular basis for Golgi maintenance and biogenesis. Curr Opin Cell Biol 16:364–372. doi: 10.1016/j.ceb.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E. 2008. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog 4:e1000216. doi: 10.1371/journal.ppat.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wessels E, Duijsings D, Niu TK, Neumann S, Oorschot VM, de Lange F, Lanke KH, Klumperman J, Henke A, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev Cell 11:191–201. doi: 10.1016/j.devcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Wessels E, Duijsings D, Lanke KH, Melchers WJ, Jackson CL, van Kuppeveld FJ. 2007. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J Virol 81:5238–5245. doi: 10.1128/JVI.02680-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belov GA, Kovtunovych G, Jackson CL, Ehrenfeld E. 2010. Poliovirus replication requires the N-terminus but not the catalytic Sec7 domain of Arf GEF GBF1. Cell Microbiol 12:1463–1479. doi: 10.1111/j.1462-5822.2010.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wessels E, Duijsings D, Lanke KH, van Dooren SH, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. Effects of picornavirus 3A proteins on protein transport and GBF1-dependent COP-I recruitment. J Virol 80:11852–11860. doi: 10.1128/JVI.01225-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL. 2013. ACBD3 interaction with TBC1 domain 22 protein is differentially affected by enteroviral and kobuviral 3A protein binding. mBio 4:e00098–e00013. doi: 10.1128/mBio.00098-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J 31:754–766. doi: 10.1038/emboj.2011.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa-Sasaki K, Sasaki J, Taniguchi K. 2014. A complex comprising phosphatidylinositol 4-kinase IIIbeta, ACBD3, and Aichi virus proteins enhances phosphatidylinositol 4-phosphate synthesis and is critical for formation of the viral replication complex. J Virol 88:6586–6598. doi: 10.1128/JVI.00208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shinoda Y, Fujita K, Saito S, Matsui H, Kanto Y, Nagaura Y, Fukunage K, Tamura S, Kobayashi T. 2012. Acyl-CoA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER-Golgi membrane contact sites. FEBS Lett 586:3024–3029. doi: 10.1016/j.febslet.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Bang S, Park S, Shi H, Kim SF. 2015. Acyl-CoA-binding domain containing 3 modulates NAD+ metabolism through activating poly (ADP-ribose) polymerase 1. Biochem J 469:189–198. doi: 10.1042/BJ20141487. [DOI] [PubMed] [Google Scholar]
- 16.Fan J, Liu J, Culty M, Papadopoulos V. 2010. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog Lipid Res 49:218–234. doi: 10.1016/j.plipres.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J Virol 86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teoule F, Brisac C, Pelletier I, Vidalain PO, Jegouic S, Mirabelli C, Bessaud M, Combelas N, Autret A, Tangy F, Delpeyroux F, Blondel B. 2013. The Golgi protein ACBD3, an interactor for poliovirus protein 3A, modulates poliovirus replication. J Virol 87:11031–11046. doi: 10.1128/JVI.00304-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klima M, Toth DJ, Hexnerova R, Baumlova A, Chalupska D, Tykvart J, Rezabkova L, Sengupta N, Man P, Dubankova A, Humpolickova J, Nencka R, Veverka V, Balla T, Boura E. 2016. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci Rep 6:23641. doi: 10.1038/srep23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorobantu CM, Ford-Siltz LA, Sittig SP, Lanke KH, Belov GA, van Kuppeveld FJ, van der Schaar HM. 2015. GBF1- and ACBD3-independent recruitment of PI4KIIIbeta to replication sites by rhinovirus 3A proteins. J Virol 89:1913–1918. doi: 10.1128/JVI.02830-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorobantu CM, van der Schaar HM, Ford LA, Strating JR, Ulferts R, Fang Y, Belov G, van Kuppeveld FJ. 2014. Recruitment of PI4KIIIβ to coxsackievirus B3 replication organelles is independent of ACBD3, GBF1, and Arf1. J Virol 88:2725–2736. doi: 10.1128/JVI.03650-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei X, Xiao X, Zhang Z, Ma Y, Qi J, Wu C, Xiao Y, Zhou Z, He B, Wang J. 2017. The Golgi protein ACBD3 facilitates Enterovirus 71 replication by interacting with 3A. Sci Rep 7:44592. doi: 10.1038/srep44592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balla A, Tuymetova G, Toth B, Szentpetery Z, Zhao X, Knight ZA, Shokat K, Steinbach PJ, Balla T. 2008. Design of drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry 47:1599–1607. doi: 10.1021/bi7017927. [DOI] [PubMed] [Google Scholar]
- 24.Brown-Augsburger P, Vance LM, Malcolm SK, Hsiung H, Smith DP, Heinz BA. 1999. Evidence that enviroxime targets multiple components of the rhinovirus 14 replication complex. Arch Virol 144:1569–1585. doi: 10.1007/s007050050611. [DOI] [PubMed] [Google Scholar]
- 25.Greninger AL. 2015. Picornavirus-host interactions to construct viral secretory membranes. Prog Mol Biol Transl Sci 129:189–212. doi: 10.1016/bs.pmbts.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards AL, Soares-Martins JA, Riddell GT, Jackson WT. 2014. Generation of unique poliovirus RNA replication organelles. mBio 5:e00833-00813. doi: 10.1128/mBio.00833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Schaar HM, van der Linden L, Lanke KH, Strating JR, Purstinger G, de Vries E, de Haan CA, Neyts J, van Kuppeveld FJ. 2012. Coxsackievirus mutants that can bypass host factor PI4KIIIbeta and the need for high levels of PI4P lipids for replication. Cell Res 22:1576–1592. doi: 10.1038/cr.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibaut HJ, van der Schaar HM, Lanke KH, Verbeken E, Andrews M, Leyssen P, Neyts J, van Kuppeveld FJ. 2014. Fitness and virulence of a coxsackievirus mutant that can circumnavigate the need for phosphatidylinositol 4-kinase class III beta. J Virol 88:3048–3051. doi: 10.1128/JVI.03177-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strating JR, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, Leyssen P, van der Schaar HM, Lanke KH, Thibaut HJ, Ulferts R, Drin G, Schlinck N, Wubbolts RW, Sever N, Head SA, Liu JO, Beachy PA, De Matteis MA, Shair MD, Olkkonen VM, Neyts J, van Kuppeveld FJ. 2015. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep 10:600–615. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Schaar HM, Leyssen P, Thibaut HJ, de Palma A, van der Linden L, Lanke KH, Lacroix C, Verbeken E, Conrath K, Macleod AM, Mitchell DR, Palmer NJ, van de Poe H, Andrews M, Neyts J, van Kuppeveld FJ. 2013. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIbeta. Antimicrob Agents Chemother 57:4971–4981. doi: 10.1128/AAC.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPhail JA, Ottosen EH, Jenkins ML, Burke JE. 2017. The molecular basis of Aichi virus 3A protein activation of phosphatidylinositol 4 kinase IIIβ, PI4KB, through ACBD3. Structure 25:121–131. doi: 10.1016/j.str.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Klima M, Chalupska D, Różycki B, Humpolickova J, Rezabkova L, Silhan J, Baumlova A, Dubankova A, Boura E. 2017. Kobuviral non-structural 3A proteins act as molecular harnesses to hijack the host ACBD3 protein. Structure 25:219–230. doi: 10.1016/j.str.2016.11.021. [DOI] [PubMed] [Google Scholar]
- 33.Wu Z, Yang F, Zhao R, Zhao L, Guo D, Jin Q. 2009. Identification of small interfering RNAs which inhibit the replication of several Enterovirus 71 strains in China. J Virol Methods 159:233–238. doi: 10.1016/j.jviromet.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Lei X, Xiao X, Yang C, Lu W, Huang Z, Leng Q, Jin Q, He B, Meng G, Wang J. 2015. Reciprocal regulation between Enterovirus 71 and the NLRP3 inflammasome. Cell Rep 12:42–48. doi: 10.1016/j.celrep.2015.05.047. [DOI] [PubMed] [Google Scholar]
- 35.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84:8051–8061. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang Z, Li L, Lei X, Zhou H, Zhou Z, He B, Wang J. 2014. Enterovirus 68 3C protease cleaves TRIF to attenuate antiviral responses mediated by Toll-like receptor 3. J Virol 88:6650–6659. doi: 10.1128/JVI.03138-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong Z, Yang X, Yang G, Zhang L. 2014. Hepatitis C virus NS5A competes with PI4KB for binding to ACBD3 in a genotype-dependent manner. Antiviral Res 107:50–55. doi: 10.1016/j.antiviral.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Yin Y, Xu Y, Ou Z, Su L, Xia H. 2015. A simple and highly repeatable viral plaque assay for enterovirus 71. J Basic Microbiol 55:538–541. doi: 10.1002/jobm.201400330. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Z, Cao M, Guo Y, Zhao L, Wang J, Jia X, Li J, Wang C, Gabriel G, Xue Q, Yi Y, Cui S, Jin Q, Wang J, Deng T. 2014. Fragile X mental retardation protein stimulates ribonucleoprotein assembly of influenza A virus. Nat Commun 5:3259. doi: 10.1038/ncomms4259. [DOI] [PubMed] [Google Scholar]
- 40.Zhou Z, Jia X, Xue Q, Dou Z, Ma Y, Zhao Z, Jiang Z, He B, Jin Q, Wang J. 2014. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc Natl Acad Sci U S A 111:E245–E254. doi: 10.1073/pnas.1316941111. [DOI] [PMC free article] [PubMed] [Google Scholar]