

Abstract
Autophagy targets intracellular molecules, damaged organelles, and invading pathogens for degradation in lysosomes. Recent studies have identified autophagy receptors that facilitate this process by binding to ubiquitinated targets, including NDP52. Here, we demonstrate that the small guanosine triphosphatase Rab35 directs NDP52 to the corresponding targets of multiple forms of autophagy. The active GTP‐bound form of Rab35 accumulates on bacteria‐containing endosomes, and Rab35 directly binds and recruits NDP52 to internalized bacteria. Additionally, Rab35 promotes interaction of NDP52 with ubiquitin. This process is inhibited by TBC1D10A, a GAP that inactivates Rab35, but stimulated by autophagic activation via TBK1 kinase, which associates with NDP52. Rab35, TBC1D10A, and TBK1 regulate NDP52 recruitment to damaged mitochondria and to autophagosomes to promote mitophagy and maturation of autophagosomes, respectively. We propose that Rab35‐GTP is a critical regulator of autophagy through recruiting autophagy receptor NDP52.
Keywords: autophagy, NDP52, Rab35, TBK1
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Introduction
Macroautophagy, hereafter referred to as autophagy, is an evolutionarily conserved cellular process by which cytoplasmic components are delivered to and degraded in lysosomes to promote cellular homeostasis, stress response, development, and nutrient recycling (Levine et al, 2011; Mizushima & Komatsu, 2011). Autophagy is initiated by formation of a phagophore that encapsulates a portion of the cytoplasm, and elongates to form an autophagosome, a double‐membrane vesicle. This process depends on several autophagy‐related (ATG) proteins. The outer membrane of the autophagosome fuses with the lysosome to form an autolysosome, in which the cargo is degraded by lysosomal hydrolases and recycled. Autophagy has been traditionally characterized as a non‐selective, wholesale degradation system that occurs constitutively (basal autophagy), but is also inducible by stimuli such as depletion of amino acids (starvation‐induced autophagy). However, a growing body of evidence now suggests that autophagy can also selectively target invading microorganisms (xenophagy), damaged mitochondria (mitophagy), and protein aggregates (Khaminets et al, 2016). These selective modes are also constitutive, as well as inducible in response to cellular stress (Weidberg et al, 2011). Notably, dysregulation of autophagy is associated with cancer, infectious disease, neurodegenerative disorders such as Parkinson's disease, and inflammatory disorders such as Crohn's disease (Mizushima & Komatsu, 2011).
Selective autophagy is regulated by cargo receptors that interact both with ubiquitinated targets in the cytoplasm and with ATG8 (LC3/GABARAP) in the phagophore. These interactions are mediated by a ubiquitin‐binding domain and an LC3‐interacting region in receptors, respectively (Stolz et al, 2014). The receptors optineurin (OPTN), NDP52 (nuclear dot protein 52 kDa, also known as CALCOCO2), and TAX1BP1 recognize damaged mitochondria, while p62/SQSTM1, OPTN, NDP52, and TAX1BP1 also recognize bacteria (Khaminets et al, 2016). In particular, NDP52 has been demonstrated to be the primary autophagy receptor for bacteria, which are coated with ubiquitin (Thurston et al, 2009). On the other hand, NDP52 activates autophagy during Salmonella infection by binding galectin 8, a protein that accumulates on damaged vacuoles containing bacteria (Thurston et al, 2012). NDP52 also promotes autophagosome maturation (Tumbarello et al, 2012; Verlhac et al, 2015). Furthermore, PINK1‐ and Parkin‐mediated mitophagy depends on NDP52, and localization of NDP52 to damaged mitochondria is regulated by TBK1 (TANK‐binding kinase 1) activity (Heo et al, 2015). However, whether the multiple roles of NDP52 are universally regulated remains to be established.
It has been shown that Rab guanosine triphosphatases (Rab GTPases), key regulators of intracellular membrane trafficking and organelle biogenesis, control starvation‐induced and selective autophagy (Nozawa et al, 2012; Haobam et al, 2014; Szatmari & Sass, 2014). Rabs are activated and inactivated by guanine nucleotide exchange factors (GEFs) and GTPase‐activating proteins (GAPs), respectively. Activated Rab interacts with various effectors to elicit distinct downstream events. Rab‐specific GAPs that contain a Tre2‐Bub2‐Cdc16 (TBC) domain, referred to as TBC/RabGAPs, inactivate Rab via a dual‐finger mechanism (Pan et al, 2006). Based on the presence of the TBC domain, 44 putative TBC/RabGAPs have been identified in the human genome (Frasa et al, 2012). Among these, TBC1D5, TBC1D14, and TBC1D25 (Rab33B‐GAP) regulate starvation‐induced autophagy (Itoh et al, 2011; Carroll et al, 2013; Popovic & Dikic, 2014; Lamb et al, 2016), while TBC1D15 (Rab7‐GAP) and TBC1D17 control mitophagy (Yamano et al, 2014). Nevertheless, regulation of selective autophagy by Rabs and TBC/RabGAPs remains poorly understood.
In this study, we identify TBC/RabGAPs and the corresponding Rabs that affect the autophagosome formation against bacterial pathogen Group A Streptococcus (GAS), a target of selective autophagy. We demonstrate that Rab35 controls GAS degradation by xenophagy through recruiting NDP52 and is also a master regulator of multiple forms of autophagy.
Results
TBC1D10A suppresses xenophagy
To comprehensively screen for TBC/RabGAPs that modulate selective autophagy during GAS infection, we first engineered HeLa cells to overexpress EmGFP‐tagged TBC/RabGAPs and mCherry‐tagged LC3, a marker of autophagic membranes, and infected these cells with GAS. We examined the efficiency of autophagosome formation against GAS 4 h post‐infection, at which point autophagy was highest. Of the 30 TBC/RabGAPs tested, overexpression of TBC1D2, 14, and 22A significantly increased autophagosome formation. In contrast, overexpression of TBC1D10A, 10B, 18, 23, 25, and RN‐Tre significantly suppressed autophagosome formation (Fig 1A and Appendix Fig S1).
Figure 1. TBC1D10A negatively regulates NDP52 during xenophagy.

-
AScreening for TBC/RabGAPs that affect autophagosome formation during GAS infection. HeLa cells overexpressing EmGFP‐tagged TBC/RabGAP and mCherry‐tagged LC3 were infected with GAS for 4 h, and the percentage of cells that formed autophagosomes was determined by confocal microscopy.
-
BAutophagosome formation in HeLa cells overexpressing EmGFP‐TBC1D10A catalytic mutants R160K or D157A, and infected with GAS.
-
C, DHeLa cells expressing indicated FLAG‐TBC1D10 constructs were infected with GAS and analyzed by immunoblotting with indicated antibodies. Data in (D) are mean ± SEM from three independent experiments of LC3‐II 4 h post‐infection and normalized to actin.
-
E, FBacterial invasion (E) and viability (F) of GAS in HeLa cells overexpressing TBC1D10A and TBC1D10A R160K.
-
GRecruitment of indicated proteins to invading bacterial cells, as quantified by confocal microscopy.
-
HConfocal micrographs of NDP52 recruitment to GAS 4 h post‐infection in HeLa cells expressing indicated EmGFP‐TBC1D10A constructs. Scale bars, 10 μm.
To test if the GAP activity of TBC1D10A is necessary to inhibit autophagosome formation, we constructed TBC1D10A mutants, in which a conserved arginine required for GAP activity had been replaced with lysine (R160K), or in which a conserved glutamic acid in the TBC domain had been replaced with alanine (D157A). Wild‐type TBC1D10A reduced autophagosome formation 2 h and 4 h post‐infection (Fig 1B and Appendix Fig S2A). In contrast, the R160K and D157A mutants did not (Fig 1B and Appendix Fig S2A), indicating that GAP activity is essential to inhibit autophagosome formation during GAS infection.
To confirm that TBC1D10A suppresses GAS autophagy, we next investigated the accumulation of phosphatidylethanolamine‐conjugated forms of LC3 (LC3‐II). Conversion of a cytosolic form (LC3‐I) to LC3‐II, which is specifically targeted to autophagosomal membranes, is commonly used to quantify the number of autophagosomes (Kabeya et al, 2000). Infection with GAS increased LC3‐II with time in cells overexpressing FLAG or FLAG‐tagged R160K. However, LC3‐II did not increase in cells overexpressing FLAG‐tagged wild‐type TBC1D10A (Fig 1C and D). These results suggest that decreased autophagy in infected cells overexpressing wild‐type TBC1D10A is due to autophagosome synthesis.
Autophagy and degradation by lysosomal proteases can efficiently eliminate invading GAS (Nakagawa et al, 2004; Nozawa et al, 2012). Thus, we examined the relationship between TBC1D10A and the invasive efficiency of GAS, as well as its survival 6 h post‐infection. While overexpression of TBC1D10A and the R160K mutant did not affect invasive efficiency (Fig 1E), survival was ~1.5 times higher in cells overexpressing TBC1D10A than in control cells and cells overexpressing R160K (Fig 1F). Taken together, the data indicate that GAP activity in TBC1D10A is necessary to inhibit autophagosome formation and subsequent degradation of GAS.
TBC1D10A inhibits recruitment of NDP52 to invading bacteria
Group A Streptococcus invades host epithelial cells through endocytosis. This bacterium produces streptolysin O, a pore‐forming toxin that damages the endosomal membrane. Damaged endosomal membranes are then recognized by cytosolic galectin 8 (O'Seaghdha & Wessels, 2013), while bacteria exposed to the cytosol are coated with ubiquitin, and targeted to autophagosomes via autophagy receptors, including p62, NDP52, and OPTN (O'Seaghdha & Wessels, 2013). To determine whether TBC1D10A inhibits these pathways to suppress autophagosome formation, we examined the recruitment of autophagy markers in cells overexpressing TBC1D10A. Overexpression did not alter the frequency of cells in which GAS was coated with galectin 8, ubiquitin, p62, or OPTN (Fig 1G and Appendix Fig S2B), suggesting that TBC1D10A did not affect the escape of GAS from endosomes to the cytoplasm. However, the frequency of cells with NDP52‐coated bacteria significantly decreased in cells overexpressing TBC1D10A, but not in cells overexpressing R160K and D157A mutants (Fig 1G and H). To confirm that NDP52 is involved in GAS autophagy, we generated NDP52 knockout HeLa cells by CRISPR/Cas9 genome editing (Appendix Fig S3A). Autophagosome formation was significantly decreased in these knockout cells (Appendix Fig S3B and C), suggesting that NDP52 is required to form autophagosomes in response to GAS infection. These findings indicate that TBC1D10A GAP activity inhibits the recruitment of NDP52 to GAS.
Binding of NDP52 with galectin 8 and ubiquitin is required to recruit NDP52 to bacteria during Salmonella infection, and the NDP52 residues L374 and D439 are essential for such interactions, respectively (Thurston et al, 2012, 2016). The D439K mutation significantly decreased recruitment to GAS, while the L374A mutation did not (Appendix Fig S3D and E), suggesting that NDP52‐ubiquitin binding, but not NDP52‐galectin 8 binding, is required for NDP52 recruitment to GAS.
Rab35 regulates xenophagy and recruitment of NDP52 to bacteria
GTPase‐activating protein activity of TBC1D10A against Rab27A and Rab35 is known to regulate melanosome transport (Itoh & Fukuda, 2006) and exosome secretion (Hsu et al, 2010), respectively. Thus, we generated Rab27A and Rab35 knockout cell lines (Fig EV1A–C) and examined NDP52 recruitment and autophagosome formation in these knockout cells. Knockout of Rab35, but not Rab27A, significantly suppressed NDP52 recruitment and autophagosome formation in response to invading bacteria (Figs 2A–D and EV1D). To further confirm these results, NDP52 recruitment and autophagosome formation were examined in HeLa cells in which Rab35 was depleted by miR‐RNAi knockdown (Fig EV1E). Consistent with the results for the Rab35 knockout, similar inhibitory effects were obtained in these cells (Fig EV1F and G). In contrast, Rab35 knockout did not reduce recruitment of galectin 8, ubiquitin, p62, and OPTN (Fig 2E and F). These results suggest that the inhibitory effects of NDP52 recruitment on GAS and autophagosome formation by TBC1D10A are specifically mediated by Rab35. In addition, we investigated whether Rab35 is involved in the NDP52‐ubiquitin interaction by using an in situ proximity ligation assay (PLA) (Leuchowius et al, 2011). Notably, compared to the wild‐type cells, fewer PLA signals in the cytoplasm were observed in Rab35 knockout cells (Fig 2G and H), suggesting that Rab35 promotes the interaction between NDP52 and ubiquitin. Finally, knockout of NDP52 and Rab35 suppressed LC3‐II accumulation in response to GAS (Fig 2I and J). These results suggest that Rab35 and NDP52 are involved in autophagosome biogenesis during GAS infection.
Figure EV1. Analysis of knockdown and knockout of Rab35.
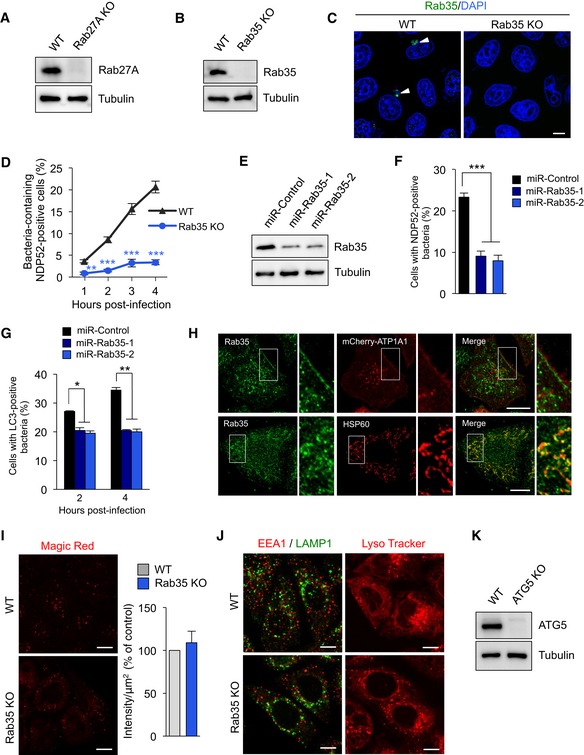
-
A, BKnockout of Rab27 (A) and Rab35 (B) in HeLa cells.
-
CHeLa wild‐type and Rab35 knockout cells were infected with GAS for 4 h, fixed, immunostained with anti‐Rab35 antibody, and stained with DAPI. Arrowheads indicate Rab35‐positive GAS.
-
DTime course of Rab35‐dependent NDP52 recruitment to GAS. HeLa wild‐type and Rab35 knockout cells expressing mCherry‐NDP52 were infected with GAS. The percentages of cells with NDP52‐positive GAS were quantified.
-
EmiR‐RNAi knockdown of Rab35.
-
F, GControl (miR‐Control) and Rab35‐knocked down (miR‐Rab35) HeLa cells expressing mCherry‐NDP52 (F) or mCherry‐LC3 (G) were infected with GAS. Cells were analyzed with confocal microscopy and quantified the percentages of cells with NDP52‐positive or LC3‐positive GAS.
-
HSubcellular localization of endogenous Rab35.
-
IHeLa wild‐type and Rab35 knockout cells were treated with Magic Red Cathepsin B (Magic Red CatB, in red) for 2 h. Quantification of the intensity of the Magic Red Cathepsin B signal, presented as a percentage of control (wild‐type cells).
-
JWild‐type and Rab35 knockout HeLa cells were immunostained with against EEA1 and LAMP1 (left images) or treated with Lysotracker (100 nM) for 90 min (right images).
-
KKnockout of ATG5 in HeLa cells.
Figure 2. Rab35 regulates NDP52 recruitment to invading bacteria.
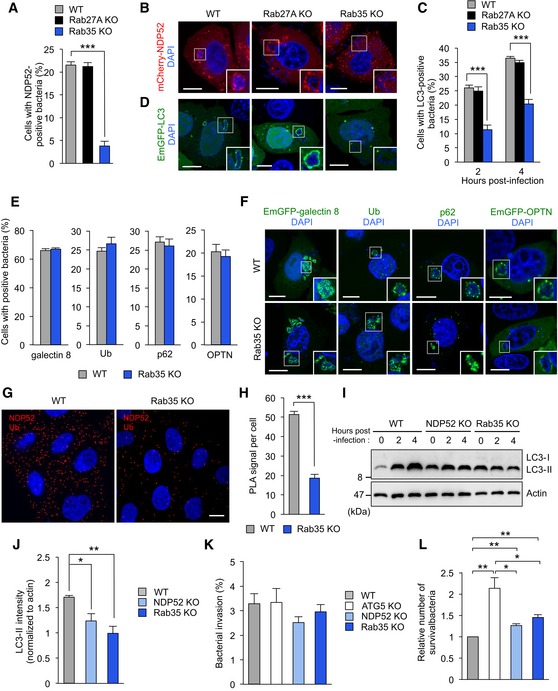
-
A, BQuantification (A) and confocal micrographs (B) of mCherry‐NDP52 recruited to GAS at 4 h post‐infection in wild‐type, Rab27, and Rab35 knockout HeLa cells. Scale bars, 10 μm.
-
C, DAutophagosome formation (C) and confocal micrographs (D) in wild‐type, Rab27, and Rab35 knockout HeLa cells expressing EmGFP‐LC3 and infected with GAS for 4 h. Scale bars, 10 μm.
-
E, FQuantification (E) and confocal micrographs (F) of galectin 8, ubiquitin (Ub), p62, and OPTN recruited to GAS at 4 h post‐infection in wild‐type and Rab35 knockout HeLa cells. Scale bars, 10 μm.
-
G, HHeLa wild‐type and Rab35 knockout cells were stained with NDP52 and ubiquitin primary antibodies to assess NDP52‐Ub binding by Duolink proximity ligation assay. Dots (red) indicate NDP52‐Ub complexes. Scale bars, 10 μm.
-
I, JImmunoblotting of LC3 in wild‐type, NDP52, and Rab35 knockout cells infected with GAS (I). Data in (J) are mean ± SEM from three independent experiments of LC3‐II 4 h post‐infection and normalized to actin.
-
K, LInvasion (K) and viability (L) of GAS in wild‐type, ATG5, NDP52, and Rab35 knockout cells.
While it has been shown that Rab35 regulates endocytosis, neurite outgrowth, exosome release, cytokinesis, and actin organization (Chaineau et al, 2013), its role in autophagy is unknown. We next examined where endogenous Rab35 localizes under basal condition. Endogenous Rab35 was localized on mitochondria and directly under the plasma membrane (Fig EV1H). Because Rab35 was reported to be involved in trafficking the cation‐independent mannose 6‐phosphate receptor (Cauvin et al, 2016), lysosomal enzymatic activity, endosome/lysosome morphology, and lysosomal pH were examined in Rab35 knockout cells. Rab35 knockout did not impair lysosomal enzymatic activity in HeLa cells (Fig EV1I) and did not alter endosome/lysosome morphology and lysosomal pH (Fig EV1J).
We next investigated bactericidal activity in NDP52, Rab35, and autophagy‐deficient ATG5 knockout cells (Fig EV1K). As expected, GAS invasion was comparable between wild‐type and knockout cells (Fig 2K), but survival was significantly higher in the latter (Fig 2L). The number of surviving GAS cells was higher in ATG5 knockout cells than in NDP52 and Rab35 knockout cells (Fig 2L). Taken together, the results indicate that Rab35 is required for NDP52‐mediated autophagosome formation and bactericidal activity.
To determine whether impaired autophagosome formation and NDP52 recruitment to GAS in Rab35 knockout cells are due to loss of GTP‐bound Rab35, we overexpressed EmGFP‐tagged Rab35, as well as Rab35 Q67A, a constitutively active GTP‐bound form, and Rab35 S22N, a dominant negative GDP‐bound form. Overexpression of either mutant significantly decreased autophagosome formation (Fig EV2A and B) and NDP52 recruitment (Fig EV2C and D) in response to GAS. These results suggest that an appropriate level of Rab35‐GTP and/or Rab35 GTPase activity is required to regulate NDP52. Thus, we constructed cell lines stably expressing Rab35 mutants in the knockout background, which were made to express a similar level of Rab35 compared with endogenous Rab35 by using lentiviral vectors (Fig EV2E), and tested whether these cell lines showed rescue of the inhibition of NDP52 recruitment and autophagosome formation. Stable expression of EmGFP‐tagged Rab35 and Rab35 Q67A fully rescued this inhibition, whereas stable expression of Rab35 S22N did not (Fig EV2F–H). These results suggest that GTP‐bound Rab35 regulates NDP52 recruitment to GAS.
Figure EV2. Effects of Rab35 mutants on autophagosome formation and recruitment of NDP52 to bacteria.
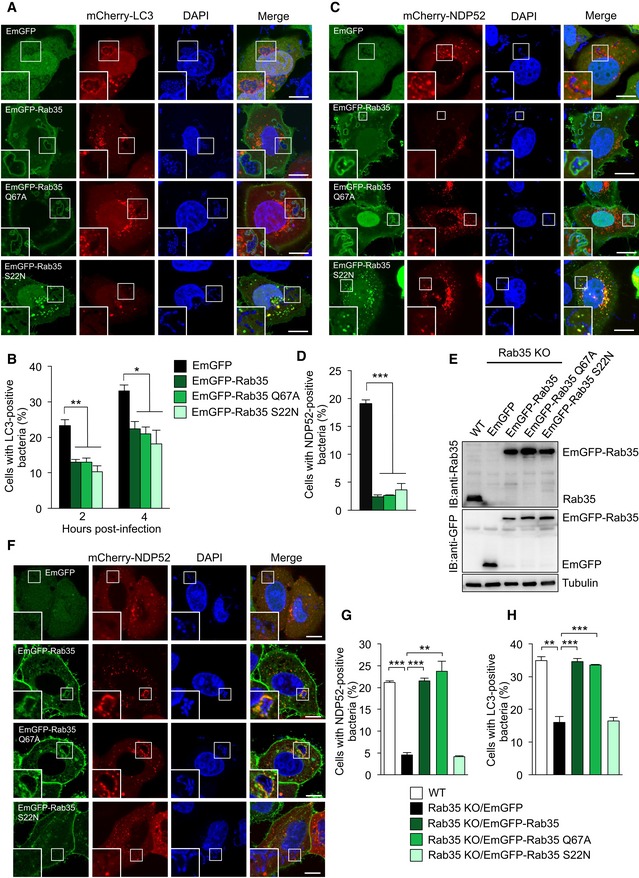
-
A, BRepresentative confocal micrographs (A) and autophagosome formation (B) in HeLa cells expressing mCherry‐LC3 along with EmGFP, EmGFP‐Rab35, EmGFP‐Rab35 Q67A, or EmGFP‐Rab35 S22N, infected for 4 h with GAS, and stained with DAPI.
-
C, DRepresentative confocal micrographs (C), and NDP52‐tagged bacteria (D) in HeLa cells expressing mCherry‐NDP52 along with EmGFP, EmGFP‐Rab35, EmGFP‐Rab35 Q67A, or EmGFP‐Rab35 S22N, infected for 4 h with GAS, and stained with DAPI.
-
EWestern blotting in HeLa wild‐type and Rab35 knockout cells stably expressing EmGFP, EmGFP‐Rab35, EmGFP‐Rab35 Q67A, or EmGFP‐Rab35 S22N.
-
F, GHeLa wild‐type and Rab35 knockout cells stably expressing EmGFP, EmGFP‐Rab35, EmGFP‐Rab35 Q67A, or EmGFP‐Rab35 S22N were transfected with mCherry‐NDP52 and infected with GAS for 4 h. Confocal images (F) and quantification of NDP52 recruitment to GAS (G).
-
HHeLa wild‐type and Rab35 knockout cells stably expressing EmGFP, EmGFP‐Rab35, EmGFP‐Rab35 Q67A, or EmGFP‐Rab35 S22N were transfected with mCherry‐LC3 and infected with GAS for 4 h.
NDP52 and Rab35 are required to activate TBK1 and recruit p‐TBK1S172 to bacteria
Optineurin and NDP52 are required to activate TBK1 during mitophagy (Heo et al, 2015). In turn, TBK1 phosphorylates S403 in p62 and S177 in OPTN to facilitate binding with ubiquitin chains and LC3, respectively (Matsumoto et al, 2011, 2015; Wild et al, 2011; Pilli et al, 2012). Therefore, we examined TBK1 activation in NDP52 and Rab35 knockout cells infected with GAS, as measured by phosphorylation of S172 in TBK1. In wild‐type cells, the ratio of p‐TBK1S172/TBK1 gradually increased to 2.5‐fold 4 h post‐infection (Appendix Fig S4A and B). In contrast, phosphorylated TBK1 was significantly lower in knockout cells than in wild‐type cells (Appendix Fig S4A and B). Further, NDP52 and Rab35 knockout decreased p‐TBK1S172 recruitment to GAS (Appendix Fig S4C and D). These results suggest that NDP52 and Rab35 are required to activate TBK1 and to recruit p‐TBK1S172 to invading bacteria.
Activated Rab35 directly interacts with NDP52 via the zinc‐finger domain
Immunofluorescence analysis shows that endogenous Rab35 is colocalized with endogenous NDP52 recruited to GAS in wild‐type or ATG5 knockout HeLa cells (Fig 3A). These results indicate that Rab35 colocalizes with NDP52 independently of autophagy. Supporting the interaction between Rab35 and NDP52, we found that antibodies against Rab35 coprecipitated endogenous NDP52 from lysates of both uninfected and infected cells (Fig 3B). To determine whether interaction of NDP52 with ubiquitin is required for interaction of NDP52 with Rab35, we investigated the interaction between the NDP52 D439K mutant and Rab35. Immunoprecipitation demonstrated that the EmGFP‐NDP52 D439K mutant interacted with FLAG‐Rab35 (Appendix Fig S5A), suggesting that NDP52‐ubiquitin binding is not essential for NDP52‐Rab35 binding.
Figure 3. Rab35 interacts with the zinc‐finger domain in NDP52.
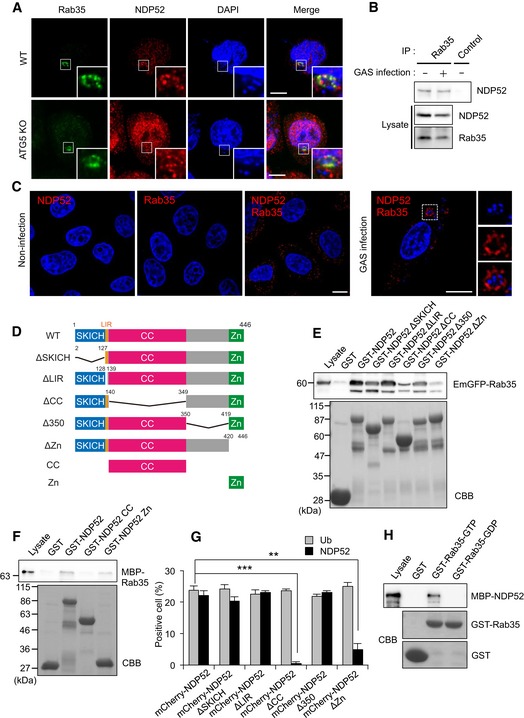
- Wild‐type and ATG5 knockout cells were infected with GAS for 4 h and immunostained with anti‐NDP52 and anti‐Rab35. Scale bars, 10 μm.
- Coimmunoprecipitation of endogenous Rab35 and NDP52 in uninfected HeLa cells and HeLa cells infected with GAS for 4 h.
- HeLa cells infected with or without GAS were stained with Rab35 and NDP52 primary antibodies to assess Rab35‐NDP52 binding by Duolink proximity ligation assay. Dots (red) indicate Rab35‐NDP52 complexes. Scale bars, 10 μm.
- Domain organization and deletion mutants of NDP52.
- HEK293T cells were transfected with EmGFP‐Rab35 and analyzed by pull‐down assay using immobilized GST or indicated GST‐NDP52 constructs. Rab35 was detected with anti‐GFP.
- Beads coated with GST or indicated GST‐NDP52 constructs were incubated with MBP‐Rab35 and immunoblotted with an antibody against MBP.
- Quantification of ubiquitin‐ and NDP52‐tagged bacteria, as measured by confocal microscopy. Data are mean ± SEM of three independent experiments. Data were tested by two‐tailed Student's t‐test: **P < 0.01, ***P < 0.001.
- Beads coated with GST or GST‐Rab35 loaded with GDP or GTP were incubated with MBP‐NDP52 and immunoblotted with an anti‐MBP antibody.
Source data are available online for this figure.
We further validated the Rab35‐NDP52 interaction using PLA. PLA signals were scattered in the cytoplasm in non‐infected cells (Fig 3C, left panels), suggesting that interaction is observed between Rab35 and NDP52 in basal level. On the other hand, clear PLA signals surrounded GAS during infection (Fig 3C, right panels). These observations indicate that the interaction between Rab35 and NDP52 accumulates around GAS‐containing vacuoles during infection.
Human NDP52 contains a skeletal muscle and kidney‐enriched inositol phosphatase carboxyl homology domain (SKICH, aa 1–127) at the amino terminus, an LC3‐interacting region (LIR, aa 134–136), a coiled‐coil domain (CC, aa 140–349), and a ubiquitin‐binding zinc‐finger domain (Zn, aa 420–446, Fig 3D; von Muhlinen et al, 2012). Thus, we tested the interaction of EmGFP‐Rab35 overexpressed in HEK293T cells with purified GST‐tagged NDP52 and deletion mutants ΔSKICH, ΔLIR, ΔCC, Δ350, and ΔZn. While GST‐NDP52 robustly interacted with EmGFP‐Rab35, the deletion mutants ΔCC and ΔZn bound EmGFP‐Rab35 only weakly (Fig 3E). To quantify the interaction between Rab35 and NDP52 deletion mutants, we performed a fluorescence protein fragment complementary assay. In this assay, green fluorescence is observed upon formation of a complex between a fusion protein tagged with the N‐terminal region of monomeric Kusabira Green (mKGN) and another fusion protein tagged with the C‐terminal region (mKGC; Kerppola, 2006). We found that co‐expression of mKGN‐NDP52 and mKGC‐Rab35 elicited green fluorescence. ΔSKICH, ΔLIR, ΔCC, Δ350, and ΔZn generated fluorescence intensity similar to that of wild‐type NDP52, whereas the fluorescence was drastically decreased when ΔCC and ΔZn were expressed (Appendix Fig S5B and C). These results suggest that the NDP52‐Rab35 interaction involves the coiled‐coil and zinc‐finger domains in NDP52. To verify the direct interaction between Rab35 and NDP52, purified GST‐NDP52, GST‐NDP52 140‐349 fragment (GST‐NDP52 CC), and GST‐NDP52 420‐446 fragment (GST‐NDP52 Zn; Fig 3D) were tested their binding with MBP‐Rab35. MBP‐Rab35 interacted with GST‐NDP52 and GST‐NDP52 Zn, but not with GST‐NDP52 CC (Fig 3F). These results suggest that Rab35 directly binds to the zinc‐finger domain in NDP52. On the other hand, recruitment of mCherry‐tagged ΔCC and ΔZn to GAS was dramatically decreased relative to mCherry‐NDP52, even though ubiquitin labeling was unaffected (Fig 3G and Appendix Fig S5D), suggesting that the coiled‐coil and zinc‐finger domains in NDP52 are both required for recruitment to GAS.
To determine whether the Rab35‐NDP52 interaction depends on the nucleotide‐bound status of Rab35, we performed fluorescence protein fragment complementary assay. In particular, GTP‐locked Q67A mutant generated intense fluorescence, while co‐expression of NDP52 and GDP‐locked S22N mutant did not (Appendix Fig S5E and F). Additionally, we demonstrated that recombinant MBP‐NDP52 specifically bound to GTP‐loaded GST‐Rab35, but not GDP‐loaded GST‐Rab35 (Fig 3H). These results suggest that GTP‐bound Rab35 directly interacts with NDP52.
Rab35‐dependent autophagy degrades NDP52
Since autophagy receptors such as p62 and NBR1 are degraded along with the cargo (Bjorkoy et al, 2005; Kirkin et al, 2009), to test if NDP52 is similarly degraded during autophagy, wild‐type HeLa and ATG5 knockout cells, expressing NDP52 with a dual EmGFP‐mCherry tag, were infected with GAS for 4 h. Since EmGFP is rapidly quenched in acidic lysosomes while mCherry remains fluorescent in acid, entry of NDP52 into acidic lysosome can be monitored by selective decrease in EmGFP signals (Kimura et al, 2007). EmGFP fluorescence surrounding bacteria was partly lost in approximately 60% (Fig 4A and B) of wild‐type cells. On the other hand, EmGFP was detected without loss of fluorescence in knockout cells (Fig 4A and B). In addition, GAS infection elicited time‐dependent degradation of endogenous NDP52 and p62 in wild‐type cells. Intriguingly, degradation was impaired in cells from which ATG5, NDP52, and Rab35 had been knocked out (Fig 4C–E). Moreover, expression of NDP52 and p62 was significantly higher in ATG5 knockout cells than in wild‐type cells (Fig 4C–E). Collectively, these data strongly suggest that NDP52 is degraded by autophagy in a Rab35‐dependent manner.
Figure 4. NDP52 is targeted by Rab35‐dependent autophagy.
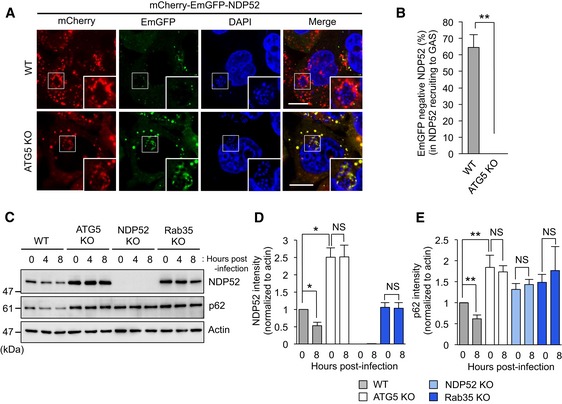
-
AConfocal micrographs of wild‐type HeLa and ATG5 knockout cells expressing mCherry‐EmGFP‐NDP52 and infected with GAS for 4 h. The reporter appears yellow in autophagosomes due to fluorescence from mCherry and EmGFP, but appears red in autolysosomes due to acid quenching of GFP fluorescence. Scale bars, 10 μm.
-
BAutolysosomal NDP52 (red fluorescence in panel A) as a percentage of total NDP52 (red and yellow fluorescence in panel A). Data are mean ± SEM of more than 30 cells.
-
C–ETime course of NDP52 and p62 expression in wild‐type, ATG5, NDP52, and Rab35 knockout HeLa cells infected with GAS. Expression was normalized to actin and plotted in (D and E).
Rab35 regulates recruitment of NDP52 to damaged mitochondria
NDP52 is also known to be the primary receptor for PINK1‐ and Parkin‐mediated mitophagy (Lazarou et al, 2015). To confirm recruitment of NDP52 to damaged mitochondria, HeLa cells expressing EmGFP‐Parkin were treated with CCCP, a compound that depolarizes and damages mitochondria to facilitate mitophagy. mCherry‐NDP52 clearly colocalized with damaged mitochondria and EmGFP‐Parkin in wild‐type cells (Fig 5A). Notably, the NDP52 mutants ΔSKICH, ΔLIR, and Δ350 were also recruited to damaged mitochondria, whereas ΔCC and ΔZn were not (Appendix Fig S6A), suggesting that NDP52‐ubiquitin and NDP52‐Rab35 interactions are necessary to recruit NDP52 to depolarized mitochondria. On the other hand, endogenous Rab35 was clearly colocalized with mCherry‐NDP52 in HeLa cells transiently expressing EmGFP‐Parkin and exposed to CCCP, implying that Rab35 is involved in Parkin‐mediated mitophagy (Fig 5B). Immunoelectron microscopy analysis revealed that Rab35 localized on the mitochondrial membrane under the mitochondrial depolarization condition with CCCP (Fig 5C). Wild‐type Rab35 and the GTP‐locked Q67A mutant were detected in damaged mitochondria, whereas the GDP‐locked S22N mutant was not (Appendix Fig S6B), suggesting that Rab35 localizes to depolarized mitochondria in a GTP‐dependent manner. In contrast, knockout of NDP52 did not affect Rab35 recruitment to depolarized mitochondria, suggesting that NDP52 is not involved in Rab35 recruitment to depolarized mitochondria (Fig EV3A).
Figure 5. Rab35 regulates NDP52 translocation to damaged mitochondria.
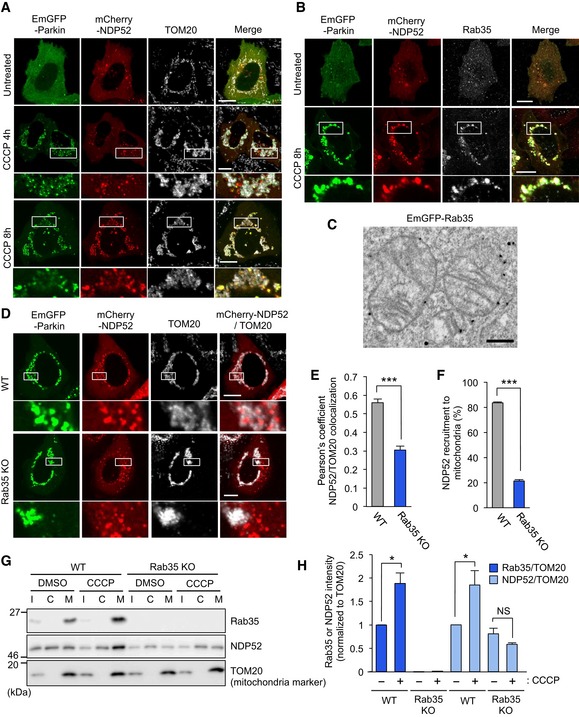
-
A, BConfocal micrographs of NDP52 in HeLa cells transfected with EmGFP‐Parkin and mCherry‐NDP52, exposed to 10 μM CCCP for 4 or 8 h, and immunostained with antibodies against mitochondrial TOM20 (A) or Rab35 (B). Scale bars, 10 μm.
-
CImmunoelectron micrograph of HeLa cells transfected with EmGFP‐Rab35 and mCherry‐Parkin, exposed to 10 μM CCCP for 3 h, and labeled with anti‐GFP antibodies followed by gold particles. Scale bars, 500 nm.
-
D–FRepresentative confocal micrographs (D), Pearson's coefficient between NDP52 and TOM20 (E), and NDP52 recruitment to damaged mitochondria (F) in wild‐type and Rab35 knockout cells transfected with EmGFP‐Parkin and mCherry‐NDP52, treated with 10 μM CCCP for 8 h, and immunostained with anti‐TOM20. Scale bars, 10 μm. Data in (E) are mean ± SEM from > 10 cells, and data in (F) are mean ± SEM from > 100 cells in three independent experiments.
-
G, HSubcellular fractionation of endogenous Rab35 and NDP52. HeLa cells expressing EmGFP‐Parkin were treated with 10 μM CCCP or DMSO and fractionated. I, C, and M in Western blot images indicate input, cytosol‐rich supernatant, and mitochondria‐rich membrane pellet, respectively (G). Rab35 and NDP52 intensities were normalized to TOM20 in the mitochondria‐rich membrane pellet and quantified (H). Data in (H) are mean ± SEM from three independent experiments.
Figure EV3. Involvement of Rab35 and TBC1D10A in NDP52 recruitment to damaged mitochondria.

-
AConfocal micrographs of mCherry‐Rab35 recruited to depolarized mitochondria in HeLa wild‐type (WT) and NDP52 knockout cells expressing EmGFP‐Parkin, exposed to CCCP for 8 h, and immunostained with antibodies against mitochondrial TOM20.
-
B, CHeLa cells expressing EmGFP‐Parkin along with mCherry‐NDP52 treated with antimycin A (4 μM) and oligomycin (10 μM) for 8 h and immunostained with anti‐TOM20. Representative confocal micrographs (B) and quantification (C) of NDP52 recruitment to mitochondria.
-
DHeLa cells were treated with CCCP for the indicated times and analyzed by Western blot against NDP52, Rab35, and actin.
-
E, FHeLa cells expressing EmGFP‐Parkin, mCherry‐NDP52, and FLAG, FLAG‐TBC1D10A, or FLAG‐TBC1D10A R160K were treated with antimycin A and oligomycin for 8 h, and immunostained with anti‐TOM20 and anti‐FLAG. Representative confocal micrographs (E) and quantification (F) of NDP52 recruitment to mitochondria.
Importantly, we found that 8 h after CCCP treatment, NDP52 clearly colocalized with mitochondria in wild‐type HeLa cells, but not in Rab35 knockout cells, as visualized by confocal microscopy and quantified by Pearson's correlation coefficient (Fig 5D and E). The percentage of cells with NDP52 recruited to depolarized mitochondria was significantly reduced in knockout cells (Fig 5F). Similarly, induction of mitochondrial depolarization by antimycin A and oligomycin (AO) treatment in Rab35 knockout cells also inhibited NDP52 recruitment to depolarized mitochondria (Fig EV3B and C). To further confirm the recruitment of NDP52 and Rab35 to damaged mitochondria, we analyzed mitochondrial and cytosolic fractions by Western blot against NDP52 and Rab35. Both NDP52 and Rab35 significantly increased after CCCP treatment in the mitochondria‐enriched fraction (Fig 5G and H). Moreover, CCCP‐induced accumulation of NDP52 in the mitochondria‐enriched fraction was impaired in Rab35 knockout cells (Fig 5G and H). The total expression of NDP52 in wild‐type cells decreased with time after CCCP treatment, whereas that of Rab35 did not change (Fig EV3D). Collectively, these data indicate that Rab35 efficiently recruits NDP52 to depolarized mitochondria.
We examined whether TBC1D10A is involved in NDP52 recruitment during mitophagy. Overexpression of TBC1D10A significantly suppressed NDP52 recruitment to depolarized mitochondria after AO treatment, whereas expression of the R160K mutant did not (Fig EV3E and F), suggesting that GAP activity of TBC1D10A negatively regulates NDP52 recruitment to damaged mitochondria.
Rab35 regulates NDP52‐mediated autophagosome maturation in starvation‐induced and basal autophagy
NDP52 was reported to facilitate autophagosome maturation via a complex that includes myosin VI, TRAF6‐binding protein, and OPTN under the basal condition (Tumbarello et al, 2012). TBK1, which interacts with NDP52, is necessary for autophagosome maturation during starvation‐induced autophagy, as well as Mycobacterium tuberculosis var. bovis BCG‐induced autophagy (Pilli et al, 2012). Thus, we speculated that Rab35 may be involved in starvation‐induced and basal autophagy by spatially regulating NDP52. NDP52 was detected in basal and starvation‐induced autophagosomes (Fig EV4A). The zinc‐finger domain was dispensable to target NDP52 to autophagosomes, while the coiled‐coil domain was required (Fig EV4A). In wild‐type HeLa cells stably expressing GFP‐LC3, fluorescent puncta increased in response to nutrient starvation (Fig 6A and B). In NDP52 and Rab35 knockout cells, fluorescent puncta accumulated even under basal conditions (Fig 6A and B). Moreover, knockout of NDP52 and Rab35 increased the abundance of LC3‐II under basal conditions (Fig 6C and D). LC3‐II levels increased in these cells when autophagosome–lysosome fusion and lysosomal degradation were blocked using bafilomycin A1, and similar levels of LC3‐II were accumulated among wild‐type and knockout cells (Fig 6C and D). These results suggest that autophagic degradation is impaired in NDP52 or Rab35 knockout cells.
Figure EV4. Recruitment of NDP52 to starvation‐induced and basal autophagosomes.
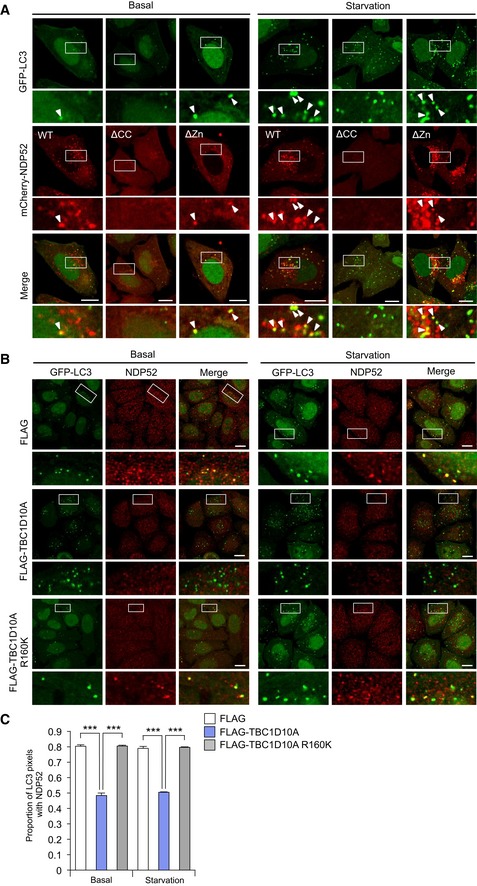
-
AConfocal micrographs of autophagosomes in HeLa cells stably expressing GFP‐LC3 along with mCherry‐NDP52, mCherry‐NDP52ΔCC, or mCherry‐NDP52ΔZn, and cultured in regular (basal) or starvation medium for 2 h. Scale bars, 10 μm. Arrowheads indicate the NDP52‐positive LC3 puncta.
-
B, CHeLa cells stably expressing GFP‐LC3 along with FLAG, FLAG‐TBC1D10A, or FLAG‐TBC1D10A R1660K were cultured in regular (basal) or starvation medium for 2 h and immunostained for NDP52. Confocal images (B) and proportion of LC3 puncta colocalized with NDP52 from at least 30 randomly selected cells were quantified by Mander's coefficient M1 (C). Scale bars, 10 μm. Data were tested by two‐tailed Student's t‐test: ***P < 0.001. Error bars indicate mean ± SEM.
Figure 6. Rab35 regulates NDP52‐mediated autophagosome maturation in starvation‐induced and basal autophagy.
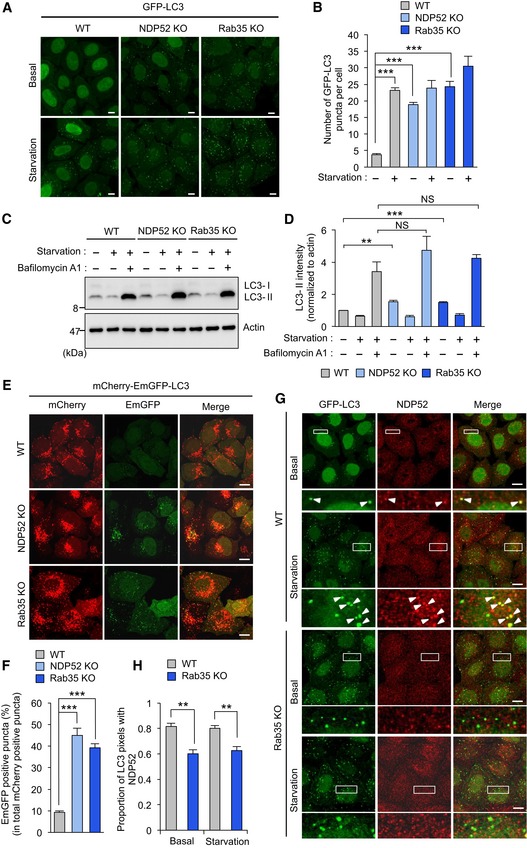
-
A, BConfocal micrographs of wild‐type, NDP52, and Rab35 knockout cells stably expressing GFP‐LC3 and cultured for 2 h in regular (basal condition) or starvation medium (A). LC3‐labeled autophagosomes from at least 45 randomly selected cells were then quantified (B).
-
C, DWild‐type, NDP52, and Rab35 knockout cells were cultured in regular or starvation medium for 2 h and analyzed by immunoblotting (C). LC3‐II was quantified and normalized to actin (D).
-
E, FRepresentative confocal micrographs (E) and percentage of EmGFP‐fluorescent puncta from at least 10 randomly selected fields were quantified (F) in wild‐type HeLa, NDP52, and Rab35 knockout cells stably expressing mCherry‐EmGFP‐LC3.
-
G, HRepresentative confocal micrographs (G), and proportion of LC3 puncta colocalized with NDP52 from at least 30 randomly selected fields were quantified by Mander's coefficient M1 (H) in wild‐type and Rab35 knockout cells stably expressing GFP‐LC3 and cultured in regular or starvation medium for 2 h, and immunostained with anti‐NDP52. Arrowheads indicate the NDP52‐positive LC3 puncta.
To demonstrate if NDP52 and Rab35 are involved in autophagosome maturation, we examined autophagic flux in HeLa cells stably expressing an mCherry‐EmGFP‐LC3 tandem reporter, which appears yellow in autophagosomes due to combined fluorescence from mCherry and EmGFP, but appears red in autolysosomes due to acid quenching of EmGFP fluorescence. In wild‐type cells, many red puncta were observed under basal conditions, along with a smaller number of yellow puncta (Fig 6E). However, knockout of NDP52 or Rab35 generated many yellow puncta, and significantly increased the ratio of autophagosomes to autolysosomes (Fig 6E and F), suggesting that transition from autophagosomes to autolysosomes was suppressed. Moreover, many LC3‐containing autophagic structures in wild‐type cells also contained endogenous NDP52 under basal and starvation conditions (Fig 6G). Additionally, a significantly smaller proportion of LC3 vesicles contained endogenous NDP52 in Rab35 knockout cells (Fig 6G and H), demonstrating that recruitment of NDP52 to basal or starvation‐induced autophagosomes is also regulated by Rab35. Collectively, our results indicate that Rab35 regulates autophagosome maturation in starvation‐induced and basal autophagy by efficiently recruiting NDP52.
Moreover, we examined whether TBC1D10A is involved in NDP52 recruitment in basal and starvation‐induced autophagy. Overexpression of TBC1D10A significantly reduced endogenous NDP52 recruitment to basal and starvation‐induced autophagosomes, whereas overexpression of R160K mutant did not, suggesting that GAP activity of TBC1D10A negatively regulates NDP52 recruitment to basal and starvation‐induced autophagosomes (Fig EV4B and C).
TBK1 facilitates the interaction of NDP52 with Rab35
To investigate the mechanism of Rab35‐mediated recruitment of NDP52 during autophagy, we determined the subcellular localization of Rab35 during GAS infection. Endogenous Rab35 was colocalized with endogenous galectin 3, a marker of endosomes damaged by invading pathogens (Paz et al, 2010; Fig EV5A). This process depends on GTP‐bound form of Rab35, since wild‐type Rab35 and the GTP‐locked Q67A mutant were recruited to GAS, whereas the GDP‐locked S22N mutant was not (Fig EV2A and F). Immunoelectron microscopy analysis revealed that Rab35 was located at vacuoles surrounding GAS, but was not double‐membrane autophagic structures (Fig EV5B). Furthermore, live cell imaging showed that decoration of GAS cells by Rab35 preceded appearance of galectin 3 (Fig EV5C). Collectively, these results suggest that prior to galectin recruitment, Rab35 localizes in a GTP‐dependent manner on bacteria‐containing endosomes.
Figure EV5. Rab35 localizes to bacteria‐containing endosomes prior to membrane damage.

-
AHeLa cells were infected with GAS for 4 h and stained with anti‐galectin 3 and anti‐Rab35. Scale bars, 10 μm.
-
BImmunoelectron microscopy analysis revealed colloidal gold particles, indicating the presence of GFP‐Rab35 around invading GAS. Scale bars, 0.5 μm.
-
CLive cell images of HeLa cells expressing EmGFP‐Rab35 and mCherry‐galectin 3, or mCherry‐Rab35, and EmGFP‐galectin 3 during GAS infection. Arrowheads indicate Rab35‐positive GAS. Scale bars, 2 μm.
-
D, EHeLa cells expressed mCherry‐NDP52 or‐Rab35 were treated with DMSO or 4 μM BX795 for 24 h and infected with GAS for indicated times. The percentages of cells with NDP52 (D)‐ or Rab35 (E)‐positive bacteria were quantified.
-
F, GHEK293T cells transfected with mKGN‐NDP52 and mKGC‐Rab35 or mKGN‐NDP52ΔZn and mKGC‐Rab35 were treated or untreated with BX795, and the green fluorescence was analyzed by microscopy. Representative micrographs (F) and quantification of fluorescence intensity (G). Scale bars, 10 μm. White lines indicate the outline of the cells.
Since TBK1 regulates NDP52 recruitment to invading M. tuberculosis and damaged mitochondria during xenophagy and mitophagy, respectively (Watson et al, 2012; Heo et al, 2015), we speculated that TBK1 may also be required for Rab35‐mediated NDP52 recruitment. Immunoprecipitation demonstrated that EmGFP‐Rab35 interacted with FLAG‐NDP52 and that this interaction is blocked by BX795, an inhibitor of TBK1 (Fig 7A), suggesting that TBK1 kinase activity promotes NDP52‐Rab35 interaction. BX795 also inhibited the recruitment of NDP52 to GAS without affecting Rab35 recruitment (Figs 7B and C, and EV5D and E). Furthermore, the fluorescence protein fragment complementary assay showed that the interaction of NDP52 with Rab35 was inhibited by BX795 (Fig EV5F and G). Notably, the interaction of NDP52ΔZn with Rab35 was also reduced, suggesting that a region of NDP52 other than the Zn domain facilitates the interaction with Rab35 via TBK1 activity (Fig EV5F and G). Consistent with an idea that TBK1 activation is required for the interaction of NDP52 with Rab35, knockout of TBK1 (Fig EV6A) also prevented the interaction of NDP52 with Rab35 (Fig 7D and E, and EV6B) and suppressed the recruitment of NDP52 to GAS, as well as the subsequent formation of autophagosomes (Figs 7F–I and EV6C and D). However, TBK1 knockout did not alter galectin 8, ubiquitin, and Rab35 to GAS (Fig 7F and G). Similarly, TBK1 knockout impaired NDP52 recruitment to damaged mitochondria without inhibiting Rab35 recruitment (Fig EV6E). Additionally, TBK1 knockout inhibited effective autophagic flux (Fig EV6F and G) and NDP52 recruitment to basal and starvation‐induced autophagosomes (Fig EV6H and I). Taken together, these findings indicate that TBK1 is involved in NDP52 recruitment to autophagic targets by promoting the interaction of NDP52 with Rab35.
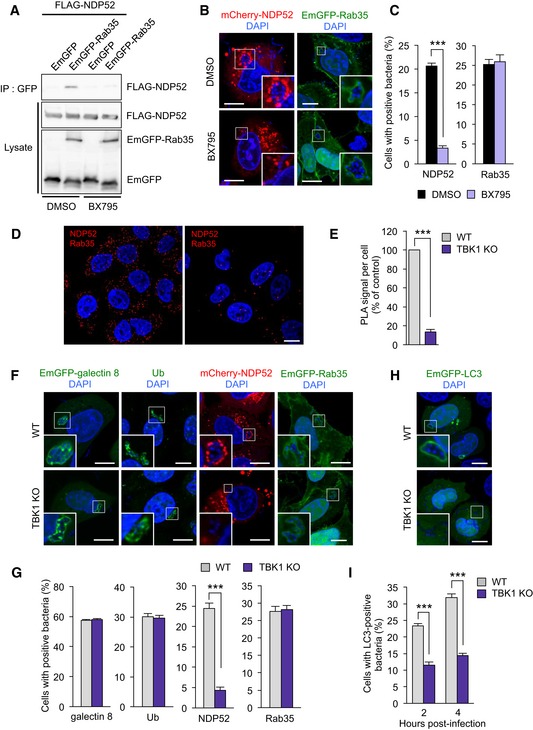
Figure 7. TBK1 regulates NDP52 recruitment to bacteria by facilitating NDP52‐Rab35 interaction.
-
ACoimmunoprecipitation of EmGFP‐Rab35 and FLAG‐NDP52 in HEK293T cells treated with DMSO or BX795.
-
B, CHeLa cells expressing mCherry‐NDP52 or EmGFP‐Rab35 were treated with DMSO or 4 μM BX795 for 24 h and infected with GAS for 4 h. Representative confocal micrographs (B) and percentages of cells with NDP52‐ or Rab35‐tagged bacteria (C). The error bars indicate the mean ± SEM from three independent experiments.
-
D, EWild‐type or TBK1 knockout HeLa cells were stained with Rab35 and NDP52 primary antibodies to assess Rab35‐NDP52 binding using Duolink‐PLA. Representative confocal micrographs (D) and quantification of PLA dots per cell (E). Data in (E) are mean ± SEM from > 50 cells in three independent experiments.
-
F, GConfocal micrographs (F) and quantification (G) of galectin 8, ubiquitin (Ub), NDP52, and Rab35 recruited to GAS 4 h post‐infection in wild‐type and TBK1 knockout HeLa cells. The error bars indicate the mean ± SEM from three independent experiments.
-
H, IConfocal micrographs (H) and quantification (I) of LC3 recruited to GAS in wild‐type and TBK1 knockout HeLa cells. The error bars indicate the mean ± SEM from three independent experiments.
Figure EV6. TBK1 regulates NDP52 recruitment to GAS and mitochondria by promoting Rab35‐NDP52 interaction.
-
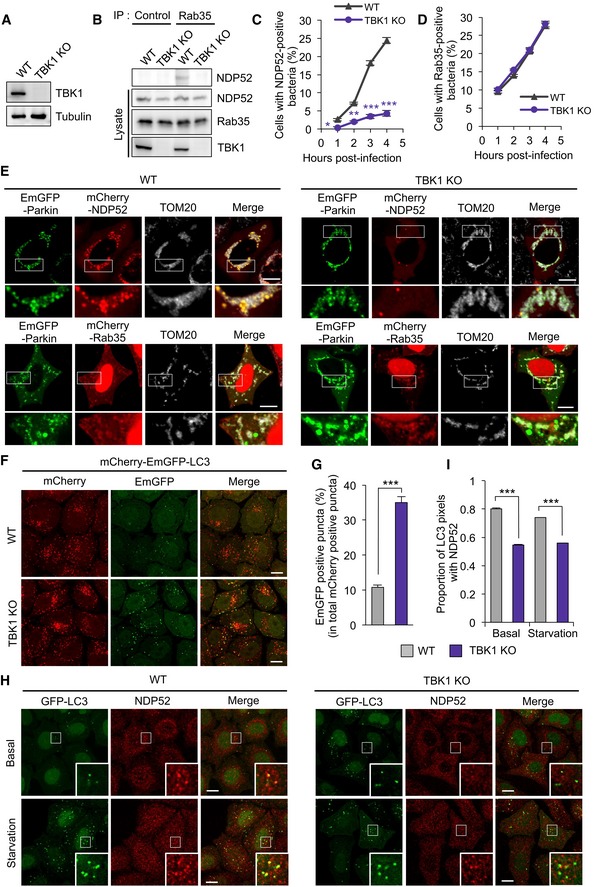
AKnockout of TBK1 in HeLa cells.
-
BCoimmunoprecipitation of endogenous Rab35 and NDP52 in wild‐type or TBK1 knockout HeLa cells.
-
C, DHeLa cells expressed mCherry‐NDP52 or‐Rab35 were treated with DMSO or 4 μM BX795 for 24 h and infected with GAS for indicated times. The percentages of cells with NDP52 (C)‐ or Rab35 (D)‐positive bacteria were quantified.
-
EWild‐type and TBK1 knockout HeLa cells expressed EmGFP‐Parkin and either mCherry‐NDP52 or ‐Rab35 were treated with 10 μM CCCP for 8 h. Cells were fixed and immunostained with antibody against TOM20.
-
F, GWild‐type and TBK1 knockout HeLa cells stably expressing mCherry‐EmGFP‐LC3 were cultured under starvation for 2 h. Representative confocal images (F) and quantification (G) of EmGFP‐positive puncta.
-
H, IWild‐type and TBK1 knockout HeLa cells stably expressing GFP‐LC3 were cultured under nutrient (basal) or starvation condition for 2 h. Cells were fixed and immunostained with an antibody against NDP52. Representative confocal micrographs (H) and proportion of LC3 puncta colocalized with NDP52 from at least 30 randomly selected fields were quantified by Mander's coefficient M1 (I).
Discussion
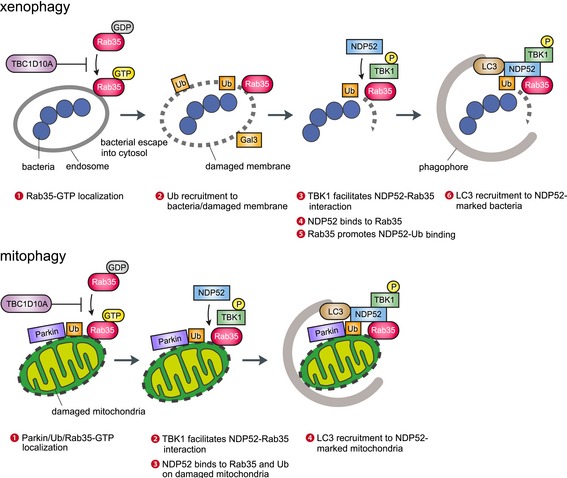
Autophagy attacks multiple envelope architectures that contain damages or pathogens, but how it can distinguish between targets and nontargets remains unclear. Here, we provide evidence that Rab35 plays a critical role in this process through recruiting the autophagy receptor NDP52. The working model for these processes is presented in Fig 8. Upon GAS infection, Rab35, also a regulator of endocytosis (Chaineau et al, 2013), accumulates on bacteria‐containing endosomes ahead of galectin and in a GTP‐dependent manner. This process is suppressed by TBC1D10A, GAP of Rab35 (Hsu et al, 2010). Active GTP‐bound form of Rab35 then directly recruits NDP52 in a manner dependent on TBK1 activity. In addition, Rab35 facilitates the interaction of NDP52 with ubiquitin. This pathway was not only limited to GAS‐induced xenophagy. Rab35 and TBK1 promote targeting NDP52 to damaged mitochondria in mitophagy, and TBC1D10A negatively controls this process by exchanging GTP‐Rab35 with GDP‐Rab35 (Fig 8). Similarly, Rab35 and TBK1 also regulate autophagosome maturation in starvation‐induced and basal autophagy, also via NDP52, and TBC1D10A negatively regulates this process.
Figure 8. Model of targeting for autophagosome through Rab35 and TBK 1‐mediated NDP52 recruitment in xenophagy and mitophagy.
Experiments with knockout cells demonstrated that NDP52 is involved in the autophagy response to GAS infection, in agreement with studies indicating that NDP52 helps clear bacteria from the cytosol (Thurston et al, 2009). However, galectin 8 binding is not required for NDP52 recruitment to GAS, in contrast to the cellular response against Salmonella. This difference might be due to the ability of Salmonella to damage endosomes via SPI‐1 type III secretion system without completely disintegrating the encapsulating multilamellar structures (Zheng et al, 2009), while GAS secretes streptolysin O that disrupts the structural integrity of endosome membranes (Nakagawa et al, 2004). Importantly, these experiments also demonstrated that Rab35 and its inactivation by TBC1D10A regulate autophagosome biogenesis by recruiting NDP52 to invading bacteria. However, we cannot explain at present why Rab35 overexpression elicits the same effects as Rab35 knockdown or knockout. We note that a similar observation was recently reported for Rab40C, overexpression and knockdown of which both repressed production of melanin (Yatsu et al, 2015). As overexpression of Rab35, Rab35 Q67A, and Rab35 S22N suppressed recruitment of NDP52 to bacteria and autophagosomes and stable expression of Rab35 and Rab35 Q67A rescued this inhibition in Rab35 knockout cells, NDP52 subcellular localization may indeed be finely tuned to the balance and precise cycling between the GTP‐bound active and the GDP‐bound inactive form of Rab35, as well as to its abundance.
We also found that NDP52 directly interacts with the GTP‐bound form of Rab35. Because NDP52 binds the motor protein myosin VI to facilitate autophagosome–lysosome fusion (Tumbarello et al, 2012), NDP52 would serve as a Rab35 effector in regulating autophagosome transport dependent on myosin VI. We also demonstrated that the zinc‐finger domain in NDP52 directly interacts with Rab35, and that TBK1 activity is required for facilitating the interaction. Furthermore, we also revealed that a region of NDP52 other than the zinc‐finger domain is required for the NDP52‐Rab35 interaction via TBK1 activity. Given that the coiled‐coil domain is believed to be involved in NDP52 dimerization (Korioth et al, 1995), it may increase the avidity of the interaction via TBK1 activity. Recently, TBK1 was reported to phosphorylate the coiled‐coil and SKICH domains in NDP52 (Richter et al, 2016). In contrast, the coiled‐coil and zinc‐finger domains were required for recruitment of NDP52 in xenophagy and mitophagy. Although the zinc‐finger domain in NDP52 directly binds with Rab35, the zinc‐finger domain was not required to recruit NDP52 to autophagosomes under basal and starvation conditions, as previously reported (Tumbarello et al, 2012). The phosphorylation of the coiled‐coil domain in NDP52 via TBK1 may play an important role in the interaction of NDP52 with Rab35.
Although TBK1 is already known to phosphorylate NDP52 (Heo et al, 2015; Richter et al, 2016), it is unclear whether TBK1 indirectly or directly phosphorylates NDP52 and promotes the NDP52‐Rab35 interaction. Intriguingly, TBK1 was recently reported to directly phosphorylate SMCR8, an interaction partner of C9ORF72 and WDR41 (GEF of Rab39B), and these proteins thereby control autophagic flux (Sellier et al, 2016). It may also phosphorylate Rab35 or other related proteins. Thus, TBK1 substrates and the corresponding phosphorylation sites would have to be identified and validated in future studies.
We found that Rab35 regulates multiple forms of autophagy by recruiting NDP52 in human cells. However, we do not know whether NDP52 and Rab35 function in the same manner in vivo. We note that mouse NDP52 (mNDP52) is a truncated form lacking the C‐terminal zinc‐finger domain (Tumbarello et al, 2015), which interacts with ubiquitin and Rab35. This suggests that analysis of NDP52 and Rab35 in mice might not be informative regarding human NDP52 and Rab35 function, although mouse models are otherwise generally useful for in vivo studies. Interestingly, NDP52 can bind to phosphorylated tau via SKICH domain and facilitates autophagy‐mediated degradation of tau in mouse (Jo et al, 2014), suggesting that NDP52 recognizes different targets through different domains. NDP52 might be controlled by other regulator besides Rab35 in mouse.
The missense mutation V248A in NDP52 was recently implicated in several diseases, including Crohn's disease (Ellinghaus et al, 2013). This mutation also increases the risk of spontaneous bacterial peritonitis in patients with alcoholic liver cirrhosis (Lutz et al, 2016). Nevertheless, this point mutant is effectively recruited to damaged mitochondria (Lazarou et al, 2015) and GAS (data not shown), even though the mutation is in the coiled‐coil domain. Thus, genomewide association studies and further analysis of NDP52 regulation are needed to clarify the pathogenesis of autophagy‐associated disease.
In conclusion, our data unveil the functional significance of Rab35 in multiple forms of autophagy through recruiting NDP52. Our discovery of the Rab35‐NDP52 axis may help elucidate the pathogenesis of autophagy‐associated diseases, as well as in identifying therapeutic targets.
Materials and Methods
Cell culture and transfection
HeLa and HEK293T cells were maintained in 5% CO2 and 37°C in Dulbecco's modified Eagle's medium (Nacalai Tesque) supplemented with 10% fetal bovine serum (Gibco) and 50 μg/ml gentamicin (Nacalai Tesque). To induce starvation, cells were incubated in Hanks' balanced salt solution (HBSS(−), Nacalai Tesque) without serum. Polyethylenimine (Polyscience) and Lipofectamine 3000 (Invitrogen) were used for transfection.
Group A Streptococcus
Group A Streptococcus strain JRS4 (M6+ F1+) was grown in Todd‐Hewitt broth (BD Diagnostic Systems, Sparks, MD) supplemented with 0.2% yeast extract, as described previously (Nakagawa et al, 2004).
Antibodies and other reagents
Rabbit antibodies against Rab35 (Proteintech, 11329‐2AP), NDP52 (Cell Signaling Technology, 9036), NDP52 (Abcam, ab68588), LC3B (D11; Cell Signaling Technology, 3868), TBK1 (EP611Y; Abcam, ab40676), phosphorylated TBK1 (Ser172; D52C2; Cell Signaling Technology, 5483), ATG5 (D1G9; Cell Signaling Technology, 8540), p62 (H‐290; Santa Cruz Biotechnology, sc‐25575), EEA1 (B45B10; Cell Signaling Technology, 3288), TOM20 (Abcam, ab78547), and β‐Actin (D6A8; Cell Signaling Technology, 8457) were used as primary antibodies, along with mouse antibodies against Rab27A (BD Pharmingen, 558532), NDP52 (4H5; OriGene Technologies, TA501971), galectin 3 (B2C10; BD Pharmingen, 556904), p62 (D‐3; Santa Cruz Biotechnology, sc‐28359), multi‐ubiquitin (FK2; Nippon Bio‐Test Laboratories, MFK‐004), LAMP1 (H4A3; Santa Cruz Biotechnology, sc‐20011), HSP60 (LK‐1; Enzo Life Sciences, ADI‐SPA‐806), FLAG (M2; Sigma‐Aldrich, A2220), GFP (GF200; Nacalai Tesque, 04363‐24), MBP (New England Biolabs), and α‐tubulin (Sigma‐Aldrich, T6199). Rabbit Immunoglobulin Fraction (Dako, X0903) was used as negative control for immunoprecipitation. HRP‐conjugated anti‐rabbit IgG (Jackson Immuno Research Laboratories), anti‐mouse IgG (Jackson Laboratories), and anti‐rabbit IgG (Conformation Specific; L27A9; Cell Signaling Technology, 5127) were used as secondary antibodies for immunoblotting. Cy5‐conjugated goat anti‐rabbit IgG (Jackson ImmunoResearch Laboratories) was used as secondary antibody for immunostaining, along with anti‐mouse or anti‐rabbit IgG conjugated to AlexaFluor 488, and 594, anti‐mouse IgG conjugated to AlexaFluor 350 (Molecular Probes/Invitrogen), and anti‐mouse IgG conjugated to AlexaFluor 647 (Jackson ImmunoResearch Laboratories). BX795, bafilomycin A1, and LysoTracker Red DND‐99 solution were procured from Invitrogen. Carbonyl cyanide m‐chlorophenyl hydrazone (CCCP), antimycin A, and oligomycin were obtained from Nacalai Tesque, Santa Cruz Biotechnology, and Calbiochem, respectively. Cathepsin B substrate Magic Red was purchased from Immunochemistry Technologies.
Plasmids
Human TBC/RabGAPs, Rab35, NDP52, OPTN, galectin 3, galectin 8, Parkin, and ATP1A1 were amplified by PCR from HeLa, KYSE, and HEK293T total mRNA, and cloned into pcDNA‐6.2/N‐EmGFP‐DEST, pcDNA‐6.2/N‐3xFLAG‐DEST, pcDNA‐6.2/N‐mCherry‐DEST, pGEX‐6P‐1‐DEST, and pLenti6/V5‐DEST using Gateway (Invitrogen) technology. The resulting constructs are N‐terminally fused with corresponding tags. TBC1D10A, Rab35, and NDP52 were mutated by site‐directed mutagenesis using PrimeSTAR Mutagenesis Basal Kit (Takara). pBABE‐puro‐GFP‐LC3 (plasmid 22405), which was generated by Fung et al (2008), was purchased from Addgene. NDP52, NDP52ΔCC, and NDP52ΔZn N‐terminally fused to mCherry were cloned into NotI/BamHI sites in pMEI‐5 neo (Takara) to generate retroviruses.
BLOCK‐iT Pol II miR‐RNAi Expression Vector Kit (Invitrogen) was used to knock down Rab35, using 5′‐CACGATCGGAGTGGATTTCAA‐3′ (Rab35‐1), and 5′‐GAGACGGAAGATGCCTACAAA‐3′ (Rab35‐2) as target sequences. Double‐stranded miRNA sequences were ligated to pcDNA‐6.2‐GW/miR (Invitrogen) according to the supplier's instructions and transfected into HeLa cells as described. pcDNA6.2‐GW/miR‐neg (Invitrogen) was used as miRNA control. Knockdown was confirmed by immunoblotting.
Generation of knockout lines by CRISPR/Cas9
CRISPR/Cas9 (Mali et al, 2013) was used to knock out NDP52, Rab35, TBK1, and Rab27A as described previously (Oda et al, 2016). Briefly, CRISPR guide RNAs were designed to target an exon common to all splicing variants of the gene of interest (5′‐AAGCAGAACTCAGACATGC‐3′ for NDP52, 5′‐TCTTCAAGCTGCTCATCAT‐3′ for Rab35, 5′‐GAGCACTTCTAATCATCTG‐3′ for TBK1, and 5ʹ‐ TATTTCTCTGCGAGTGCTA‐3′ for Rab27A). HeLa cells were transfected with vector hCAS9 (Addgene 41815) and a gRNA‐hyg vector containing the CRISPR target sequence. Untransfected cells were removed by selection on 300 μg/ml hygromycin B (Nacalai Tesque) and 750 μg/ml geneticin (G418, Nacalai Tesque). Single colonies were expanded, and depletion of the targeted gene was confirmed by immunoblotting. As secondary screen for some knockout lines, genomic DNA was isolated, and target regions were amplified by PCR and sequenced to confirm presence of the desired frameshift insertions and deletions.
Generation of stable cell lines
Stable cell lines were generated by retroviral expression as previously described (Haobam et al, 2014). Briefly, Plat‐E cells (kindly provided by T. Kitamura, The University of Tokyo) were transiently transfected using FuGENE HD reagent with constructs based on pBABE‐puro (Addgene, 1764) or pMEI‐5 neo (Takara) and cultured for 48 h. The resulting supernatant containing retroviruses was collected and used to infect HeLa cells. Uninfected cells were removed by selection on 2 μg/ml puromycin (Invitrogen) or 800 μg/ml geneticin (G418, Nacalai Tesque).
Additionally, constructs based on pLenti6/V5‐DEST were produced using ViraPower lentiviral expression system (Invitrogen), following the manufacturer's protocol. Briefly, 293 FT cells were co‐transfected using Lipofectamine 2000 (Invitrogen) with pLenti‐mCherry‐EmGFP‐LC3 and packaging plasmids (Invitrogen), and cultured for 48 h. The culture supernatant was then collected and used to infect HeLa cells. After 24 h, uninfected cells were removed by selection on 5 μg/ml blasticidin (Invitrogen).
Bacterial infection
Cells were infected with GAS as described previously (Nakagawa et al, 2004). Briefly, cell cultures in media without antibiotics were infected for 1 h at multiplicity of infection 100. Infected cells were washed with PBS and treated with 100 μg/ml gentamicin for an appropriate period to kill bacteria that were not internalized.
Bacterial invasion and viability assay
HeLa cells were cultured in a 24‐well plate at 5 × 104 cells/well, and infected with GAS as described. After 1 h, cells were treated with gentamicin for 1 or 5 h. Cells were lysed in sterile distilled water, and serial dilutions of lysates were plated on agar plates containing Todd‐Hewitt broth and 0.2% yeast extract. Bacterial invasion efficiency was determined as the ratio of intracellular live GAS recovered at 2 h to total intracellular and adherent GAS recovered at 1 h. Surviving bacteria were quantified as the ratio of bacteria recovered at 6 h to those recovered at 2 h.
Immunoblotting and immunoprecipitation
Cells were harvested, washed with PBS, and lysed for 30 min on ice in lysis buffer containing 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1% Triton X‐100, and proteinase inhibitor cocktail (Nacalai Tesque). Lysates were then centrifuged, and supernatants were pre‐cleared for 30 min at 4°C with Protein G Sepharose Fast Flow (GE Healthcare Life Sciences). After brief centrifugation, supernatants were reacted for 2 h at 4°C with appropriate antibodies and incubated in Protein G Sepharose beads, with mixing, for another 1 h at 4°C. Immunoprecipitates were collected by brief centrifugation, washed five times with lysis buffer, and analyzed by immunoblotting as described previously (Nakagawa et al, 2004).
Protein expression and purification
GST fusion proteins constructed in pGEX‐6P‐1 (GE Healthcare Life Sciences) and MBP fusion proteins in pMAL‐c5x (NEB) were transformed into Escherichia coli BL21 (DE3) cells, which were then cultured at 37°C in LB medium supplemented with 100 μg/ml ampicillin, and induced for 3 h at 37°C with 0.3 mM isopropyl β‐D‐thiogalactopyranoside (Nacalai Tesque). Cells were harvested by centrifugation, washed with PBS, lysed in 40 mM Tris–HCl pH 7.5, 5 mM EDTA, and 0.5% Triton X‐100, sonicated, and cleared by centrifugation. The resulting supernatant was incubated with Glutathione Sepharose 4 Fast Flow (GE Healthcare Life Sciences) for 2 h at 4°C. After several washes with buffer, and beads were used directly in pull‐down assays.
Pull‐down assay
HEK293T cells were transfected with expression constructs encoding the protein of interest. After 24–48 h, cells were lysed in 50 mM HEPES pH 7.4, 250 mM NaCl, 10 mM MgCl2, 1% Triton X‐100, and proteinase inhibitor cocktail (Nacalai Tesque). These lysates or bacterial lysates expressing MBP fusion proteins were incubated for 2–4 h with beads pre‐coated with GST fusion proteins. Beads were washed five times with buffer and analyzed by immunoblotting.
In vitro GTP/GDP loading and binding assay
GST‐Rab35 was bound to a Glutathione Sepharose 4 Fast Flow column (GE Healthcare Life Sciences) for 2 h at 4°C. The beads were washed three times with nucleotide depletion buffer (20 mM Tris pH 7.5, 1 mM DTT, 20 mM EDTA, 50 mM NaCl, 5% glycerol, 0.1% Triton X‐100) and incubated for 20 min at room temperature to deplete the GTPases of GDP and GTP. To load GST‐Rab35 with GDP or GTP, aliquots were washed three times with GDP loading buffer (20 mM Tris, 1 mM DTT, 10 mM MgCl2, 50 mM NaCl, 5% glycerol, 0.1% Triton X‐100, 200 μM GDP), or GTP loading buffer (20 mM Tris, 1 mM DTT, 10 mM MgCl2, 50 mM NaCl, 5% glycerol, 0.1% Triton X‐100, 20 μM GTP) and incubated for 25 min at room temperature. The beads were incubated with bacterial lysates expressing MBP‐NDP52 proteins for 2 h at 4°C. Beads were washed five times with lysis buffer and analyzed by immunoblotting.
In situ proximity ligation assay
Proximity ligation assay was performed with Duolink (Olink bioscience). HeLa cells grown on coverslips were infected with or without GAS for 4 h, fixed in 4% paraformaldehyde for 15 min, washed, permeabilized with 0.1% Triton X‐100, and blocked with 2% BSA blocking buffer for 1 h at room temperature. Cells were probed overnight at 4°C with mouse anti‐NDP52 and rabbit anti‐Rab35 antibodies diluted in 2% BSA blocking buffer. As negative control, cells were incubated in antibody diluent with either anti‐NDP52 or anti‐Rab35 antibody. Labeling with secondary antibodies, ligation, and signal amplification were done as recommended by the manufacturer. Proximity ligation assay dots were imaged with an FV1000 confocal microscope (Olympus).
Fluorescent protein fragment complementation assay
NDP52 and Rab35 cDNA sequences were cloned into the BamHI/XhoI sites in vectors phmKGN‐MN, phmKGN‐MC, phmKGC‐MN, and phmKGC‐MC (Coral Hue Fluo‐chase Kit; Amalgaam). The N‐terminal or C‐terminal fragment of monomeric Kusabira Green (mKG) was fused to either NDP52 or Rab35. To examine protein–protein interactions, mKGN‐ and mKGC‐fused proteins were expressed pairwise in HEK293T cells. After 48 h, cells were imaged by fluorescence microscopy. Fluorescence intensity was calculated in ImageJ from > 30 cells as signal intensity of cell/area of cell – signal intensity of background/area of background.
Fluorescence microscopy
Cells were washed with PBS, fixed for 15 min with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton in PBS for 10 min, washed with PBS, and blocked at room temperature for 1 h with skim milk (5% skim milk, 2.5% goat serum, 2.5% donkey serum, and 0.1% gelatin in PBS) or BSA (2% BSA and 0.02% sodium azide in PBS). Cells were then probed at room temperature for 1 h with primary antibody diluted in blocking solution, washed with PBS, and labeled with secondary antibody. To visualize bacterial and cellular DNA, cells were stained with DAPI (Dojindo). Confocal fluorescence micrographs were acquired with an FV1000 laser‐scanning microscope (Olympus). Fluorescence micrographs of fluorescent protein fragment complementation were acquired in AxioVision Rel 4.6 software using a Zeiss AxioImager A1 microscope equipped with an AxioCam MRc5 camera (Carl Zeiss).
Immunoelectron microscopy
Cells expressing EmGFP‐Rab35 were infected with GAS, fixed with 4% paraformaldehyde and 0.1% glutaraldehyde in PBS pH 7.4, and blocked for 30 min at room temperature with 2% normal donkey serum and 0.01% Photo‐Flo (Kodak) in PBS. Cells were probed overnight at 4°C with rabbit polyclonal antibody against GFP (ab6556; Abcam) diluted in blocking buffer and labeled overnight at 4°C with goat anti‐rabbit IgG Fab fragment conjugated to 1.4 nm Nanogold (Nanoprobes) and diluted in blocking buffer without Photo‐Flo. Samples were then washed with PBS and fixed with 1% glutaraldehyde in PBS, and gold particles were silver‐enhanced for 6 min with HQ Silver Kit, as described by the manufacturer (Nanoprobes). The sections were post‐fixed with 1% osmium tetraoxide in 0.1 M phosphate buffer, dehydrate ethanol series, and embedded in epoxy‐resin Luveak (Nacalai Tesque) and polymerized at 60°C for 3 days. The samples were cut into ultrathin sections (70 nm) on an ultramicrotome (EM UC6; Leica). The ultrathin sections were mounted on mesh grids and stained with uranyl acetate and lead citrate and examined with electron microscopic H‐7650 (Hitachi).
Cell fractionation
HeLa cells expressing EmGFP‐Parkin cells were treated with 10 μM CCCP for 8 h to depolarize the mitochondria and suspended in Chappell‐Perry buffer (0.15 M KCl, 20 mM HEPES‐NaOH pH 8.1, 5 mM MgCl2) supplemented with protease inhibitors (Nacalai Tesque). Cells were lysed by six passages through a 26‐gauge needle and a 1‐ml syringe and cleared by centrifugation at 1,000 × g for 7 min. The supernatant was centrifuged at 10,000 × g for 10 min to separate the mitochondria‐rich fraction from the cytosol‐rich fraction. Fractions were then analyzed by conventional immunoblotting techniques.
Statistical analysis
Cells containing LC3, other autophagy factors were quantified through direct visualization in a confocal microscope. Unless indicated otherwise, 200 GAS‐infected cells were examined per treatment in each experiment, and at least three independent experiments were performed for each trial. Values, including those plotted, are mean ± SEM. Data were tested by two‐tailed Student's t‐test. P‐values less than 0.05 were considered to indicate statistical significance and are marked *P < 0.05, **P < 0.01, ***P < 0.001, and NS for not significant.
Author contributions
AM‐N, TN, and IN conceived the study, provided reagents, and wrote the paper. AM‐N, TN, KO‐F, and HK performed experiments and analyzed data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This work was supported in part by a Grant‐in‐Aid for Scientific Research (25293370, 15K15130, 26462776, 16H05188, and 17K19552), by a Grant‐in‐Aid for JSPS Fellows (14J05847), by the Research Program on Emerging and Re‐emerging Infectious Diseases (17824541) and J‐PRIDE (17fm0208030h0001) from the Japan Agency for Medical Research and Development, AMED, and by Daiichi Sankyo Foundation of Life Science (to I. N.).
The EMBO Journal (2017) 36: 2790–2807
Contributor Information
Takashi Nozawa, Email: nozawa.takashi.4r@kyoto-u.ac.jp.
Ichiro Nakagawa, Email: nakagawa.ichiro.7w@kyoto-u.ac.jp.
References
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll B, Mohd‐Naim N, Maximiano F, Frasa MA, McCormack J, Finelli M, Thoresen SB, Perdios L, Daigaku R, Francis RE, Futter C, Dikic I, Braga VM (2013) The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev Cell 25: 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauvin C, Rosendale M, Gupta‐Rossi N, Rocancourt M, Larraufie P, Salomon R, Perrais D, Echard A (2016) Rab35 GTPase triggers switch‐like recruitment of the lowe syndrome lipid phosphatase OCRL on newborn endosomes. Curr Biol 26: 120–128 [DOI] [PubMed] [Google Scholar]
- Chaineau M, Ioannou MS, McPherson PS (2013) Rab35: GEFs, GAPs and effectors. Traffic 14: 1109–1117 [DOI] [PubMed] [Google Scholar]
- Ellinghaus D, Zhang H, Zeissig S, Lipinski S, Till A, Jiang T, Stade B, Bromberg Y, Ellinghaus E, Keller A, Rivas MA, Skieceviciene J, Doncheva NT, Liu X, Liu Q, Jiang F, Forster M, Mayr G, Albrecht M, Hasler R et al (2013) Association between variants of PRDM1 and NDP52 and Crohn's disease, based on exome sequencing and functional studies. Gastroenterology 145: 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM (2012) Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat Rev Mol Cell Biol 13: 67–73 [DOI] [PubMed] [Google Scholar]
- Fung C, Lock R, Gao S, Salas E, Debnath J (2008) Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 19: 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haobam B, Nozawa T, Minowa‐Nozawa A, Tanaka M, Oda S, Watanabe T, Aikawa C, Maruyama F, Nakagawa I (2014) Rab17‐mediated recycling endosomes contribute to autophagosome formation in response to Group A Streptococcus invasion. Cell Microbiol 16: 1806–1821 [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C, Morohashi Y, Yoshimura S, Manrique‐Hoyos N, Jung S, Lauterbach MA, Bakhti M, Gronborg M, Mobius W, Rhee J, Barr FA, Simons M (2010) Regulation of exosome secretion by Rab35 and its GTPase‐activating proteins TBC1D10A‐C. J Cell Biol 189: 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Fukuda M (2006) Identification of EPI64 as a GTPase‐activating protein specific for Rab27A. J Biol Chem 281: 31823–31831 [DOI] [PubMed] [Google Scholar]
- Itoh T, Kanno E, Uemura T, Waguri S, Fukuda M (2011) OATL1, a novel autophagosome‐resident Rab33B‐GAP, regulates autophagosomal maturation. J Cell Biol 192: 839–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV (2014) Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 5: 3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK (2006) Visualization of molecular interactions by fluorescence complementation. Nat Rev Mol Cell Biol 7: 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 3: 452–460 [DOI] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33: 505–516 [DOI] [PubMed] [Google Scholar]
- Korioth F, Gieffers C, Maul GG, Frey J (1995) Molecular characterization of NDP52, a novel protein of the nuclear domain 10, which is redistributed upon virus infection and interferon treatment. J Cell Biol 130: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C, Tooze SA (2016) TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J 35: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchowius KJ, Weibrecht I, Soderberg O (2011) In situ proximity ligation assay for microscopy and flow cytometry. Curr Protoc Cytom Chapter 9: Unit 9.36 [DOI] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469: 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz P, Kramer B, Kaczmarek DJ, Hubner MP, Langhans B, Appenrodt B, Lammert F, Nattermann J, Hoerauf A, Strassburg CP, Spengler U, Nischalke HD (2016) A variant in the nuclear dot protein 52kDa gene increases the risk for spontaneous bacterial peritonitis in patients with alcoholic liver cirrhosis. Dig Liver Dis 48: 62–68 [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 44: 279–289 [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Shimogori T, Hattori N, Nukina N (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 24: 4429–4442 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F (2012) LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 48: 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T (2004) Autophagy defends cells against invading group A Streptococcus . Science 306: 1037–1040 [DOI] [PubMed] [Google Scholar]
- Nozawa T, Aikawa C, Goda A, Maruyama F, Hamada S, Nakagawa I (2012) The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell Microbiol 14: 1149–1165 [DOI] [PubMed] [Google Scholar]
- Oda S, Nozawa T, Nozawa‐Minowa A, Tanaka M, Aikawa C, Harada H, Nakagawa I (2016) Golgi‐resident GTPase Rab30 promotes the biogenesis of pathogen‐containing autophagosomes. PLoS One 11: e0147061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Seaghdha M, Wessels MR (2013) Streptolysin O and its co‐toxin NAD‐glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog 9: e1003394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Eathiraj S, Munson M, Lambright DG (2006) TBC‐domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual‐finger mechanism. Nature 442: 303–306 [DOI] [PubMed] [Google Scholar]
- Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, Leffler H, Poirier F, Prevost MC, Lafont F, Sansonetti P (2010) Galectin‐3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 12: 530–544 [DOI] [PubMed] [Google Scholar]
- Pilli M, Arko‐Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V (2012) TBK‐1 promotes autophagy‐mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Dikic I (2014) TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep 15: 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113: 4039–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb‐Cheynel I, Oulad‐Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet‐Berguerand N (2016) Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin‐2 to induce motor neuron dysfunction and cell death. EMBO J 35: 1276–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Szatmari Z, Sass M (2014) The autophagic roles of Rab small GTPases and their upstream regulators: a review. Autophagy 10: 1154–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin‐coated bacteria. Nat Immunol 10: 1215–1221 [DOI] [PubMed] [Google Scholar]
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, Kaul A (2016) Recruitment of TBK1 to cytosol‐invading Salmonella induces WIPI2‐dependent antibacterial autophagy. EMBO J 35: 1779–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick‐Jones J, Buss F (2012) Autophagy receptors link myosin VI to autophagosomes to mediate Tom1‐dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol 14: 1024–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick‐Jones J, Buss F (2015) The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog 11: e1005174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, Faure M (2015) Autophagy receptor NDP52 regulates pathogen‐containing autophagosome maturation. Cell Host Microbe 17: 515–525 [DOI] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, Cox JS (2012) Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA‐sensing pathway. Cell 150: 803–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Elazar Z (2011) Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 80: 125–156 [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ (2014) Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 3: e01612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsu A, Shimada H, Ohbayashi N, Fukuda M (2015) Rab40C is a novel Varp‐binding protein that promotes proteasomal degradation of Varp in melanocytes. Biol Open 4: 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH (2009) The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 183: 5909–5916 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7