

ABSTRACT
Epstein-Barr virus (EBV) establishes a stable latent infection that can persist for the life of the host. EBNA1 is required for the replication, maintenance, and segregation of the latent episome, but the structural features of EBNA1 that confer each of these functions are not completely understood. Here, we have solved the X-ray crystal structure of an EBNA1 DNA-binding domain (DBD) and discovered a novel hexameric ring oligomeric form. The oligomeric interface pivoted around residue T585 as a joint that links and stabilizes higher-order EBNA1 complexes. Substitution mutations around the interface destabilized higher-order complex formation and altered the cooperative DNA-binding properties of EBNA1. Mutations had both positive and negative effects on EBNA1-dependent DNA replication and episome maintenance with OriP. We found that one naturally occurring polymorphism in the oligomer interface (T585P) had greater cooperative DNA binding in vitro, minor defects in DNA replication, and pronounced defects in episome maintenance. The T585P mutant was compromised for binding to OriP in vivo as well as for assembling the origin recognition complex subunit 2 (ORC2) and trimethylated histone 3 lysine 4 (H3K4me3) at OriP. The T585P mutant was also compromised for forming stable subnuclear foci in living cells. These findings reveal a novel oligomeric structure of EBNA1 with an interface subject to naturally occurring polymorphisms that modulate EBNA1 functional properties. We propose that EBNA1 dimers can assemble into higher-order oligomeric structures important for diverse functions of EBNA1.
IMPORTANCE Epstein-Barr virus is a human gammaherpesvirus that is causally associated with various cancers. Carcinogenic properties are linked to the ability of the virus to persist in the latent form for the lifetime of the host. EBNA1 is a sequence-specific DNA-binding protein that is consistently expressed in EBV tumors and is the only viral protein required to maintain the viral episome during latency. The structural and biochemical mechanisms by which EBNA1 allows the long-term persistence of the EBV genome are currently unclear. Here, we have solved the crystal structure of an EBNA1 hexameric ring and characterized key residues in the interface required for higher-order complex formation and long-term plasmid maintenance.
KEYWORDS: DNA-binding domain, EBNA1, Epstein-Barr virus, OriP, cooperative binding, hexameric ring, oligomers, episome maintenance, viral latency
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous human herpesvirus that establishes long-term latent infection in more than 90% of the human population (1–6). Latent infection by EBV is estimated to be responsible for over 200,000 cancers worldwide, including subtypes of Burkitt's lymphoma, diffuse large B-cell lymphoma, primary central nervous system lymphoma, and Hodgkin's lymphoma as well as gastric carcinoma and nasopharyngeal carcinoma (NPC) (1–4). Epstein-Barr virus nuclear antigen 1 (EBNA1) is the only viral protein consistently expressed in all EBV-associated tumors and is essential for viral DNA replication and episome maintenance during the proliferation of latently infected cells (1–4, 6). EBNA1 has no known enzymatic activity, and how EBNA1 initiates and maintains the EBV genome is still not fully understood (2–4). The DNA-binding domain (DBD) of EBNA1 has been crystallized in the presence and absence of DNA (7, 8) and shares structural features with its orthologs, including latency-associated nuclear antigen (LANA) from Kaposi's sarcoma-associated herpesvirus (KSHV) and E2 from human papillomavirus (HPV) (7–15).
The DNA replication and episome maintenance functions of EBNA1 correspond to its ability to bind to different DNA elements within the EBV origin of plasmid replication (OriP). The family of repeats (FR) is composed of 20 tandem 30-bp repeats and is required for EBNA1 to perform metaphase chromosome tethering and transcriptional enhancer activities (2, 3, 16, 17). The dyad of symmetry (DS), located ∼1 kb from the FR, is composed of four EBNA1-binding sites and enables EBNA1 to initiate DNA replication (2, 3, 16, 17). The spacing and phasing of EBNA1-binding sites at the DS are known to be critical for cooperative binding and replication function (4, 18, 19). EBNA1 is also known to interact with both elements at the same time through a DNA-looping mechanism (20, 21). The cooperative binding of EBNA1 at OriP is likely to be critical for both replication and episome maintenance function.
EBNA1 is known to have multiple forms of cooperative binding at OriP. The dimer-dimer interaction at the DS is essential for the initiation of DNA replication, and this cooperativity is mediated, in part, through predicted interactions in the DNA-binding domain (8). Cooperative interactions that enable DNA looping between distal EBNA1 sites are known to be mediated through the amino-terminal linking domains of EBNA1, which are capable of forming homotypic Zn-hook interactions (22, 23). Cooperative interactions between EBNA1 DNA-binding domains at the FR are not yet known. Recently, a higher-order oligomeric form of the KSHV ortholog LANA was shown to mediate cooperative binding at the KSHV terminal repeats (9, 10, 15). LANA was found to form multiple different oligomeric assemblies, including a multimeric ring structure with an interior surface capable of incorporating double-stranded DNA (dsDNA) and proteins (9, 10, 15). Alternative oligomeric forms of viral origin-binding proteins, such as simian virus 40 (SV40) T antigen and papillomavirus E1, are known to correspond to changes in functional activities (24, 25). Although cooperative interactions of EBNA1 dimers with the DS element are predicted from structural studies, relatively little is known about the oligomeric state and function of EBNA1 complexes binding to the FR elements. Here, we solved a new crystal structure of the DNA-binding domain of EBNA1 that revealed a hexameric ring structure or trimer of dimers. We investigated the functional significance of higher-order EBNA1 oligomers using site-directed mutagenesis of residues at the trimer interface and examined the activity in several biochemical and cell-based assays. This study demonstrates the functional significance of the oligomeric state of EBNA1 in regions of the EBV genome important for plasmid replication and episome maintenance.
RESULTS
An X-ray crystal structure of the EBNA1 DBD shows the formation of a higher-order hexameric oligomer.
To gain further insights into the structural properties of the EBNA1 DBD and its potential interaction interfaces, we crystallized the EBNA1 DBD consisting of amino acids 468 to 619 (using strain B95-8 coordinates). EBNA1 (residues 468 to 619) crystallized in the P1 triclinic space group and was solved by molecular replacement using the structure reported under Protein Data Bank (PDB) accession no. 1VHI as the search model (7). The crystals diffracted to 1.9 Å, but a complete data set (>92% complete) was collected at 2.2 Å, and the structure was refined to an Rfree value of 0.275 (Table 1).
TABLE 1.
Crystallographic information
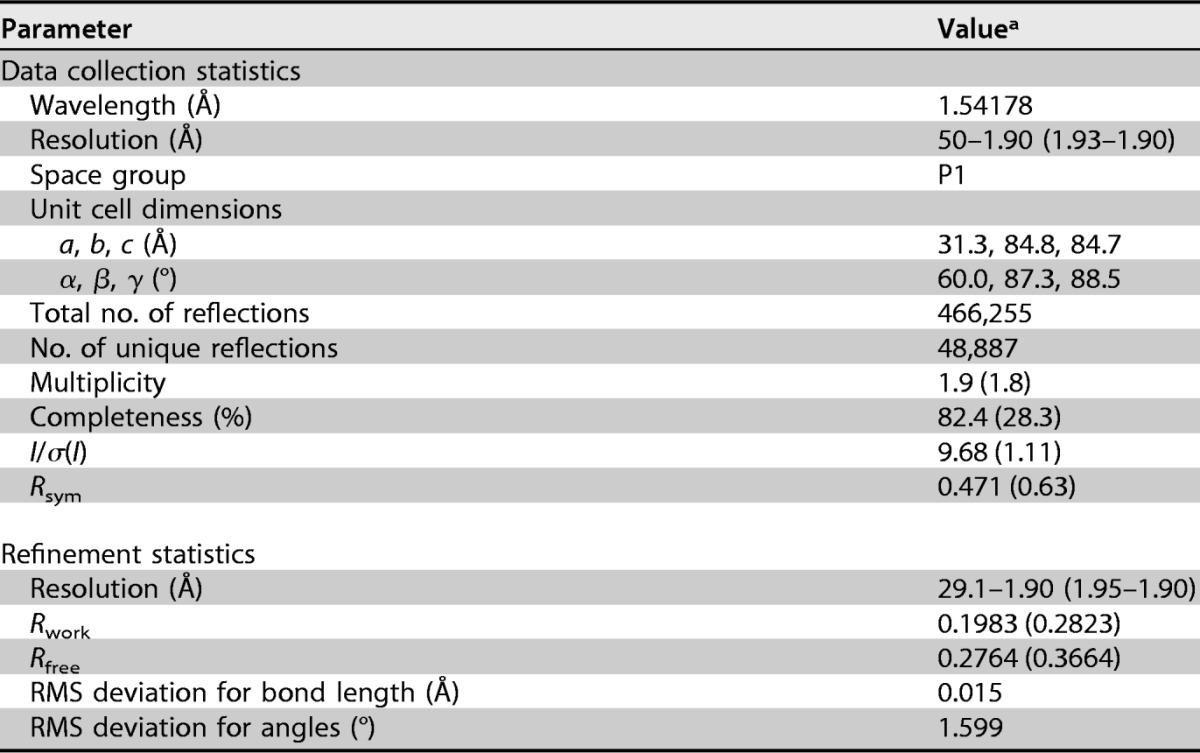
aValues in parentheses represent the highest-resolution shell.
The overall structure was different from those reported previously in that it consisted of a trimer of dimers forming a ring with the DNA-binding region aligning on one common side (Fig. 1A and G). The oligomeric (trimer) interface is composed primarily of amino acids Arg496, Gln530, Leu582, Met584, and Thr585 (Fig. 1B). The DNA-binding surface is positioned on the one surface of the hexagonal wheel enriched in basic amino acids implicated in DNA contacts (Fig. 1D and E). The opposing surface of the hexagonal wheel is enriched in acidic amino acids and may explain the electrostatic forces driving the stacking of hexagonal wheels (Fig. 1F and G).
FIG 1.

Crystal structure of the EBNA1 hexameric ring. (A) Structure showing that three EBNA1 dimers interact to form a hexameric ring. Each monomer is a different color and designated by a different chain ID in the PDB (accession no. 5WMF) (green, chain A; cyan, chain B; yellow, chain C; salmon, chain E; gray, chain F). (B) Closeup view of the trimer interface highlighted by a black box, with key residues labeled. (C) Closeup view of C-terminal residues (residues 608 to 612) stabilizing PGP loops, highlighted by a black-dashed box. (D) The surface electrostatic potential of the EBNA1 hexamer (red, acidic; blue, basic; white, neutral) shows the bottom basic patch of residues that would hypothetically interact with the DNA, and the top view (180° rotation) of the EBNA1 hexamer opposite the DNA-binding surface is generally acidic, which is opposite of what is seen with LANA. (E) Superposition of EBNA1 (PDB accession no. 1B3T) (8) with the EBNA1 dimer within a hexamer shows where DNA would bind in comparison to the location of the key interface residues, highlighted as black sticks. (F) Crystal structure of the double-EBNA1-hexamer stacking pattern showing the interaction of the basic region (blue) of one hexamer with the acidic region (red) of the apposing hexamer. (G) Same as panel E but with the superimposition of DNA (PDB accession no. 1B3T) (8) highlighting DNA binding to the basic patch of one of the two hexamers.
The dimer structures superimpose well on the DNA-bound structure (PDB accession no. 1B3T), with a root mean square (RMS) deviation for Cα of 0.36 Å (Fig. 2A). The dimers are largely superimposable upon each other, with RMS deviations of 0.32 Å (chains A and B compared with chains C and D) and 0.23 Å (chains A and B compared with chains E and F) (Fig. 2B). The most substantial differences between the dimers are found in the flexible but highly conserved winged-loop regions, termed PGP loops based on the amino acid composition (8) (Fig. 1C and 2B). The C terminus (residues 608 to 612) from one monomer stabilizes the PGP loop from the other monomer (Fig. 1C). The PGP loops largely fill in the hexameric middle region, as shown in an electrostatic surface representation (Fig. 1D and F). A complex network of residues is located at the interface of the trimers of a single hexagonal wheel as well as between hexagonal stacks, mediated largely by PGP loop packing between wheels.
FIG 2.
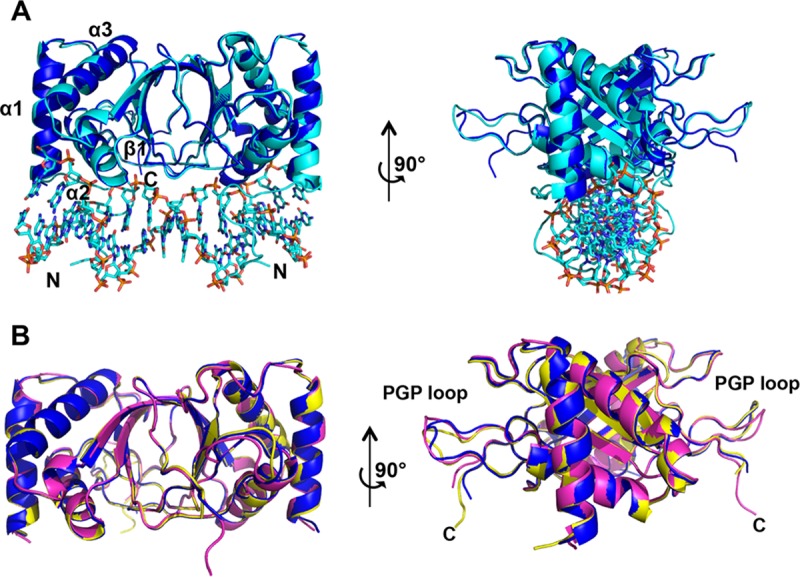
PGP loop variations of the EBNA1 hexamer. (A) Superposition of the EBNA1 dimer from the hexamer (chains A and B) (dark blue) with the EBNA1 dimer bound to DNA (cyan) (PDB accession no. 1B3T) and rotated 90°. (B) Comparison of all three EBNA1 dimer PGP loops within the context of the hexamer. Dark blue, chains A and B; yellow, chains C and D; magenta, chains E and F.
The trimer interface point mutants alter complex formation and DNA binding.
The EBNA1 trimer interface consists of residues Arg496, Gln530, Leu582, Met584, and Thr585 (Fig. 1B). The intricate hydrogen-bonding network reveals that the side-chain nitrogens of Arg496 hydrogen bond through water molecules with both the main-chain oxygens of Met584 and Thr585 of the adjacent EBNA1 dimer. Gln530 stabilizes Arg496 on the same EBNA1 molecule through both main-chain and side-chain interactions and additionally hydrogen bonds with Met584 on the adjacent EBNA1 dimer. The Leu582 main chain hydrogen bonds to stabilize with the Thr585 side-chain oxygen on the same molecule. Comparison with the EBNA1 DBD crystallized in the presence of DNA (PDB accession no. 1B3T) (7) shows that the EBNA1 trimer interface residues are not directly involved in DNA binding, and the DNA-binding surface is below the hexameric ring (Fig. 1E).
To investigate the role of the trimer interface in cooperative EBNA1-EBNA1 binding, we created and purified a series of interface point mutants of the EBNA1 DBD (positions 459 to 607) (R496A, R496E, Q530A, Q530E, L582P, L582F, T585A, and T585P) to compare complex formation and DNA binding with those of wild-type (WT) EBNA1 (B95-8) (Fig. 3). The mutants and WT EBNA1 migrated on denaturing gels, in agreement with the predicted molecular mass of the DBD of 16 kDa (Fig. 3A). Previous studies have shown that EBNA1 dimers can physically link to each other on multiple sites within the DS and FR to form a multi-EBNA1 complex (4, 26–28). We next performed cross-linking experiments with glutaraldehyde to link free amines in the absence of DNA. Cross-linked EBNA1 produced a laddering of complexes with the dominant species at 32 kDa, consistent with EBNA1 dimers (Fig. 3B). WT EBNA1 and most of the mutants formed cross-linked dimers of similar intensities, consistent with the fact that these hexamer interface mutations do not alter the EBNA1 dimer interface. WT EBNA1 formed larger complexes, including a collection of very large oligomers that required brief heating to enter the gel. Complexes marked as higher-order complexes included a band with an apparent molecular mass of 100 kDa, consistent with the predicted molecular mass of a hexamer (96 kDa). Larger, more diffuse complexes migrating at >100 kDa were observed for WT EBNA1. Interface point mutations, especially Q530E, L582P, L582F, T585A, and T585P, formed significantly fewer of these higher-order complexes than did WT EBNA1 (Fig. 3C). These findings suggest that EBNA1 can form higher-order complexes in solution and that mutations in the hexameric ring interface compromise the formation of higher-order complexes.
FIG 3.
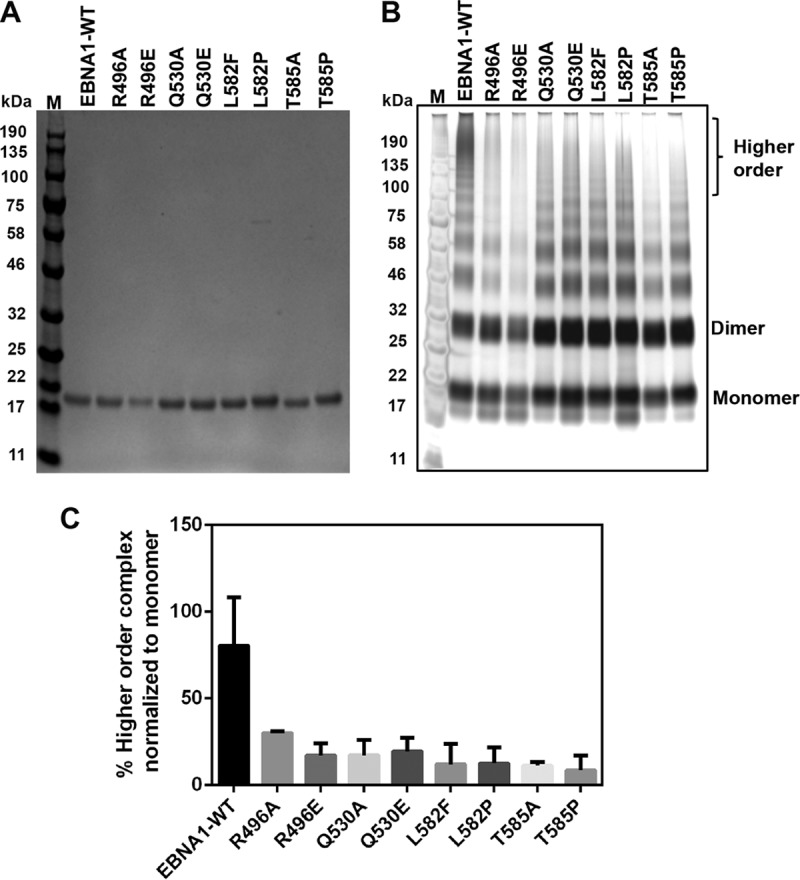
The EBNA1 hexameric interface modulates higher-order complex formation. (A) Tagless WT EBNA1 and mutants (residues 459 to 607) were purified to similar extents, as shown on the Coomassie-stained denaturing gel (left). (B) Silver staining shows increased levels of higher-order complexes stabilized in the presence of glutaraldehyde in WT EBNA1 versus the mutants. M, molecular marker. (C) Quantification of higher-order bands, indicated by brackets in panel B, normalized to the total signal for each respective lane in the cross-linked gel in panel A. The mutants were defective in higher-order complex formation, with a P value of <0.0001 by using one-way analysis of variance to compare the WT to all of the mutants.
To assess the role of the trimer interface in EBNA1 DNA binding, we evaluated the mutants for their ability to bind cooperatively to OriP DNA in electrophoretic mobility shift assays (EMSAs). Previous studies have shown cooperative binding for EBNA1 at the DS element (4), while other studies suggest cooperative binding to the FR (29, 30). We assayed the binding of the EBNA1 DBD to either a single 18-bp FR-binding site (1FR) or a 60-bp tandem pair of sites (2FR) (Fig. 4A and B). Comparison of the binding of the WT EBNA1 DBD to 1FR versus 2FR revealed cooperative binding properties for the paired sites from 2FR (Fig. 4B). The data were fitted by using a one-site binding model, and we found that the KD (equilibrium dissociation constant) for the WT EBNA1 DBD decreased 2-fold for one FR site in the presence of another FR site, and also, the Hill coefficient was >1, which is indicative of cooperative binding. EBNA1 hexamer interface mutants also bound FR DNA efficiently but with measurably different properties than those of the WT EBNA1 DBD (Fig. 4C to F). Interestingly, all the mutants except for L582P and T585A bound to the double-FR fragment with higher affinities than did the WT EBNA1 DBD (Fig. 4F). In agreement with data from our cross-linking studies and based on predicted structural disruptions in helix α3, the L582P mutant had a greatly reduced binding affinity. It should also be noted that the L582P mutant was not expressed efficiently in bacteria. However, the L582P mutant retained the ability to bind DNA albeit with a lower affinity than that of the WT EBNA1 DBD, indicating that this mutant retains sufficient structure to bind DNA and does not form random and unfolded aggregates.
FIG 4.

Hexameric interface mutants alter EBNA1 DNA-binding affinity and cooperativity for binding tandem sites at the FR. (A) EMSA measuring the WT EBNA1 DBD cooperatively binding to a double-FR DNA fragment (60-mer) at the indicated concentrations. (B) Quantification (right) showing that the EBNA1 DBD binds with high affinity to a single FR site in the context of two FR sites. (C) Mutants were titrated onto the 2FR DNA as described above for panel A and compared with the WT EBNA1 DBD. (D) Graph showing quantification of proteins bound to both FR sites. (E) Data were fit to a one-site-specific-binding model with a Hill slope in GraphPad Prism. KD values and Hill coefficients are displayed below the graph.
Mutations T585A and T585P resulted in two very different effects on DNA binding. The T585A mutant showed a DNA-binding affinity and cooperativity Hill coefficients similar to those of WT EBNA1, and both the T585A and T585P mutants formed similar complexes in the DNA cross-linking studies. The T585P mutant showed the highest affinity for the double-FR site (1.8-fold compared to the WT) and had the highest Hill coefficient (2.6-fold compared to the WT), suggesting that the T585P mutant has greater cooperativity than WT EBNA1 on the FR fragment (Fig. 4F and G). Interestingly, T585P is a naturally occurring polymorphism in EBNA1 and therefore is of considerable interest for further functional analysis.
To understand why the T585P mutant had a higher affinity for the DNA, we hypothesized that T585P forms more stable, lower-order complexes on the DNA. To test this hypothesis, we assayed the stability of EBNA1-DNA complexes by challenging preformed complexes with a molar excess of competitor DNA in EMSAs (Fig. 5). Under these EMSA conditions, we observed that the WT EBNA1 DBD forms higher-order protein-DNA complexes than those formed by the T585P mutant, as seen by the formation of a slower-mobility complex (Fig. 5A and C). Quantification of DNA-bound complexes revealed that WT EBNA1 and the T585P mutant form approximately the same number of complexes, although T585P saturates the DNA more than does the WT EBNA1 DBD at the same concentration of protein (Fig. 5A and B). The addition of competitor DNA resulted in a greater reduction of complex formation in the WT EBNA1 DBD, indicating that WT EBNA1-DNA complexes were more dynamic and more readily competed with unlabeled competitor DNA molecules (Fig. 5D). In contrast, T585P complexes formed more stable lower-order complexes and were not as easily displaced by the competitor DNA.
FIG 5.

EBNA1 forms dynamic higher-order complexes (HC) on 2FR DNA compared to the T585P mutant. (A) WT EBNA1 or the T585P mutant was titrated on DNA and allowed to form complexes for 30 min. On the right part of gel, the addition of identical but unlabeled 2FR DNA (740 nM) initiated the exchange of EBNA1 and the T585P mutant between the two sets of FR DNA molecules. (B) Quantification of total complexes (higher-order complex plus 2FR plus 1FR). (C) Quantification of higher-order complexes alone. (D) Quantification of all complexes as described above for panel B but in the presence of competitor DNA.
The trimer interface is not required for DNA replication but is essential for plasmid maintenance.
To determine if the trimer interface had a physiological role in living cells, we investigated if the interface was required for plasmid replication and maintenance. EBNA1 trimer interface mutants were engineered into plasmids containing both OriP and an expression cassette for an otherwise full-length EBNA1 protein lacking the glycine-alanine repeats (amino acids [aa] 101 to 324), as this repeat domain is known to have a minimal role in DNA replication or episome maintenance function (16, 17). As a control, we included a plasmid containing OriP alone and lacking EBNA1. Plasmid replication was evaluated at 72 h posttransfection by measuring levels of DpnI-resistant DNA (Fig. 6A). We found that all interface mutants, with the exception of L582P, were expressed at protein levels similar to those of WT EBNA1 (Fig. 6B). With the exception of the L582P mutant, most of the trimer interface mutants showed replication efficiencies similar to those of WT EBNA1 (Fig. 6C). A second series of plasmids with a cytomegalovirus (CMV) promoter driving EBNA1 was constructed to increase the expression of the L582P mutant and to eliminate the second BamHI site in the first series of plasmids. Using this plasmid system, we assayed the L582F, L582P, T585A, and T585P trimer interface mutants (Fig. 6D). The L582P mutant was expressed at higher levels using this plasmid system (Fig. 6E) but still showed a significant defect in plasmid replication (Fig. 6F). This is consistent with the biochemical data showing a defect in L582P DNA binding. On the other hand, T585A showed slightly elevated levels of replication compared to those of WT EBNA1, and most other mutations were not significantly affected in DNA replication.
FIG 6.

Plasmid replication assay. (A) Representative Southern blot for OriP-dependent DNA replication with the first series of trimer interface substitution mutants of EBNA1. Replication is quantified as the ratio of BamHI- and DpnI-resistant full-length DNA (right) to BamHI (2 cut sites)-digested DNA (left). (B) Representative Western blot of protein levels at 4 days posttransfection. (C) Average replication activity from multiple DNA replication assays of WT EBNA1 or mutants after transient transfection in 293T and HeLa cells, normalized to the value for WT EBNA1. (D) Representative Southern blot for the second series of OriP-CMV-EBNA1 plasmids digested with BamHI (single cut site) (left) or with both BamHI and DpnI. (E) Representative Western blot of protein levels during the replication assay. (F) Quantification of replicated DNA compared to input BamHI-digested DNA normalized to the value for WT EBNA1.
We next determined if the trimer interface was important for long-term OriP-dependent plasmid maintenance (Fig. 7). OriP-dependent plasmids expressing a hygromycin resistance gene and either WT EBNA1 or trimer interface mutants were transfected into 293T or HeLa cells, selected for hygromycin resistance, and then assayed at various time points after the removal of hygromycin selection. Southern blot assays were performed on the same number of cells at four different time points postselection (Fig. 7A). At early stages after selection, cells expressed similar levels of the EBNA1 protein, with the exception of the L582P mutant (Fig. 7B). Over time, the mutants varied in their abilities to maintain the OriP-dependent plasmid (Fig. 7A and C). After 7 days, the L582P mutant showed defects in maintenance, consistent with its lower levels of EBNA1 expression and impaired replication function. After 19 days, the T585P and Q530E mutants showed significantly reduced levels of plasmid maintenance, while the T585A mutant showed significantly enhanced plasmid maintenance (Fig. 7A and C). The decrease in EBNA1 expression for the T585P mutant at later time points is consistent with the loss of episomes carrying the EBNA1 gene. These findings indicate that mutations that alter the trimer interface, especially T585 and Q530, alter the episome maintenance function of EBNA1.
FIG 7.
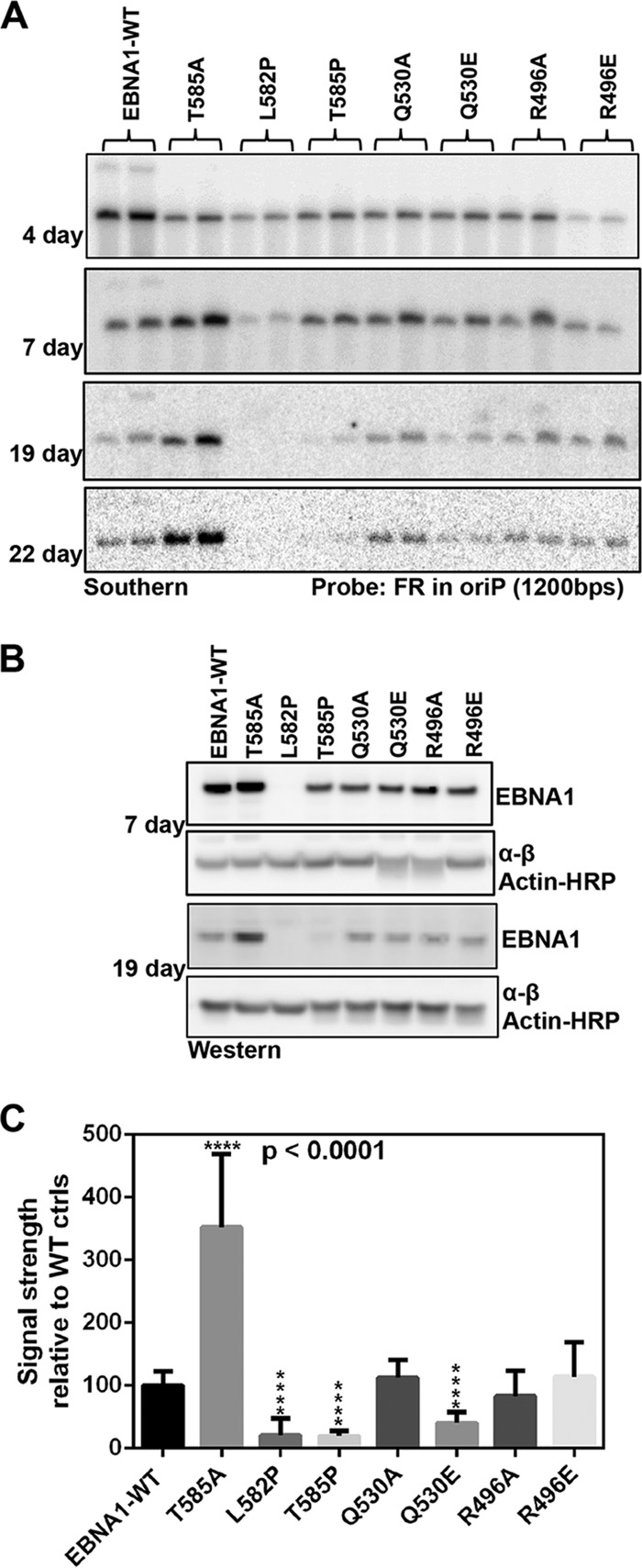
Plasmid maintenance assay. (A) Representative Southern blot of isolated WT and mutant EBNA1-OriP plasmids at the indicated days posttransfection. (B) Western blot of EBNA1 protein levels at the indicated days posttransfection during the maintenance assay. (C) Quantification of plasmid DNA normalized to the value for WT EBNA1 at ≥19 days posttransfection.
We next assessed the effects of the naturally occurring polymorphism T585P on EBNA1 binding to OriP and its ability to assemble functional chromatin at OriP in vivo by a chromatin immunoprecipitation (ChIP) assay (Fig. 8). We designed a series of primers specific for the FR and DS regions of OriP as well as a control region at cellular actin (Fig. 8A). OriP-dependent plasmids expressing Flag-WT EBNA1 or Flag-T585P were transfected into 293T cells and shown to express equivalent levels of the EBNA1 protein (Fig. 8B). EBNA1 binding was monitored by Flag-ChIP, which revealed a small but significant reduction (∼20%) in FR and DS binding for the T585P mutant relative to WT EBNA1 (Fig. 8C). Previous studies have shown that histone H3K4me3 and ORC2 assemble at functional OriP (31). We now show that the T585P mutant is compromised for the formation of H3K4me3 (Fig. 8D) and ORC2 (Fig. 8E) at DS and FR regions of OriP. No significant binding was observed for the IgG control (Fig. 8F). These findings indicate that EBNA1-DNA binding, the formation of functional chromatin, and ORC recruitment at OriP are reduced for the T585P mutant.
FIG 8.

The T585P mutation compromises EBNA1 chromatin binding and origin formation in vivo. (A) Schematic of the EBV OriP region in the plasmid and positions of primer sets used for ChIP-quantitative PCR. Numbers indicate B95-8 coordinates for primers. (B) Western blotting of 293T cells used for ChIP at 3 days posttransfection shows similar levels of WT EBNA1 and the T585P mutant. A vector with OriP lacking EBNA1 is used as a control to show the specificity of the Flag antibody for Flag-EBNA1. (C to F) Flag-EBNA1 T585P or Flag-WT EBNA1 was assayed by ChIP for Flag-EBNA1 (C), H3K4me3 (D), ORC2 (E), or control IgG (F) at DS and FR regions or for cellular actin as a control. The bar graphs represent the mean percentages of the input, and error bars represent standard errors of the means for three independent biological replicates, and within each replicate, PCR was done in duplicate. A GraphPad Prism t test was used to calculate P values (*, P < 0.05; ***, P < 0.001).
To determine whether EBNA1 oligomer interface mutations may manifest other features consistent with higher-order protein assemblies in living cells, we examined the formation of EBNA1 subnuclear foci by immunofluorescence (IF) (Fig. 9). EBNA1 foci were observed similarly in nuclei expressing WT EBNA1 and the T585P mutant from plasmids also containing OriP and the hygromycin selection marker (Fig. 9A). EBNA1 foci could be observed after detergent permeabilization and extraction prefixation, suggesting that these are stable, chromatin-like episomes containing multiple EBNA1 molecules (Fig. 9B and D). By comparing WT EBNA1 and the T585P mutant, we observed that WT EBNA1 retained significantly more cells with stable numbers of EBNA1 foci (>20 per cell) after 7 days (Fig. 9B to D). These findings suggest that the T585P mutant is deficient for the stable formation of higher-order EBNA1 subnuclear foci in vivo.
FIG 9.

The T585P mutant shows decreased focus formation in cells compared to WT EBNA1. (A) Quantification of the percentage of cells that contain at least one focus in three independent experiments. (B) Representative images of preextracted HeLa cells at 7 days posttransfection highlighting focus formation of Flag-WT EBNA1 or the T585P mutant using mouse anti-Flag-EBNA1 and Hoechst dye to stain DNA. The inset shows a closeup view of WT EBNA1 to highlight the larger number of foci (C) Quantification of the percentage of cells containing ≤3, 4 to 19, or ≥20 foci/cell for WT EBNA1 versus the T585P mutant in three independent experiments. Bar graphs show means and standard errors of the means. A GraphPad Prism t test was used to calculate P values (***, P < 0.001).
DISCUSSION
The EBV latency program requires that EBNA1 be able to both replicate its DNA and transmit the replicated DNA to its daughter cells (32). Recent studies demonstrated that EBNA1 can orchestrate episome transmission at very high efficiencies (33, 34). However, the biochemical mechanisms and structural properties that enable EBNA1 to maintain episome stability are not completely understood. Here, we provide new X-ray crystallographic evidence that EBNA1 can form a higher-ordered hexameric ring. We show that mutations and a naturally occurring polymorphism at the oligomeric interface impact the ability of EBNA1 to cooperatively bind to the FR and to support OriP-dependent episome maintenance in cells. These findings suggest that the hexameric ring is a functional trimer of dimers that is likely to assemble at the FR.
The structural and mutational studies presented here indicate that amino acid residue T585 is the critical fulcrum at the trimer interface. Genetic analyses of various EBV strains indicate that T585 is among the EBNA1 polymorphisms found frequently in both NPC tumors and Burkitt's lymphoma (35–41). Recently, our laboratory found that an NPC variant of EBNA1 containing 8 amino acid polymorphisms different from the prototypical B95-8 strain, including a T585I substitution, was defective in replication and maintenance of the viral episome (41). The EBNA1 variant was also found to be deficient in suppressing lytic cycle gene transcription and lytic DNA replication (41). We note that the Mutu strain isolated from Burkitt's lymphoma contains both T585P and L582F among 8 additional polymorphisms. These observations suggest that amino acids in the trimer interface may be commonly subjected to polymorphisms that can alter the overall stability, geometry, or dynamics of EBNA1 oligomerization. These trimer interface polymorphisms are likely to influence the function and phenotype of their respective EBV strains.
The natural polymorphism T585P in the context of otherwise B95-8 EBNA1 was found to decrease higher-order complex formation, as shown in glutaraldehyde cross-linking studies. The EBNA1 T585P mutant shows increased DNA binding to an FR fragment but is incapable of forming the higher-order protein-DNA complexes seen with WT EBNA1. Interestingly, the T585P mutant, along with most of the trimer interface point mutants, shows only minor defects in transient replication. In contrast, the T585P mutant showed a reduced ability for long-term plasmid maintenance. On the other hand, T585A, which is not observed in natural EBNA1 polymorphisms, showed DNA binding similar to that of WT EBNA1 but enhanced replication and episome maintenance. Thus, amino acid variations at T585 confer important functional differences in EBNA1 DNA binding and episome maintenance. Since episome maintenance is largely dependent on the oligomeric binding of EBNA1 to the FR element of OriP, we suggest that the hexameric interface contributes to the oligomeric geometry and packaging of EBNA1 at the FR (Fig. 10).
FIG 10.
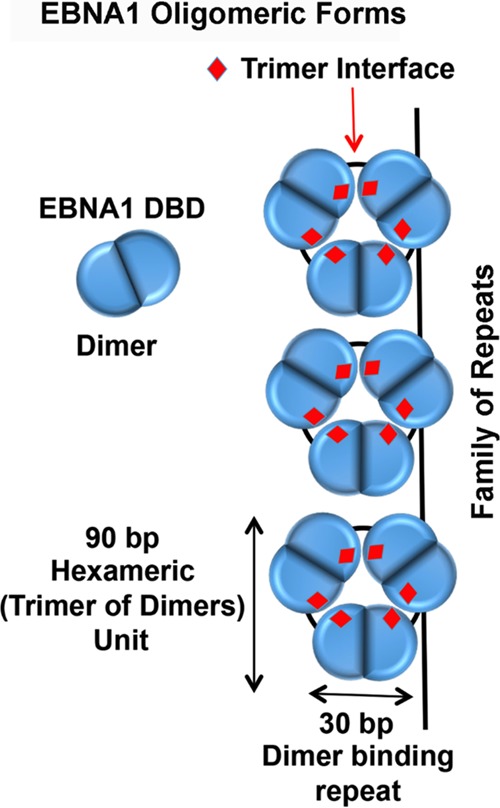
The EBNA1 DBD forms an obligate dimer that packs as a hexamer (trimer of dimers). The trimer interface (indicated in red) is critical for episome maintenance. The hexameric assembly binds DNA on the same surface above each hexameric wheel and can bind between wheels through PGP loop interactions (not modeled). One EBNA1 dimer occupies a single 30-bp FR element, and the hexamer is predicted to include 90 bp (3 FR repeat elements).
Different oligomeric forms of EBNA1 may be required for executing the various functions of EBNA1. At the DS, EBNA1 dimers bind as a pair of dimers to form a stable tetramer that recruits host cell replication machinery to initiate DNA synthesis in close proximity to the DS element. At the FR, EBNA1 forms an array of at least 6 to 20 dimers that form a higher-order structure that efficiently attaches to metaphase chromosomes and enables the faithful segregation of sister episomes to each newly divided daughter cell. Previous studies using electron microscopy (EM) have shown that EBNA1 binds as a spherical complex on the FR element (20, 21, 27, 28, 42, 43). Data from our studies are consistent with those of the previous EM studies and suggest that the critical geometry of EBNA1 at the FR is a trimer of dimers that can stack to form an array of hexagonal wheels that are likely to appear as a sphere or filament by EM.
EBNA1 binding to FR is also known to form higher-order interactions that include metaphase chromosome attachment and DNA looping involved in transcriptional enhancement (23, 44). Domains outside the EBNA1 DBD are known to contribute to these interactions, as are cellular proteins such as EBP2 (45, 46). However, the organization of EBNA1 molecules on the FR is likely to be important for all of these functions. The hexameric form of EBNA1 is strikingly similar to the multimeric forms of KSHV LANA (15). LANA has been shown to self-associate between its outer alpha helices to form a range of higher-order oligomers, including pentameric and tetrameric rings as well as multiple pentamers stacking to form a linear chain (15). These oligomeric forms of LANA were found to contribute to KSHV episome maintenance (9), similar to what we have found here for EBNA1 and OriP maintenance. One significant difference between the EBNA1 hexamer and LANA decamer is the positioning of the DNA-binding interface. The DNA-binding interface for the LANA decameric ring is along the circumference, while the EBNA1 hexameric ring binds DNA across the bottom surface area. These variances may reflect mechanistic differences between the two viral episome maintenance systems or may simply reflect the different snapshots of their dynamic conformational variations that confer their diverse functions in replication and episome maintenance.
In conclusion, our structural and functional data reveal a novel EBNA1 oligomeric isoform consisting of a trimer of dimers. The trimeric interface is organized principally through the single amino residue T585 located at the centermost region and is biologically important for the long-term maintenance of a plasmid. T585 is critical for the hydrogen-bonding network with adjacent residues as well as with residues from adjacent EBNA1 molecules. Further studies are necessary to determine whether EBNA1 multimeric complexes are associated with host chromosomes during mitosis. In addition, this novel interface may provide a new target for small-molecule inhibitors to selectively disrupt EBNA1 function in episome maintenance and latent infection.
MATERIALS AND METHODS
Expression, purification, and crystallization.
An expression construct was made by cloning an insert using B95-8 as a template for PCR amplification. The EBNA1 DNA-binding domain (positions 468 to 619) was expressed as a 6×His SUMO fusion protein in Escherichia coli [BL21-Codon-PLUS(DE3)-RIL competent cells; Agilent Technologies] by using an autoinduction procedure (47). Briefly, 500 ml of 2× Luria broth supplemented with 1 mM MgSO4, 1,000× trace metals, 50× 5052 (25% glycerol, 2.5% glucose, 10% alpha-lactose monohydrate), and 20× NPS (25 mM ammonium sulfate, 50 mM potassium phospate monobasic, 50 mM sodium phosphate dibasic) was inoculated with a 10-ml starter culture grown overnight, and the culture continued to grow at 30°C for 24 to 30 h.
Cells were harvested and resuspended in a buffer containing 50 mM Tris (pH 8.5), 1 M NaCl, 10% glycerol, 5 mM 2-mercaptoethanol, 1% Tween 20, 1 mM fresh phenylmethylsulfonyl fluoride (PMSF), and 1 mg/ml lysozyme (Sigma) and lysed by using a sonicator. After centrifugation for 40 min at 16,000 rpm in an SS-34 fixed-angle Sorvall rotor, the lysate was poured over a gravity nickel-nitrilotriacetic acid (NTA) column. After washing with 20 volumes of a solution containing 50 mM Tris (pH 8.5), 1 M NaCl, 10% glycerol, and 5 mM 2-mercaptoethanol, EBNA1 was eluted with a wash solution containing 250 mM imidazole. The eluted protein was concentrated and purified further by using a 26/60 Superdex 200-pg gel filtration column (GE Life Sciences) equilibrated with wash buffer. Fractions were collected, pooled, and incubated with SUMO protease (Ulp1) overnight at 4°C. After cleavage, the protein was passed over a second HiTrap nickel-NTA column to remove the undigested 6×His-SUMO fusion protein and SUMO protease, and the flowthrough was collected. After concentration, EBNA1 was purified further by using the Superdex gel filtration column equilibrated with a solution containing 50 mM HEPES (pH 7.5), 500 mM NaCl, and 10 mM dithiothreitol (DTT). Pure (>95%) EBNA1 (positions 469 to 619) was concentrated to ∼10 mg/ml and frozen for long-term storage.
Crystals were obtained via the hanging-drop vapor diffusion method. Concentrated EBNA1 was diluted 1:1 in a solution containing 50 mM morpholineethanesulfonic acid (MES) (pH 6.5) and 800 mM sodium formate. Because the majority of the protein was precipitated under these conditions, the solution was centrifuged to remove the precipitate protein, and 4 μl of the supernatant were pipetted on to a glass coverslip and placed over a reservoir containing the precipitating solution. Crystals had a flat rectangular morphology and appeared after 1 to 2 weeks.
For biochemical assays, the EBNA1 DNA-binding domain (positions 459 to 607) and point mutants were purified in a manner similar to that described above except for some modifications. Briefly, cell pellets were resuspended in a solution containing 20 mM Tris HCl (pH 8.0), 1 M NaCl, 12.5 mM imidazole, 0.01% Tween 20, 5 mM β-mercaptoethanol, 1 mM PMSF, and a pinch of lysozyme. Cells were lysed, sonicated, and poured over nickel-NTA resin as stated above. The resin was washed with a solution containing 20 mM Tris HCl (pH 8.0), 1 M NaCl, 30 mM imidazole, and 5 mM β-mercaptoethanol. The protein was eluted in the presence of wash buffer with 500 mM imidazole. The eluted protein was incubated with SUMO protease and dialyzed overnight at 4°C in a solution containing 20 mM Tris HCl (pH 8.0), 1 M NaCl, and 5 mM β-mercaptoethanol. The purified tagless protein was obtained by pouring the cleavage reaction mixture over a HisTrap FF prepacked column (1 ml or 5 ml; GE Healthcare). In contrast to WT EBNA1, the mutants required an imidazole gradient, with the mutant proteins eluting at 30 mM imidazole.
Crystal structure determination.
Crystals were soaked in cryoprotectant containing 30% glycerol and flash frozen in liquid nitrogen. Data were collected from a single crystal kept frozen with an Oxford Cryosystems Cobra system at 100 K on a Rigaku MicroMax-007 HF rotating-anode X-ray generator (wavelength, 1.54178 Å) with VariMax optics and using a Saturn 944 HG charge-coupled-device (CCD) detector. Data were able to be indexed only in the P1 triclinic space group and had unit cell dimensions of 31.3 by 84.8 by 84.7 Å and an α of 60.0°, a β of 87.3°, and a γ of 88.5°. Three hundred sixty 1° frames were collected. Reduction and scaling of the data were done by using HKL3000. The structure was solved by molecular replacement using PHASER integrated into PHENIX (48), with the structure reported under PDB accession number 1VHI as a search model (7). Three nonoverlapping solutions were found in the electron density. Models were refined in PHENIX using simulated annealing, minimization, and B-factor refinement. Between refinement cycles, the model was manually rebuilt by using the Coot program (49). Data collection and refinement statistics are summarized in Table 1.
Cell culture and plasmids.
HeLa (ATCC) and 293T (ATCC) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 μg/ml streptomycin, and 100 U/ml penicillin. Cells were cultured in an incubator set at 37°C with 5% CO2. The mammalian expression vector for Flag-EBNA1 or hemagglutinin (HA)-EBNA1 contained B95-8 EBNA1 lacking the GA repeats (aa 101 to 324) under the control of a CMV-3×Flag or a CMV-HA promoter in a plasmid derived from pREP10 (Clontech) containing OriP, green fluorescent protein (GFP), and hygromycin resistance (41).
Plasmid maintenance assays.
Plasmid maintenance assays were described previously (41, 50). Briefly, HeLa or 293T cells were seeded into 6-well plates at concentrations of 0.75 × 105 to 1.5 × 105 cells/ml. Twenty-four hours later, cells were transfected with Lipofectamine 2000 (10 μl) and 2 μg OriP plasmids expressing either HA-EBNA1 (series 1) or CMV promoter-Flag-EBNA1 (series 2), with either B95-8 or point mutants of the EBNA1 DBD in both plasmids, as indicated. At 2 to 3 days posttransfection (dpt), equal portions of the cells for each mutant were split into 15-cm plates to continue passaging, and the remaining cells were harvested for Hirt lysis and Western blot analyses. Isolated DNA was subjected to BamHI digestion and resolved on 0.9% agarose gels. Southern blotting was performed as described above. After the first passage, 100 μg/ml hygromycin was added for 3 to 5 days. After initial selection and the first passage, cells were continuously split and harvested once plates were confluent for 20 to 30 days without selection.
Plasmid replication assays.
Plasmid DNA replication assays were described previously (41, 51). Briefly, HeLa and 293T cells (∼1 × 106 cells) were plated into 10-cm dishes. Twenty-four hours later, cells were transfected with Lipofectamine 2000 (12 μl; Invitrogen) and 4 μg OriP plasmids expressing either FLAG-EBNA1, HA-EBNA1, point mutants of both plasmids, or a control plasmid containing OriP alone. Cells were split after 48 h and then harvested at 72 h posttransfection for both episomal DNA and protein. Episomal DNA was extracted by Hirt lysis (52). The DNA pellets were dissolved in 150 μl of H2O or 10 mM Tris HCl–1 mM EDTA buffer (pH 7.6), 15 μl was subjected to restriction digestion with BamHI alone, and 135 μl was subjected to BamHI and DpnI digestion overnight at 37°C. DNA was extracted with phenol-chloroform (1:1), precipitated, electrophoresed on a 0.9% agarose gel, and transferred onto a nylon membrane (PerkinElmer) for Southern blotting. Blots were visualized and quantified by using a Typhoon 9410 PhosphorImager (GE Healthcare).
Western blots.
Whole-cell lysates were resolved by electrophoresis in 4 to 12% Tris-Bis denaturing gels (NuPage) in 1× MES-SDS running buffer (50 mM MES, 50 mM Tris, 0.1% SDS, 1 mM EDTA [pH 7.3]). Protein products were transferred onto polyvinylidene difluoride (PVDF) membranes and blotted with the following antibodies: anti-β-actin-peroxidase (AC-15) (catalog no. A3854; Sigma-Aldrich), anti-EBNA1 mouse monoclonal antibody (0211) (catalog no. ab20777; Abcam), anti-EBNA1 rabbit polyclonal antibody (in-house), and anti-Flag M2-peroxidase (horseradish peroxidase [HRP]) (catalog no. A8592; Sigma-Aldrich).
Electrophoretic mobility shift assays.
The purified tagless EBNA1 DNA-binding domain or point mutants (2.5 μl) at the indicated concentrations were titrated onto dsDNA labeled on both 5′ ends with IRDye 700 (2.5 μl; 1 nM molecules) containing either one or two FR sites where noted. The dsDNA was formed by hybridizing oligomers in DNA annealing buffer (0.9× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 140 mM Tris HCl [pH 7.6], 20 mM MgCl2, 10 mM DTT) for 5 min at 100°C on a heat block and turning the heat block off to allow slow cooling to room temperature. The following oligonucleotides were purchased from IDT Inc. in standard desalted form: 5′-ATTAGGATAGCCTATGCTACCCAGATATAGATTAGGATAGCATATGCTACCCAGATATAG-3′ and 5′-CTATATCTGGGTAGCATATGCTATCCTAATCTATATCTGGGTAGCATAGGCTATCCTAAT-3′ for two FR sites (60-mer) and 5′-GGGTAGTATATGCTATCC-3′ and 5′GGATAGCATATACTACCC-3′ for one FR site (18-mer).
Binding reactions were allowed to proceed for 30 min at room temperature in 25 μl binding buffer containing 10 mM HEPES (pH 7.5), 5 mM MgCl2, 2.5 mM DTT, 0.25% Tween, 200 mM NaCl, and 10% glycerol. Totals of 100 mM NaCl and 2 mM MgCl2 were added to the reaction mixture with the protein. DNA-protein complexes were resolved by electrophoresis (70 to 90 V) in 6% nondenaturing polyacrylamide gels (30% acrylamide-Bis solution [29:1], catalog no. 1610156; Bio-Rad) in 0.5× Tris-borate-EDTA buffer. The gels were quantified by using the Licor Odyssey infrared imager. For competition assays, WT EBNA1 or the T585P mutant at the indicated concentrations was incubated with the 2FR DNA (10 nM) for 30 min at room temperature. After 30 min, identical but unlabeled 2FR DNA (1 μl; 74 nM) or DNA annealing buffer alone (1 μl) was added, and the mixture was incubated for a further 10 min. DNA-protein complexes were resolved as stated above.
Protein cross-linking.
Protein (19 μM) was incubated for 15 min at room temperature in reaction buffer containing 20 mM Tris HCl (pH 7.5), 1 M NaCl, 30 mM imidazole (pH 7.5), and 5 μM β-mercaptoethanol. The complexes were cross-linked with glutaraldehyde (1 μl; 0.25%) for 15 min at room temperature, followed by quenching with 1 μl of 1 M Tris HCl (pH 8.0). Six microliters of 2× SDS loading dye (100 mM Tris HCl [pH 6.8], 4% [wt/vol] SDS, 0.2% [wt/vol] bromophenol blue, 20% glycerol, and 200 mM DTT) was added to the complexes, and the mixture was incubated for 2 min at 95°C. Protein complexes were resolved by electrophoresis in 4 to 12% Tris-Bis denaturing gels (NuPage) in 1× MES-SDS running buffer (50 mM MES, 50 mM Tris, 0.1% SDS, 1 mM EDTA [pH 7.3]) and visualized by using Coomassie blue or silver staining (Invitrogen).
Immunofluorescence.
HeLa cells were plated and transfected as described above for plasmid replication assays in 10-cm plates using OriP plasmids expressing Flag-EBNA1 or the T585P point mutant. A control plasmid containing OriP alone showed no background EBNA1-Flag foci (images not shown). At 7 dpt, cells were plated onto glass coverslips in 24-well plates. Coverslips were precovered with 100% FBS and removed immediately before cells were plated at 0.5 × 105 cells/well. Twenty-four hours later, cells were washed once with phosphate-buffered saline (PBS), and 1 ml of preextraction buffer (0.2% Triton X-100 and 2 mM PMSF in PBS) was added to cells on ice for 5 min. Afterwards, cells were washed once with PBS and fixed with 3.7% paraformaldehyde in PBS for 15 min at room temperature. All further steps listed below, except after the blocking step, include two washes with PBS between steps. Cells were permeabilized with 0.3% Triton X-100 in PBS for 15 min at room temperature and blocked with PBG (0.2% fish gelatin and 0.5% bovine serum albumin [BSA] in PBS). The coverslips were then blotted with the following antibodies or stains: a 1:20,000 dilution of anti-Flag M2 mouse monoclonal antibody (catalog no. F1804; Sigma-Aldrich), a 1:1,000 dilution of goat anti-mouse Alexa Fluor 594 (catalog no. 11005; Thermo Fisher Scientific), and a 1:1,000 dilution of Hoechst 33258 (Sigma-Aldrich). Finally, coverslips were mounted (Fluoromount-G, catalog no. 0100-01; Southern Biotechnology Associates Inc.) onto microscope slides and visualized with a Nikon 80i upright microscope at a ×100 magnification. Flag-EBNA1 foci were quantified by using the analyze particle method in ImageJ with thresholds set at a lower limit of 130 and an upper limit of 215. Cells were then placed into groups depending on the number of foci contained in a cell. For each experiment, the percentage of cells in each group was calculated based on the total number of cells in that group divided by the total number of cells counted for a particular experiment. Three independent experiments were carried out, with totals of 341 and 343 cells being counted for WT EBNA1 and the T585P point mutant, respectively.
Chromatin immunoprecipitation.
ChIP assays were performed as previously described (51). Briefly, 293T cells were plated and transfected as described above for the plasmid replication assay with Flag-EBNA1 or the T585P point mutant and split 24 h later. At 72 h posttransfection, cells were harvested for cross-linking as described above, and ChIP was performed by using protein A-Dynabeads or protein A-Sepharose beads (51). Antibodies used were as follows: rabbit and mouse anti-IgG (Santa Cruz Biotechnology), anti-Orc2 (catalog no. ab99277; Abcam), trimethyl-histone H3 (Lys4) (catalog no. 07-473; Millipore), anti-Oct2 (catalog no. RB-9297-P; Thermo Fisher), anti-rabbit Flag (catalog no. F7425; Sigma-Aldrich), and anti-Flag M2 mouse monoclonal antibody (catalog no. F1804; Sigma-Aldrich).
Accession number(s).
Coordinates of the hexameric EBNA1 have been deposited in the Protein Data Bank under accession number 5WMF .
ACKNOWLEDGMENTS
We thank Jayaraju Dheekollu for technical training and Kate Beishline, Abram Calderon, Lois Tolvinski, and Samantha Soldan (all from the Wistar Institute) for advice and discussion.
This work was partially supported by a Wistar training grant to J.S.D. (T32 CA09171); NIH grants RO1 CA093606, RO1 DE017336, and P30 CA010815; and a Wellcome Trust seeding drug discovery grant (WT096496).
P.M.L. has a founding interest in Vironika, LLC.
REFERENCES
- 1.Lieberman PM. 2014. Virology. Epstein-Barr virus turns 50. Science 343:1323–1325. doi: 10.1126/science.1252786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lieberman PM. 2013. Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol 11:863–875. doi: 10.1038/nrmicro3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cruickshank J, Shire K, Davidson AR, Edwards AM, Frappier L. 2000. Two domains of the Epstein-Barr virus origin DNA-binding protein, EBNA1, orchestrate sequence-specific DNA binding. J Biol Chem 275:22273–22277. doi: 10.1074/jbc.M001414200. [DOI] [PubMed] [Google Scholar]
- 4.Summers H, Barwell JA, Pfuetzner RA, Edwards AM, Frappier L. 1996. Cooperative assembly of EBNA1 on the Epstein-Barr virus latent origin of replication. J Virol 70:1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li N, Thompson S, Schultz DC, Zhu W, Jiang H, Luo C, Lieberman PM. 2010. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS One 5:e10126. doi: 10.1371/journal.pone.0010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson S, Messick T, Schultz DC, Reichman M, Lieberman PM. 2010. Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1. J Biomol Screen 15:1107–1115. doi: 10.1177/1087057110379154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bochkarev A, Barwell JA, Pfuetzner RA, Furey W Jr, Edwards AM, Frappier L. 1995. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell 83:39–46. doi: 10.1016/0092-8674(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 8.Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, Edwards AM. 1996. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84:791–800. doi: 10.1016/S0092-8674(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 9.Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog 9:e1003672. doi: 10.1371/journal.ppat.1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Ruckert J, Ritter C, Schulz TF, Luhrs T. 2013. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog 9:e1003640. doi: 10.1371/journal.ppat.1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards AM, Bochkarev A, Frappier L. 1998. Origin DNA-binding proteins. Curr Opin Struct Biol 8:49–53. doi: 10.1016/S0959-440X(98)80009-2. [DOI] [PubMed] [Google Scholar]
- 12.Knight JD, Li R, Botchan M. 1991. The activation domain of the bovine papillomavirus E2 protein mediates association of DNA-bound dimers to form DNA loops. Proc Natl Acad Sci U S A 88:3204–3208. doi: 10.1073/pnas.88.8.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enemark EJ, Stenlund A, Joshua-Tor L. 2002. Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J 21:1487–1496. doi: 10.1093/emboj/21.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hegde RS, Grossman SR, Laimins LA, Sigler PB. 1992. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature 359:505–512. doi: 10.1038/359505a0. [DOI] [PubMed] [Google Scholar]
- 15.Hellert J, Weidner-Glunde M, Krausze J, Lunsdorf H, Ritter C, Schulz TF, Luhrs T. 2015. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc Natl Acad Sci U S A 112:6694–6699. doi: 10.1073/pnas.1421804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frappier L. 2012. The Epstein-Barr virus EBNA1 protein. Scientifica (Cairo) 2012:438204. doi: 10.6064/2012/438204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leight ER, Sugden B. 2000. EBNA-1: a protein pivotal to latent infection by Epstein-Barr virus. Rev Med Virol 10:83–100. doi: 10.1002/(SICI)1099-1654(200003/04)10:2<83::AID-RMV262>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 18.Bashaw JM, Yates JL. 2001. Replication from oriP of Epstein-Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J Virol 75:10603–10611. doi: 10.1128/JVI.75.22.10603-10611.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Summers H, Fleming A, Frappier L. 1997. Requirements for Epstein-Barr nuclear antigen 1 (EBNA1)-induced permanganate sensitivity of the Epstein-Barr virus latent origin of DNA replication. J Biol Chem 272:26434–26440. doi: 10.1074/jbc.272.42.26434. [DOI] [PubMed] [Google Scholar]
- 20.Su W, Middleton T, Sugden B, Echols H. 1991. DNA looping between the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr nuclear antigen 1. Proc Natl Acad Sci U S A 88:10870–10874. doi: 10.1073/pnas.88.23.10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frappier L, Goldsmith K, Bendell L. 1994. Stabilization of the EBNA1 protein on the Epstein-Barr virus latent origin of DNA replication by a DNA looping mechanism. J Biol Chem 269:1057–1062. [PubMed] [Google Scholar]
- 22.Hussain M, Gatherer D, Wilson JB. 2014. Modelling the structure of full-length Epstein-Barr virus nuclear antigen 1. Virus Genes 49:358–372. doi: 10.1007/s11262-014-1101-9. [DOI] [PubMed] [Google Scholar]
- 23.Aras S, Singh G, Johnston K, Foster T, Aiyar A. 2009. Zinc coordination is required for and regulates transcription activation by Epstein-Barr nuclear antigen 1. PLoS Pathog 5:e1000469. doi: 10.1371/journal.ppat.1000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.VanLoock MS, Alexandrov A, Yu X, Cozzarelli NR, Egelman EH. 2002. SV40 large T antigen hexamer structure: domain organization and DNA-induced conformational changes. Curr Biol 12:472–476. doi: 10.1016/S0960-9822(02)00696-6. [DOI] [PubMed] [Google Scholar]
- 25.Schuck S, Stenlund A. 2005. Assembly of a double hexameric helicase. Mol Cell 20:377–389. doi: 10.1016/j.molcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Frappier L. 2012. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr Opin Virol 2:733–739. doi: 10.1016/j.coviro.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Harrison S, Fisenne K, Hearing J. 1994. Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J Virol 68:1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frappier L, O'Donnell M. 1991. Epstein-Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc Natl Acad Sci U S A 88:10875–10879. doi: 10.1073/pnas.88.23.10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wysokenski DA, Yates JL. 1989. Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J Virol 63:2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frappier L, O'Donnell M. 1991. Overproduction, purification, and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J Biol Chem 266:7819–7826. [PubMed] [Google Scholar]
- 31.Dheekollu J, Wiedmer A, Sentana-Lledo D, Cassel J, Messick T, Lieberman PM. 2016. HCF1 and OCT2 cooperate with EBNA1 to enhance OriP-dependent transcription and episome maintenance of latent Epstein-Barr virus. J Virol 90:5353–5367. doi: 10.1128/JVI.00239-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Middleton T, Sugden B. 1994. Retention of plasmid DNA in mammalian cells is enhanced by binding of the Epstein-Barr virus replication protein EBNA1. J Virol 68:4067–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanbo A, Sugden A, Sugden B. 2007. The coupling of synthesis and partitioning of EBV's plasmid replicon is revealed in live cells. EMBO J 26:4252–4262. doi: 10.1038/sj.emboj.7601853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindner SE, Sugden B. 2007. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid 58:1–12. doi: 10.1016/j.plasmid.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun L, Zhao Z, Liu S, Liu X, Sun Z, Luo B. 2015. Sequence variation analysis of Epstein-Barr virus nuclear antigen 1 gene in the virus associated lymphomas of northern China. PLoS One 10:e0140529. doi: 10.1371/journal.pone.0140529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z, Wang X, Strong MJ, Concha M, Baddoo M, Xu G, Baribault C, Fewell C, Hulme W, Hedges D, Taylor CM, Flemington EK. 2013. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J Virol 87:1172–1182. doi: 10.1128/JVI.02517-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santpere G, Darre F, Blanco S, Alcami A, Villoslada P, Mar Alba M, Navarro A. 2014. Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1,000 Genomes Project. Genome Biol Evol 6:846–860. doi: 10.1093/gbe/evu054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mai SJ, Ooka T, Li DJ, Zeng MS, Jiang RC, Yu XJ, Zhang RH, Chen SP, Zeng YX. 2007. Functional advantage of NPC-related V-val subtype of Epstein-Barr virus nuclear antigen 1 compared with prototype in epithelial cell line. Oncol Rep 17:141–146. [PubMed] [Google Scholar]
- 39.Feederle R, Klinke O, Kutikhin A, Poirey R, Tsai MH, Delecluse HJ. 2015. Epstein-Barr virus: from the detection of sequence polymorphisms to the recognition of viral types. Curr Top Microbiol Immunol 390:119–148. doi: 10.1007/978-3-319-22822-8_7. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Liu X, Xing X, Cui Y, Zhao C, Luo B. 2010. Variations of Epstein-Barr virus nuclear antigen 1 gene in gastric carcinomas and nasopharyngeal carcinomas from Northern China. Virus Res 147:258–264. doi: 10.1016/j.virusres.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 41.Dheekollu J, Malecka K, Wiedmer A, Delecluse HJ, Chiang AK, Altieri DC, Messick TE, Lieberman PM. 2017. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr virus episomal latency. Oncotarget 8:7248–7264. doi: 10.18632/oncotarget.14540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reischig J, Bartsch D, Polack A, Vonka V, Hirsch I. 1987. Electron microscopy of binding of Epstein-Barr virus (EBV) nuclear antigen (EBNA-1) to EBV DNA. Virology 160:498–501. doi: 10.1016/0042-6822(87)90025-0. [DOI] [PubMed] [Google Scholar]
- 43.Hirsch I, Reischig J, Benada O, Bartsch D, Brichacek B, Boguszakova L, Vonka V. 1988. Epstein-Barr virus nuclear antigen type 1 binding: electron microscopy. J Virol Methods 22:133–142. doi: 10.1016/0166-0934(88)90096-1. [DOI] [PubMed] [Google Scholar]
- 44.Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A. 2004. The amino terminus of Epstein-Barr virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J Virol 78:11487–11505. doi: 10.1128/JVI.78.21.11487-11505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nayyar VK, Shire K, Frappier L. 2009. Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2). J Cell Sci 122:4341–4350. doi: 10.1242/jcs.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapoor P, Lavoie BD, Frappier L. 2005. EBP2 plays a key role in Epstein-Barr virus mitotic segregation and is regulated by aurora family kinases. Mol Cell Biol 25:4934–4945. doi: 10.1128/MCB.25.12.4934-4945.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng Z, Lezina L, Chen CJ, Shtivelband S, So W, Lieberman PM. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol Cell 9:493–503. doi: 10.1016/S1097-2765(02)00476-8. [DOI] [PubMed] [Google Scholar]
- 51.Deng Z, Atanasiu C, Burg JS, Broccoli D, Lieberman PM. 2003. Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein-Barr virus OriP. J Virol 77:11992–12001. doi: 10.1128/JVI.77.22.11992-12001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]