

Abstract
Melanoma differentiation‐associated protein 5 (MDA5) mediates the innate immune response to viral infection. Polymorphisms in IFIH1, the gene coding for MDA5, correlate with the risk of developing type 1 diabetes (T1D). Here, we demonstrate that MDA5 is crucial for the immune response to enteric rotavirus infection, a proposed etiological agent for T1D. MDA5 variants encoded by minor IFIH1 alleles associated with lower T1D risk exhibit reduced activity against rotavirus infection. We find that MDA5 activity limits rotavirus infection not only through the induction of antiviral interferons and pro‐inflammatory cytokines, but also by promoting cell death. Importantly, this MDA5‐dependent antiviral response is specific to the pancreas of rotavirus‐infected mice, similar to the autoimmunity associated with T1D. These findings imply that MDA5‐induced cell death and inflammation in the pancreas facilitate progression to autoimmune destruction of pancreatic β‐cells.
Keywords: inflammation, innate immunity, interferons, MDA5, type 1 diabetes
Subject Categories: Immunology, Molecular Biology of Disease
Introduction
The innate immune system is the first line of defense against microbial pathogens and initiates and modulates lymphocyte activation in pathogen‐specific adaptive immunity. The retinoic acid‐inducible gene I (RIG‐I)‐like receptors (RLRs), including RIG‐I (encoded by the DDX58 gene), melanoma differentiation‐associated factor 5 (MDA5, encoded by the IFIH1 gene), and the laboratory of genetics and physiology 2 (LGP2, encoded by the DHX58 gene), detect cytosolic viral RNA. All three proteins share DECH‐box helicase domains, which are essential for RNA binding and ATP hydrolysis, and a C‐terminal domain (CTD) that participates in RNA recognition and, at least for RIG‐I, auto‐regulation. These separate RLRs have different affinities for RNA, and so respond distinctly to separate viral pathogens. Binding of RNA induces MDA5 or RIG‐I to oligomerize and subsequently induce polymerization of the adaptor mitochondrial antiviral signaling protein (MAVS). This association is mediated via the protein's mutual caspase activation and recruitment domains (CARD). Unlike RIG‐I and MDA5, LGP2 lacks the CARD and so is believed to act co‐operatively with MDA5 (Bruns et al, 2014). Polymerization of MAVS triggers a signaling cascade that culminates in the activation of the interferon‐regulating factor (IRF)3 and IRF7 and the nuclear factor‐kappa B (NFκB). These transcription factors induce the antiviral type I and III interferons (IFNs), as well as other cytokines and chemokines, and regulate cell survival (Besch et al, 2009).
In keeping with the role of the innate immune response to modulate lymphocyte activation, population studies have identified genetic polymorphisms in components of the RLR pathway that correlate with autoimmune diseases. To date, the link between RLR signaling and autoimmunity is strongest for IFIH1. Non‐synonymous (ns) single‐nucleotide polymorphisms (SNPs) in IFIH1 were correlated with the risk of developing type 1 diabetes (T1D) by genome‐wide association (GWA) scanning (Smyth et al, 2006; Todd et al, 2007; Wellcome Trust Case Control, 2007).
T1D results from progressive destruction of insulin‐producing pancreatic β‐cells. Although principally considered a genetic disease, the progression of T1D is modulated by environmental factors. Virus infections have been proposed as potential primary agents in the initiation of T1D (Hober & Sauter, 2010). The occurrence of the nsSNPs within the coding region of the IFIH1 locus, with potential to alter the protein's activity, combined with the role of MDA5 in the antiviral response, has led to speculation that virus infection is causal in disease pathology. Rotavirus (RV) is one of the leading candidate viruses linked to T1D (Honeyman et al, 2000; van der Werf et al, 2007). Several studies have explored the immune response to RV isolated from a variety of species in human and murine cells. These studies have shown in mouse embryonic fibroblasts (MEFs) and macrophages that either RLR is sufficient and is able to compensate for loss of the other against bovine and simian RV (Broquet et al, 2011; Sen et al, 2011; Di Fiore et al, 2015). However, it has also been shown that RV isolated from different species show varying abilities to replicate in heterologous and homologous hosts, and, related to this, the effectiveness of the antiviral response varies against heterologous and homologous RV strains. This variation may be critical for resultant disease, such as T1D (Feng et al, 2008). Also, while studies have investigated the function of MDA5 encoded by different alleles to ligands such as the RNA mimetic polyriboinosinic–polyribocytidylic acid (pIC), none have explained how these influence the risk of T1D. Here, we tested the role of MDA5 in the antiviral response to a human RV isolate in human and mouse cells. We also tested the capacity of the separate alleles of IFIH1 associated with different risk of T1D to respond to RV infection. Additionally, we model the Mda5‐dependent response to RV infection in vivo using the Ifih1 −/− mice.
Results
Mda5 is critical to limit RV infection in the pancreas
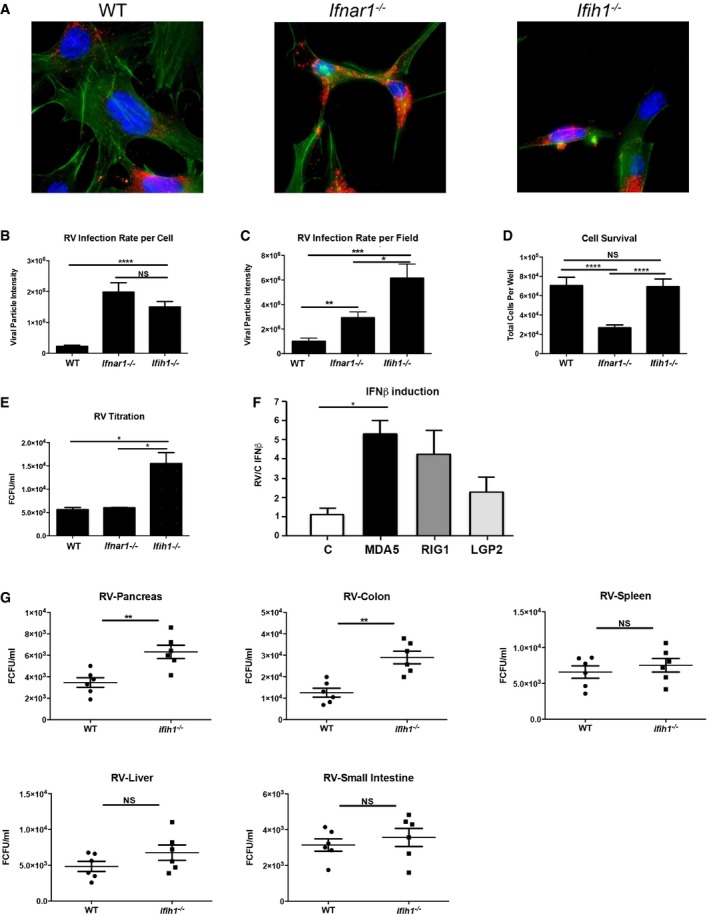
RV was isolated from an infant patient at The Royal Children's Hospital (Melbourne, Australia) and passaged through a human epithelial colorectal adenocarcinoma cell line (Caco‐2) and identified as strain SA11. To assess the dependence of Mda5 and the type I IFNs to the antiviral response to RV, we infected immortalized MEFs isolated from wild‐type (WT) mice or mice ablated for Mda5 (Ifih1 −/−) or the primary binding chain of the type I IFN receptor (Ifnar1 −/−) that is pivotal in the antiviral response (Hwang et al, 1995; Gitlin et al, 2006). Cells were infected with RV for 24 hours (h) before quantitation of intracellular virus replication by immunofluorescence detection of viral capsid proteins (Fig 1A–C). Quantitation of the viral capsid per cell shows both Ifnar1 −/− and Ifih1 −/− MEFs are significantly more permissible to RV infection than the WT cells. By this measure, the Ifnar1 −/− and Ifih1 −/− MEFs are not significantly different in their susceptibility to RV (Fig 1B). However, MEFs ablated for Mda5 showed higher virus replication, as measured by the total levels of capsid proteins produced (Fig 1C). This disparity in these measures was supposed to be due to the altered survival of the different MEFs (Fig 1D). The consequence to viral replication was confirmed by quantifying the levels of infectious RV produced from the MEFs by titration in monkey kidney cells (MA104). The Ifih1 −/− MEFs produced more infectious RV than either WT or Ifnar1 −/− MEFs (Fig 1E).
Figure 1. Mda5 is crucial to limit RV infection in the pancreas.
-
AMicrographs of WT, Ifnar1 −/−, and Ifih1 −/− MEFs infected with RV for 24 h, then assessed by immunofluorescence. The viral capsid protein is detected with an anti‐SA11 antibody (red), actin filaments are visualized with CF647 phalloidin (green), and nuclei are visualized with Hoechst stains (blue).
-
B, CRV replication in the indicated MEFs was quantitated by calculating the number and intensities of viral particles per cell (B), or per micrograph field (C).
-
DThe relative survival of WT, Ifnar1 −/−, and Ifih1 −/− MEFs after RV infection for 24 h was assessed by direct counting of the cells.
-
EA titration of virus produced in WT, Ifnar1 −/−, and Ifih1 −/− MEFs by immunofluorescence.
-
FThe relative induction of an IFNB1‐promoter firefly‐luciferase reporter 24 h after RV infection in HEK293 cells co‐transfected with control empty vector (C), MDA5‐, RIG‐I‐, or LGP2‐expressing constructs. The levels of firefly luciferase were normalized to a constitutively active Renilla luciferase and expressed relative to control cells.
-
GQuantitation of RV from the indicated tissues of WT and Ifih1 −/− mice orally infected with RV for 5 days. Viral infection was quantified by titration against MA104 cells and immunofluorescence.
Luciferase promoter reporter assays were used to compare the relative response to rotavirus infection of the different RLR. The human kidney cell line HEK293 were co‐transfected with constructs expressing MDA5, RIG‐I or LGP2, and an IFNB1‐promoter firefly‐luciferase reporter that respond to IRF3/7 and/or NFκB, as well as a constitutive β‐actin–Renilla luciferase reporter. After 24 h, the cells were infected with RV, and then 24 h later, the cells were lysed and the luciferase activity was assayed. These data show that the related MDA5 and RIG‐I induce a similar transcription response to RV infection (Fig 1F).
The role of Mda5 in RV replication was explored in vivo by infecting WT and Ifih1 −/− mice. Five‐week‐old mice were inoculated with RV by oral gavage, and then 5 days later, the levels of infectious RV in the pancreas, colon, spleen, liver, and small intestines from the mice were quantitated by titration on MA104 cells. RV was detected in all tissues sampled, although Ifih1 −/− mice produced significantly higher virus than the WT mice, specifically in their pancreas and colon (Fig 1G). These data demonstrate that despite the observed equivalence to RIG‐I in cell lines, MDA5 activity is critical to counter RV infection in vivo and this antiviral effect is important in the pancreas.
Mda5 induces IFN‐dependent and IFN‐independent anti‐RV responses
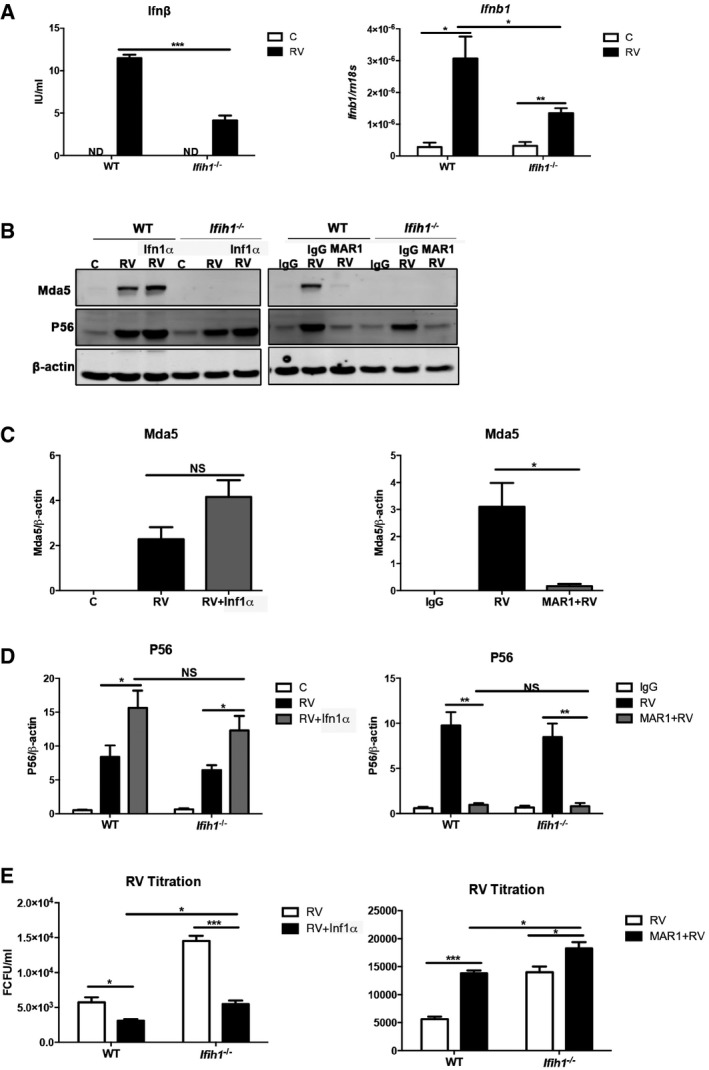
The preceding data showed divergent responses in the Ifih1 −/− compared to the Ifnar1 −/− MEFs. This indicates there is a primary role of Mda5 in the antiviral response to RV infection that is independent of type I IFN signaling. To further examine this, WT and Ifih1 −/− MEFs were infected with RV for 6 or 24 h, and then, cellular transcripts or cell supernatants were probed for the levels of Ifnβ and Ifnb1 by ELISA and quantitative real‐time PCR (qRT‐PCR), respectively. Ifnβ was induced in response to RV infection in both cells, although this was significantly attenuated in the absence of Mda5 (Fig 2A). Accordingly, robust induction of the IFN response is strongly dependent on Mda5 activity.
Figure 2. Mda5 induces IFN‐dependent and IFN‐independent anti‐RV responses.
-
AGraphs showing the levels of Ifnβ protein (on left) and the Ifnb1 transcript (on right) induced in WT and Ifih1 −/− MEFs infected with RV assessed after 24 h by ELISA or after 6 h by qRT–PCR.
-
BImmunoblots assessing the levels of Mda5 and P56 relative to β‐actin in lysates from control (C) or infected (RV) WT and Ifih1 −/− MEFs pre‐treated with Ifn1α for 16 h or, alternatively, an Ifnar1‐neutralizing (MAR1) or control (IgG) antibody for 1 h prior to infection with RV for 24 h.
-
C, DGraphs showing the quantitation of the levels of Mda5 and P56 under the identified conditions as detected by immunoblot (see also Fig EV1).
-
EA quantitation of progeny RV from WT and Ifih1 −/− MEFs treated with Ifn1α or MAR1.
To examine the impact of Mda5‐induced IFN signaling on the antiviral response, we examined RV infection in cells treated with recombinant Ifn1α alone or, alternatively, pre‐treated with an IFN‐neutralizing antibody (MAR1) (Sheehan et al, 2006). Measures of the levels of Mda5 show that its expression is induced during RV infection by IFN signaling (Figs 2B and C, and EV1). Measures of the levels of the product of the IFN‐induced protein with tetratricopeptide repeats‐1 gene (Ifit1), P56, which can also be induced directly by RLR‐dependent induction of IRF's activity, show P56 is predominantly induced by secondary IFN signaling (Fig 2D). Replication of this experiment in Ifih1 −/− MEFs demonstrates that Mda5 activity was dispensable for the induction of P56 (Figs 2B and D, and EV1). Quantitation of the RV produced from these experiments supports the earlier observation that Mda5 is critical to limit RV infection (Fig 2E). An intriguing aspect of this experiment was the impairment of the antiviral response with the Ifnar1‐neutralizing antibody that was not evident by genetically ablating the Ifnar1 receptor (Figs 1E and 2E). Together, these data show that Mda5 is required for a full response to RV and, also, that a proportion of this antiviral activity is independent of IFN signaling.
Figure EV1. Mda5 (Ifih1) and P56 induction in response to RV (related to Fig 2B–D).
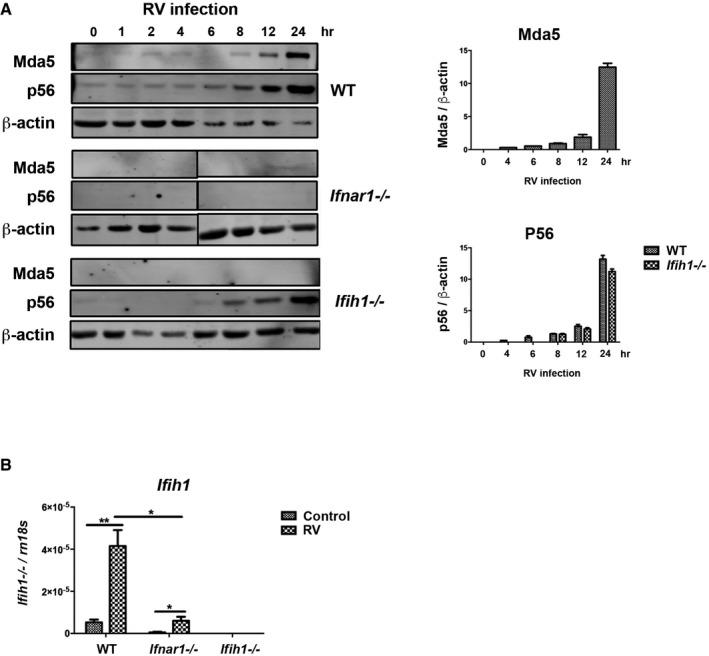
- Detection (on left) and quantitation (on right) of Mda5 and P56 induction relative to β‐actin in WT, Ifnar1 −/−, and Ifih1 −/− MEFs infected with RV for the indicated times by immunoblot.
- Quantitation of the induction of the Ifih1 mRNA in WT, Ifnar1 −/−, and Ifih1 −/− MEFs 6 h post‐infection with RV by qRT–PCR.
Mda5 induces cell death during RV infection
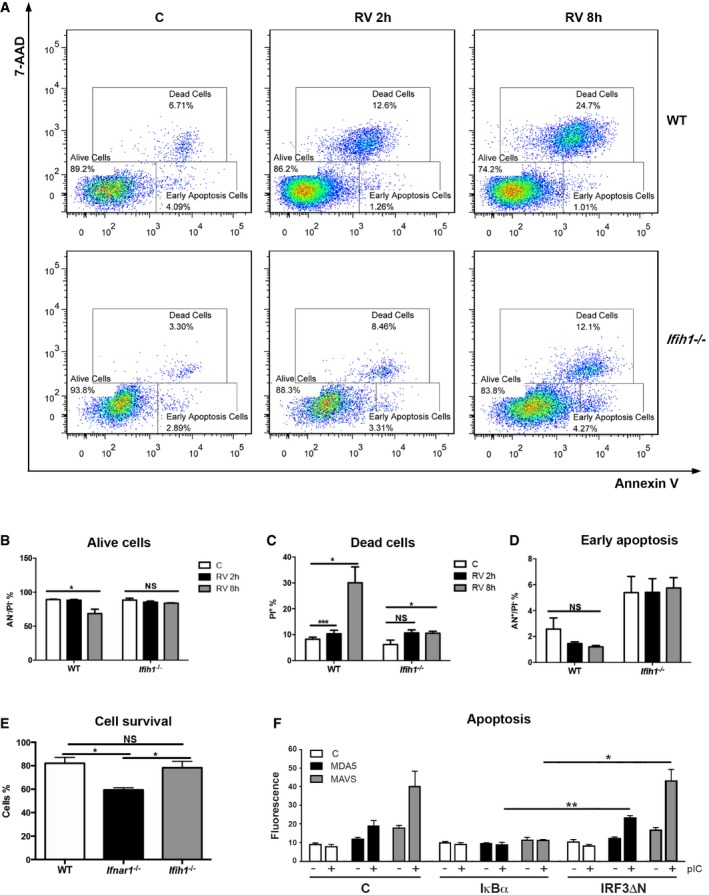
In addition to inducing IFN signaling, MDA5 regulates cell survival (Fig 1D) (Jiang & Fisher, 1993; Kang et al, 2002). To further investigate this function of Mda5, WT and Ifih1 −/− MEFs were mock‐infected or infected with RV for 2 and 8 h, and then, cell death was assessed by staining with annexin V and 7‐aminoactinomycin‐D (7‐AAD). These measures show that RV infection induces cell death in an Mda5‐dependent manner (Fig 3A). Measures of the relative intensity of the annexin V and 7‐AAD stains, to distinguish live, early apoptotic, or dead cells, show that Mda5 induced an early cell death (Fig 3B–D). Comparison of the survival of WT, Ifih1 −/−, and Ifnar1 −/− MEFs 24 h after RV infection by staining with crystal violet shows IFN signaling promotes survival (Fig 3E). These data are consistent with earlier analysis (Fig 1D) and suggest that IFN induced in response to RV infection in the absence of Mda5 (Fig 2A) is sufficient for this outcome. Transient transfection assays were conducted in HEK293 cells to predict the activity of MDA5 that induced cell death. These data show that suppressing NFκB but not IRF3 activity by expressing the IκBα inhibitor or a dominant‐negative IRF3 construct (IRF3ΔN), respectively, reduced MDA5‐dependent cell death (Figs 3F and EV2). Accordingly, MDA5 appears to induce apoptosis in HEK293 cells by activating NFκB via MAVS.
Figure 3. Mda5‐dependent activation of NFκB induces cell death.
-
AFACS plots showing annexin V and 7‐AAD staining of WT and Ifih1 −/− MEFs mock‐ or RV‐infected for 2 and 8 h.
-
B–DA quantitation of the live, dead, and early apoptotic cells by their relative 7‐AAD and annexin V staining.
-
EA quantitation of the relative survival of WT, Ifnar1 −/−, and Ifih1 −/− MEFs RV‐infected for 24 h, then assessed by staining with crystal violet and spectrophotometry.
-
FA quantitation of apoptosis in HEK293 cells co‐transfected with an empty control (C), MDA5‐, or MAVS‐expressing vectors and either control (C), IκBα, or dominant‐negative IRF3 (IRF3ΔN) constructs to repress the activity of NFκB or IRF3, respectively. These cells were either left untreated (−) or transfected (+) with FuGENE:pIC for 10 h, and then, apoptosis was assessed by measuring the nuclear accumulation of YO‐PRO by co‐localization with the nuclear Hoechst stain (see also Fig EV3).
Figure EV2. MDA5‐induced cell death in HEK293 cells is NFκB dependent (related to Fig 3F).
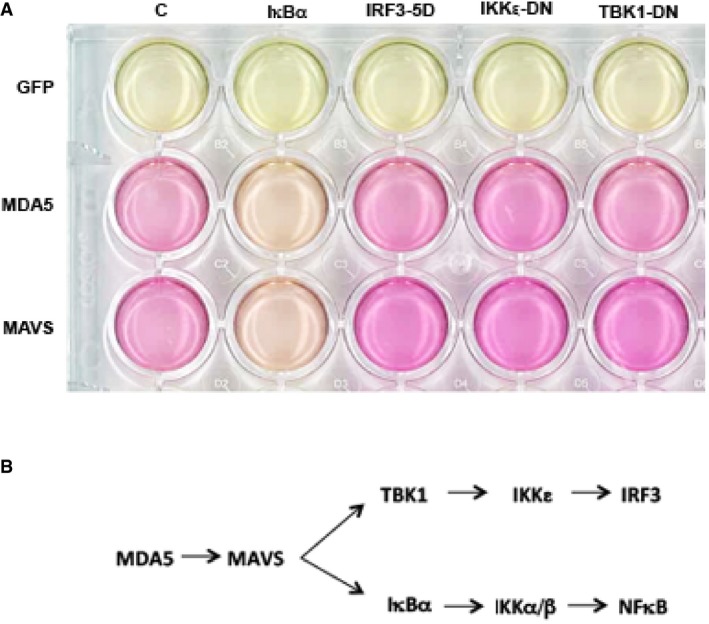
- Relative cell survival assessed by the pH‐dependent color change of culture medium supporting HEK293 cells transfected with GFP, MDA5 or MAVS expression constructs, and a control backbone vector (C) or dominant‐negative constructs of TANK‐binding kinase 1 (TBK1‐DN), IKK‐related kinase ε (IKKε‐DN), a constitutively active IRF3 construct (IRF3‐5D), or the NFκB inhibitor, IκBα, after 90 h of culture.
- A schematic of MDA5‐dependent cell signaling in the cell.
RV infection induces Mda5‐dependent inflammation in the pancreas
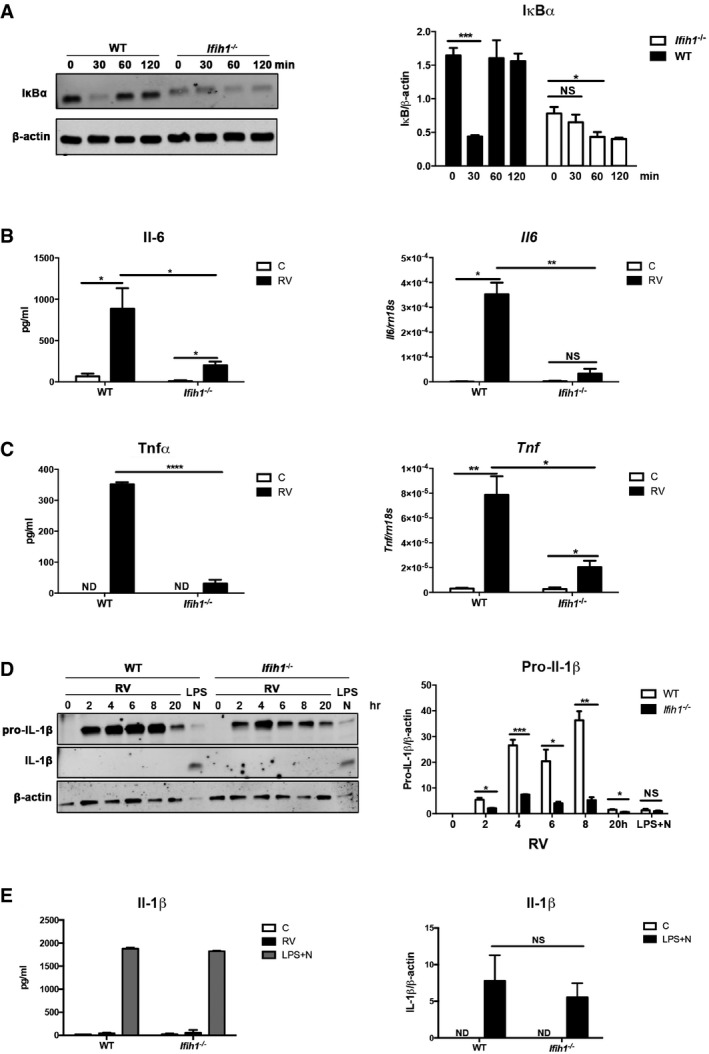
To confirm Mda5‐dependent cell signaling during RV infection, we monitored the activity of Mda5‐regulated transcription factors. NFκB activity was assessed by monitoring the degradation of IκBα, which is coordinated with translocation of NFκB to the nucleus. Western blots show reduced expression of IκBα in the Ifih1 −/− MEFs compared to the WT cells prior to addition of a stimulus (Fig 4A). Because IκBα is auto‐regulated (Sun et al, 1993), this is evidence of the influence of Mda5 on NFκB activity. Upon infection with RV, this impairment in NFκB activation was further evident as a temporal defect in IκBα degradation in the Ifih1 −/− compared to the WT MEFs (Fig 4A). RV infection also induced a modest Mda5‐dependent activation of IRF3 as detecting the translocation of the transcription factor from the cytoplasm to the nucleus in WT but not in the Ifih1 −/− MEFs by immune fluorescence (Fig EV3).
Figure 4. RV infection induces Mda5‐dependent inflammatory signaling.
-
ADetection (on left) and quantitation (on right) of NFκB activation by assessing the levels of its cytosolic inhibitor IκBα relative to the β‐actin in WT and Ifih1 −/− MEFs infected with RV for the indicated times.
-
B, CGraphs showing the relative induction of the Il‐6 and Tnfα cytokines and Il6 and Tnf transcripts in WT and Ifih1 −/− primary peritoneal macrophages infected with RV measured by ELISA after 24 h or qRT–PCR after 6 h, respectively.
-
D, EDetection (on left) and quantitation of both immature and processed Il‐1β in lysates from WT and Ifih1 −/− primary peritoneal macrophages that were infected with RV for the indicated times, or treated with lipopolysaccharide (LPS) plus the potassium ionophore nigericin (N) by ELISA (on left) and immunoblot (on right) after 20 h. The levels of Il‐1β are quantitated relative to β‐actin in the immunoblot.
Figure EV3. Mda5‐induced nuclear translocation of IRF3 in MEFs infected with RV (related to Fig 4).
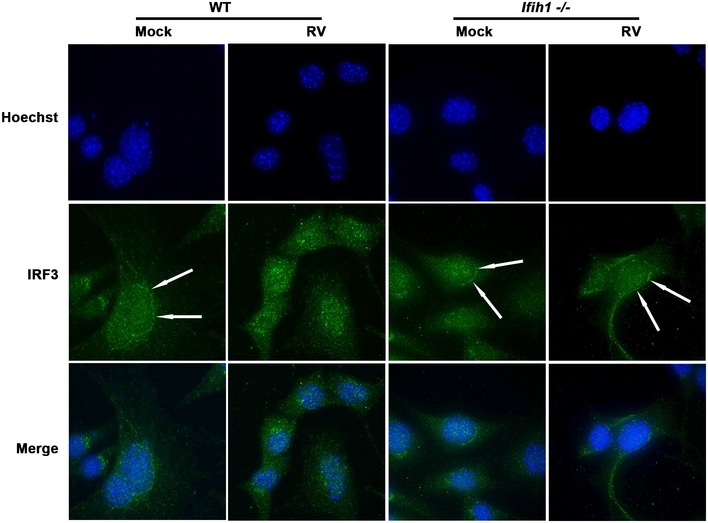
Micrographs showing the cellular distribution of IRF3 (green) in WT and Ifih1 −/− MEFs detected with an anti‐IRF3 antibody 4 h after infection with RV. Arrows indicate the more intense cytosolic, perinuclear enrichment of IRF3 in the WT and Ifih1 −/− cells before infection. This fluorescence pattern changes to a relatively more intense nuclear (blue) staining of IRF3 in RV‐infected WT cells but not infected Ifih1 −/− cells. The nucleus is visualized with the Hoechst DNA stain.
The consequence of Mda5‐dependent activity was assessed by measuring the induction of pro‐inflammatory cytokines in primary peritoneal macrophages infected with RV. The levels of the pro‐inflammatory Il‐6 and Tnfα cytokines in WT and Ifih1 −/− macrophages infected with RV for 6 or 24 h were assessed by measures of the gene transcripts and the protein by qRT–PCR and ELISA, respectively. These data show production of these cytokines in response to RV infection is Mda5 dependent (Fig 4B and C). As RIG‐I has been reported to activate the inflammasome, which has been linked to auto‐inflammatory disease (Poeck et al, 2010), we examined the role of Mda5 in this response. Figure 4D shows that pro‐Il‐1β was induced in response to RV in both WT and Ifih1 −/− peritoneal macrophages, although in keeping with the preceding findings this response is impaired in its extent and duration in the Ifih1 −/− cells. However, RV infection did not induce processing of pro‐Il‐1β as assessed by Western blot and ELISA (Fig 4D and E). Treatment of WT and Ifih1 −/− cells with the inflammasome activators lipopolysaccharide (LPS) and nigericin verifies that this pathway is functional in the Ifih1 −/− cells (Fig 4D and E). Accordingly, secondary signals that are required to form the inflammasome are either repressed or are absent in this context.
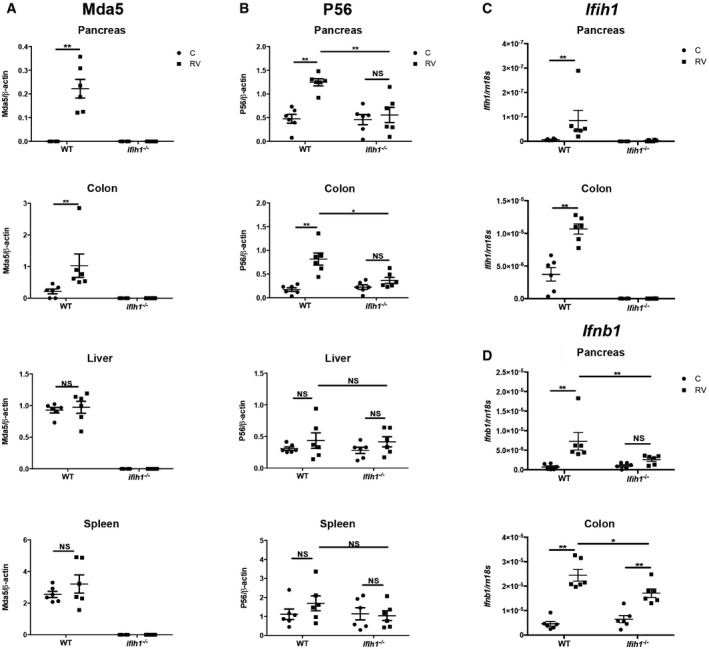
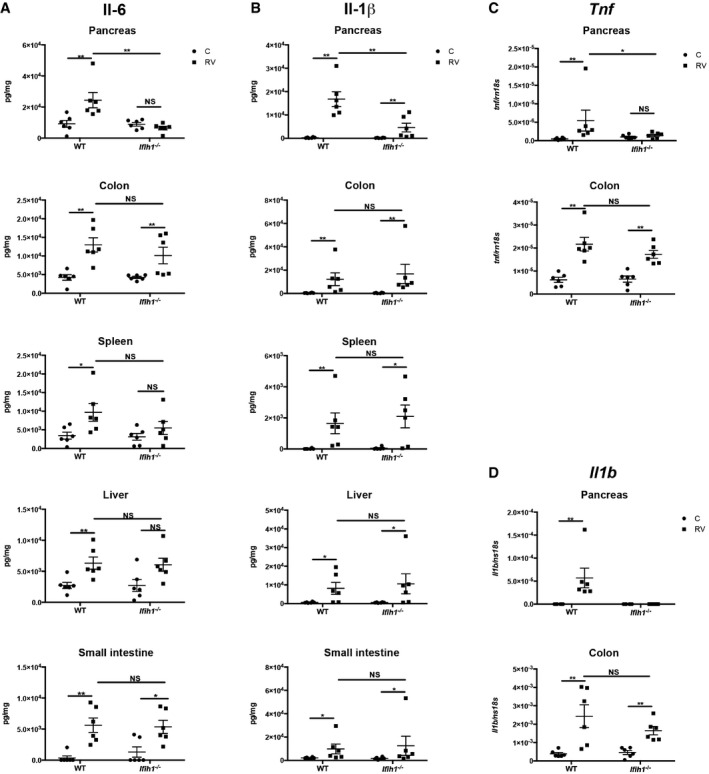
These responses were tested in vivo by assessing the levels of Mda5, P56, Il‐6, and Il‐1β by immunoblot or ELISA, and the expression of the Ifih1, Ifnb1, Tnf, and Il1b transcripts by qRT–PCR in tissues from RV‐infected WT and Ifih1 −/− mice. The levels of Mda5 and P56 with the Ifih1 and Ifnb1 transcripts show that RV infection induces IFN signaling in an Mda5‐dependent manner in the pancreas and colon (Fig 5A–D). This antiviral response is in keeping with the RV titers recorded in the tissues from WT and Ifih1 −/− mice (Fig 1G). Quantitation of Il‐6 and Il‐1β demonstrates that RV infection induces these inflammatory cytokines in all the tissues assessed (Fig 6A and B). Notably, the production of these inflammatory cytokines was Mda5 dependent only in the pancreas. Interestingly, in contrast to the response in macrophages ex vivo (Fig 4D and E), RV infection activated the inflammasome in vivo in the pancreas and this was Mda5 dependent (Fig 6B). This suggests that the stimulus that induces formation of the inflammasome is extrinsic to macrophages or, alternatively, Il‐1β is produced by another cell type. RV infection induced the Il1β and Tnf transcripts in the pancreas and colon, and, again, this was only Mda5 dependent in the pancreas (Fig 6C and D). These data demonstrate that RV infection potently induces inflammatory processes in an Mda5‐dependent manner specifically in the pancreas, thereby demonstrating a consonance with the tissue‐specific autoimmunity in T1D.
Figure 5. RV infection induces Mda5‐dependent IFN signaling in the pancreas.
-
A–DMeasures of the induction of the Mda5 and P56 proteins and the Ifih1 and Ifnb1 transcripts in the indicated tissues of 5‐week‐old WT and Ifih1 −/− mice infected with RV for 5 days, detected by ELISA or qRT–PCR, respectively. The levels of the transcripts were quantified and normalized to the levels of the 18S ribosomal RNA (Rn18s). The data are shown as mean ± SEM. The Mann–Whitney test was used to calculate the P‐values (n = 6). NS = P > 0.05, *P ≤ 0.05, **P ≤ 0.01.
Figure 6. RV infection induces Mda5‐dependent inflammation in the pancreas.
-
A–DMeasures of the induction of the Il‐6 and Il‐1β proteins and the Tnf and Il1b transcripts in the indicated tissues of 5‐week‐old WT and Ifih1 −/− mice infected with RV for 5 days as detected by ELISA or qRT–PCR, respectively. The levels of the transcripts were quantified and normalized to the levels of the 18S ribosomal RNA (Rn18s). The data are shown as mean ± SEM. The Mann–Whitney test was used to calculate the P‐values (n = 6). NS = P > 0.05, *P ≤ 0.05, **P ≤ 0.01.
Allelic variation alters MDA5 activity
To investigate the functional consequence of nsSNPs within the IFIH1 gene that correlate with the risk of T1D, we generated the corresponding amino acid variants as MDA5 expression constructs and tested their activity.
MDA5 initially self‐associates via monomers binding to RNA and then co‐operatively associates with the adaptor MAVS to propagate cell signaling (Wu et al, 2013). We sought to monitor this oligomerization through bimolecular complementation. To do this, a split‐Venus fluorophore (coded V1 and V2) was separately fused to the amino‐terminus of the products of the different IFIH1 alleles and, also, MAVS so that an association between protein partners is evidenced as Venus fluorescence in the cell. Cells transfected with MDA5 tagged with the separate halves of the split‐Venus initially produced a diffuse cytosolic fluorescence that finally condensed as a perinuclear signal (Figs 7A and EV4). The fluorescent signal in cells transfected with tagged MDA5 and MAVS or that produced by a homotypic interaction between MAVS was limited to the perinuclear region. This suggested that the protein complexes were associating with the mitochondria. Visualization of the mitochondria by staining with MitoTracker supports this (Figs 7A and EV4). Quantitation of homotypic association by assessing fluorescence produced when the Venus fluorophore was split between the products of the different IFIH1 alleles indicates that the T946 and V923 variants are equivalent to the product of the major allele, while the X627 variant has decreased oligomerization (Figs 7B and EV4). Interestingly, this pattern of fluorescence was not preserved when Venus was split between the MDA5 variants and MAVS. In this situation, the X627 and V923 variants were similar to the product of the major allele, while the T946 variant produced less fluorescence in combination with MAVS (Figs 7C and EV4).
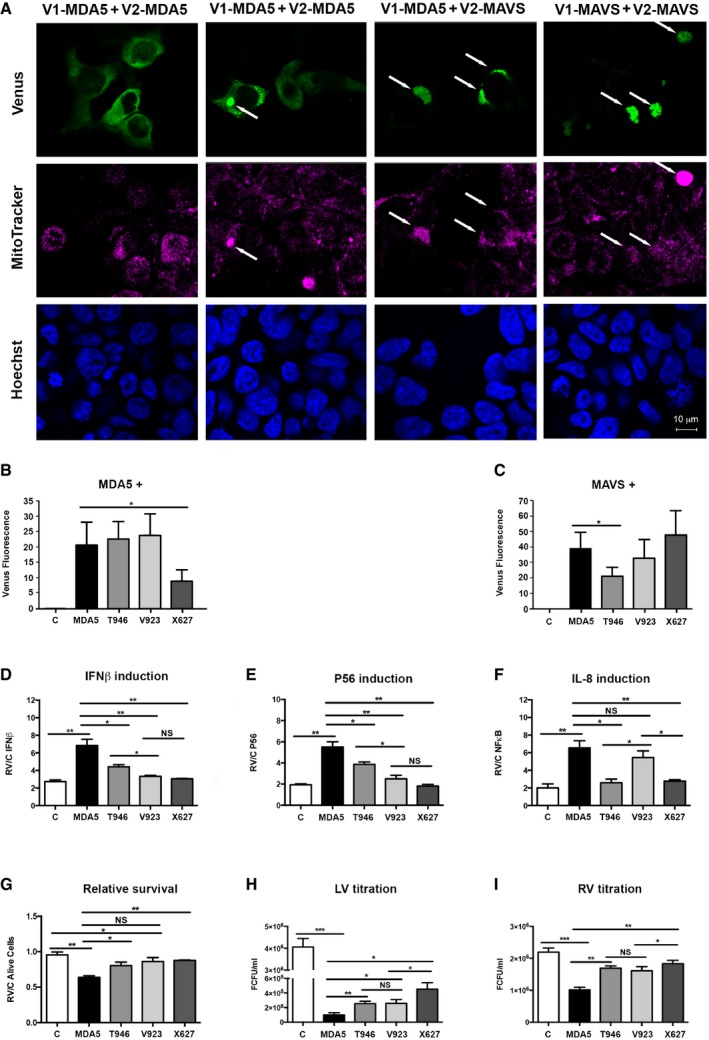
Figure 7. Allelic variation alters MDA5 activity.
-
AMicrographs showing Venus fluorescence (green) produced by homotypic interaction between split‐Venus fusion constructs of MAVS at 56 h (far right) or MDA5 after 48 and 56 h (first and second columns) and heterologous interactions between MDA5 and MAVS at 56 h (third column) in HEK293 cells. Mitochondria are visualized with MitoTracker (magenta), and cell nuclei are stained with Hoechst (blue). Arrows indicate the co‐localization of protein complexes (Venus) and mitochondria.
-
B, CGraphs quantitating the association between the major (MDA5) and minor IFIH1 alleles (T946, V923, and X627) and the complementary split‐Venus tag alone (C), MDA5 or MAVS tagged with the separate halves of the split‐Venus fluorophore and assessed by the fluorescence intensity in the cell after 48 h (n = 3) (see also Fig EV4).
-
D–FThe relative induction of IFNB1‐, IFIT1‐, and IL‐8‐promoter firefly‐luciferase reporters 24 h after mock or RV infection in HEK293 cells co‐transfected with the control LV‐GFP (C) or the indicated MDA5‐GFP constructs (n = 3). The levels of firefly luciferase were normalized to a constitutively active Renilla luciferase and expressed relative to control (GFP only) cells that did not express MDA5 (see also Fig EV5).
-
GThe relative survival of HEK293 cells expressing the indicated MDA5‐GFP or control GFP (C) constructs labeled with annexin V and 7‐AAD and assessed by FACS (n = 3) (see also Fig EV5).
-
HThe relative production of recombinant control LV‐GFP (C) or the indicated MDA5‐GFP lentivirus (LV) measured by quantitating GFP fluorescence in HEK293 cells (n = 3).
-
ITitration of RV produced from HEK293 cells transfected with the control LV‐GFP (C) or the indicated MDA5‐GFP constructs (n = 3) (see also Fig EV6).
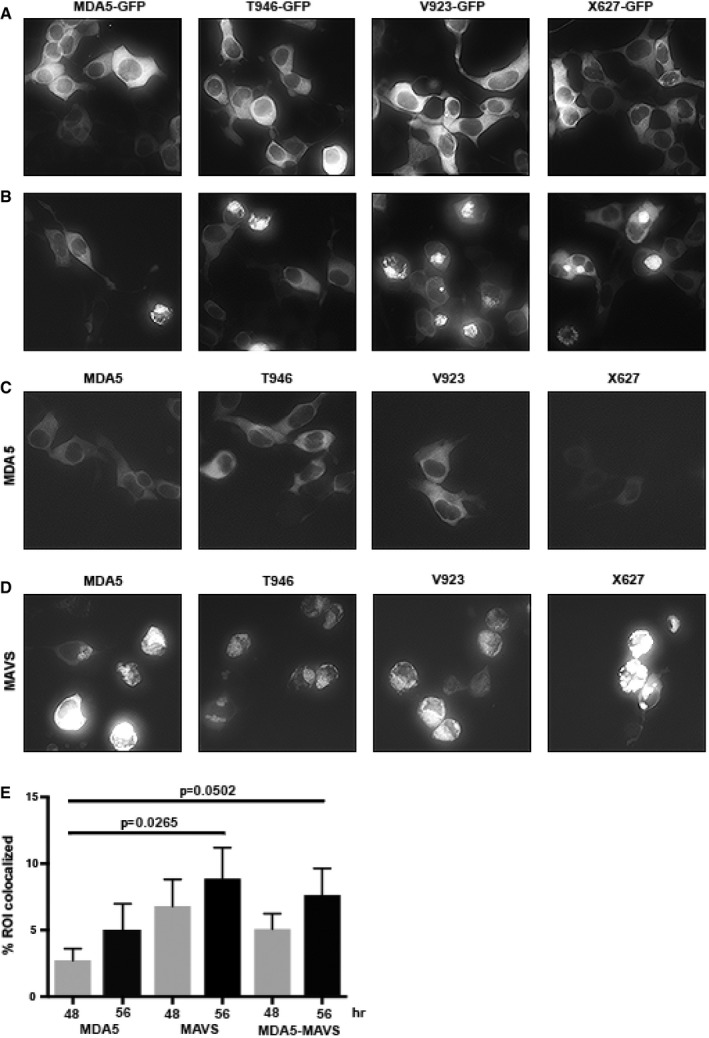
Figure EV4. Visualization of MDA5 expression and associations (related to Fig 7A–C).
-
A, BMicrographs showing GFP fluorescence in HEK293 cells transfected with the indicated LV‐MDA5‐GFP variants after 48 (A) and 56 h (B), showing that the cytosolic expressed fusion protein coalesces to a perinuclear signal.
-
C, DMicrographs showing Venus fluorescence in cells co‐transfected with split‐Venus constructs separately fused to the indicated MDA5 variants and either (C) MDA5 to detect homotropic interactions or (D) MAVS to detect hetero‐oligomerization.
-
EA quantitation of the extent of the association of protein complexes with the mitochondria assessed by measuring the co‐localization of Venus fluorescence with the signal from the MitoTracker stain as regions of interest (ROI). The data are shown as mean ± SD. Student's t‐test was used to calculate the P‐values from six measures for each protein pair at each time point. NS = P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Luciferase promoter reporter assays were used to investigate how these different SNPs affected MDA5‐dependent cell signaling. HEK293 cells were co‐transfected with the different MDA5 constructs and IFIT1‐, IFNB1‐, and IL‐8‐promoter firefly‐luciferase reporters that respond to IRF3/7 and/or NFκB and a constitutive Renilla luciferase reporter. After 24 h, the cells were infected with RV, and then 24 h later, the cells were lysed and the luciferase activity was assayed. These data show that the products of the minor alleles had reduced activity compared to that of the major IFIH1 allele (Figs 7D–F and EV5). The truncated MDA5 protein (X627) was the most impaired in its capacity to induce transcriptional activity. The MDA5 variants demonstrated varying capacity to activate different transcription factors, with the residue at position 946 in MDA5 mediating NFκB activity, whereas the residue at position 923 mediated the activity of the IRFs (Fig 7D–F). Consistent with this differential activity and our preceding data (Fig 3F), the X627 and T946 MDA5 variants also induced less cell death, while the V923 variant, which retained the ability to activate NFκB (Fig 7F), retained the activity of the dominant MDA5 isoform (Figs 7G and EV5).
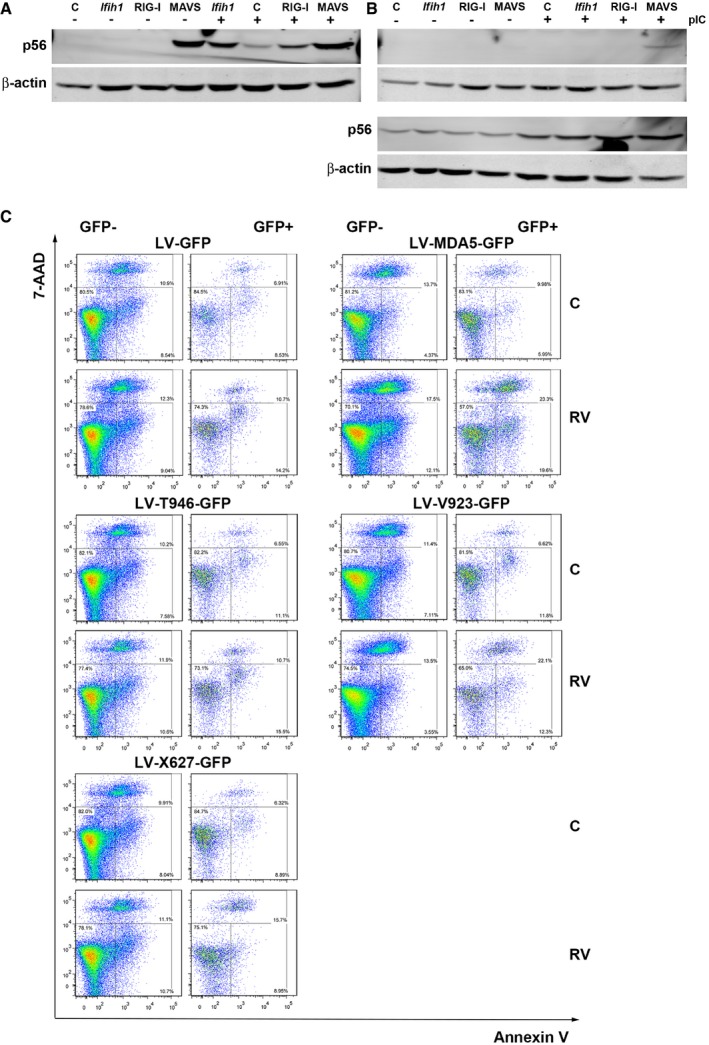
Figure EV5. Transient transfection assays with MDA5 lentiviral constructs (related to Fig 7D–G).
-
A, BImmunoblots detecting the induction of P56 to demonstrate the responsiveness of (A) HEK293 compared to (B) HEK293T (at top) and HeLa (at bottom) cells expressing MDA5 (Ifih1), RIG‐I, or MAVS when treated with FuGENE:pIC to activate the RLRs. β‐Actin is used as a loading control.
-
CFACS plots of HEK293 cells expressing LV‐GFP or the indicated LV‐MDA5‐GFP variants stained with annexin V and 7‐AAD. The data represent cellular viability in both transfected (GFP+) and non‐transfected cells (GFP−). The GFP‐, annexin V‐, and 7‐AAD‐positive cells are graphed in Fig 7G.
To assess the consequence of this for antiviral function, MDA5‐expressing lentiviral constructs were prepared with a carboxyl‐terminal green fluorescent protein (GFP) fusion. These MDA5‐expressing constructs produced lower titers of lentivirus compared to the control (GFP alone), indicating that MDA5 inhibits lentivirus production (Figs 7H and EV6). Moreover, the products of the minor alleles produced higher lentivirus titers than that of the major IFIH1 allele, indicating that the SNPs impaired this antiviral activity of MDA5 (Fig 7H). We had intended to test the consequence of polymorphisms of IFIH1 by stably expressing the different IFIH1 constructs in the Ifih1 −/− MEFs. However, consistent with our preceding data, infection of cells with the construct of the major allele of IFIH1 induced cell death after prolonged culture. Therefore, we resorted to transient transfection assays. HEK293 cells were transfected with a control GFP‐expressing plasmid or constructs of the separate MDA5 variants and then 24 h later were infected with RV. Titration of the progeny RV from the cell supernatants a further 24 h later showed that the T946, V923, and X627 variants produced more infectious RV than the product of the major IFIH1 allele (Fig 7I). The extent of this impairment was not equivalent, with the nsSNP that generated a truncated MDA5 protein (X627) least effective in controlling RV infection.
Figure EV6. MDA5‐induced cell death (related to Fig 7H and I).
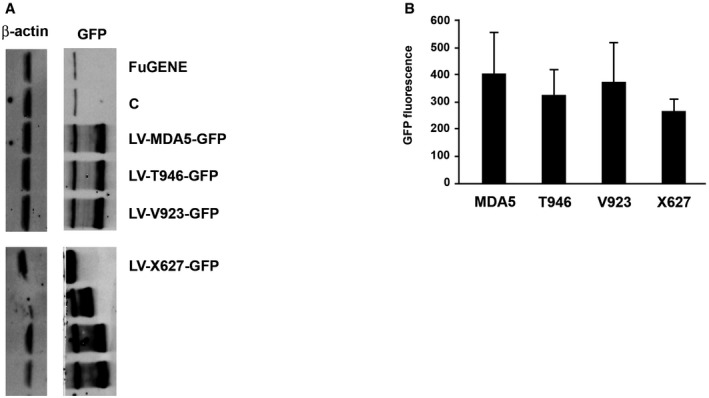
- Immunoblot detection of the expression of the indicated LV‐MDA5‐GFP constructs in HEK293 cells using an anti‐GFP antibody. Unmarked lanes are mutant MDA5 constructs that were used as additional controls in the study that are not described in the text.
- A graph showing the relative GFP fluorescence produced in HEK293 cells transfected with the indicated LV‐MDA5‐GFP constructs. The data is shown as mean ± the SD (n = 3).
Together, these data demonstrate that the T1D‐related IFIH1 SNPs differently affect the function of MDA5 to reduce the protein's activity and limit the antiviral response against RV infection.
Discussion
Although T1D is strongly influenced by genetics, there is evidence that disease progression is also impacted by environmental factors. Virus infection has been proposed as an etiological agent for T1D. The recognition that polymorphisms within the antiviral IFIH1 gene correlate with the risk of developing T1D appears to corroborate this proposition (Kato et al, 2006; Smyth et al, 2006). Explanations of how viral infection might advance autoimmunity include the following: viral molecular mimicry, although this is not supported by the association of T1D with genetic variability in the class II (as opposed to the class I) HLA receptors; epitope spreading; lymph node priming; expansion of populations of auto‐reactive cells; and insulitis. Our data support the latter by demonstrating that Mda5 induces inflammation and cell death in response to RV. The tissue‐specific innate immune response that is regulated by Mda5 identifies a consonance with subsequent autoimmunity in T1D.
We show here that ablating Mda5 increased the susceptibility to RV infection and reduced the innate immune response. This, with the demonstration that the SNPs in IFIH1 that are associated with reduced risk of T1D reduced the antiviral activity of MDA5, argues against the proposition that persistent viral infection induces autoimmunity (Dotta et al, 2007; Richardson et al, 2009; Op de Beeck & Eizirik, 2016). In fact, persistent virus infection has been associated with the repression of cell‐mediated immunity as T cells become exhausted from continuous T‐cell receptor stimulation from persistent antigen (Gruener et al, 2001; Day et al, 2006; Angelosanto et al, 2012; Dyavar Shetty et al, 2012). Rather than viral infection per se, these findings argue that it is the antiviral response that induces immune pathology. This is consistent with genetic studies linking the pathway with autoimmune diseases.
Previous reports had associated the IRF7 locus with the risk of T1D and identified that the condition is strongly associated with IFN signaling (Heinig et al, 2010). Polymorphisms in other IFN‐regulated genes that regulate MDA5 activity have also been associated with T1D. A SNP identified in the IFN‐regulated 2′‐5′‐oligoadenylate synthase 1 (OAS1) that increased enzyme activity suggested that, like MDA5, OAS1 activity is pernicious in T1D. Significantly, OAS1 activity produces RNA substrates, via ribonuclease L, that activate MDA5 (Malathi et al, 2007). Genetic studies have identified gain‐of‐function mutations in IFIH1 and loss‐of‐function mutations in the RNA‐specific adenosine deaminase 1 (ADAR1), which modifies endogenous RNAs to prevent auto‐activation of MDA5 (Liddicoat et al, 2015), in patients with interferonopathies who show elevated IFN signaling and autoimmunity (Enevold et al, 2014; Rice et al, 2014; Rutsch et al, 2015).
Corresponding with the pathophysiology associated with excess MDA5 activity in humans, it was shown that increasing Mda5 levels in transgenic mice accelerated the production of switched autoantibodies on a lupus‐susceptible background (Crampton et al, 2012). Equivalently, reducing the expression of Mda5 through heterozygosity increased regulatory compared to effector T cells at sites of autoimmunity (Lincez et al, 2015). Notably, ablating Mda5 expression protected β‐cells in the non‐obese diabetic mouse (Lincez et al, 2015). Although earlier experiments had demonstrated that forced expression of type I IFNs in the murine pancreas induced immune destruction of the β‐cells (Stewart et al, 1993), ablating the Ifnar1 receptor in the non‐obese diabetic mouse did not alter disease progression (Carrero et al, 2013). Accordingly, type I IFN signaling is not essential. This mutation does not preclude the involvement of type III IFNλs, which induce an analogous response to type I IFNs via separate receptors. Consistent with this, pancreatic islets produce IFNλs in response to viral infection and these cytokines have been demonstrated to be essential to control RV infection and to augment T‐ and B‐cell responses during viral infection (Pott et al, 2011; Lind et al, 2013; Misumi & Whitmire, 2014). Furthermore, a SNP within the locus that encodes the IFNλ genes (IL29, IL28A, and IL28B) was associated with increased seroconversion following virus challenge (Egli et al, 2014).
T1D is not currently regarded as an interferonopathy, but the former discussion argues that it should be. Although other interferonopathies have been associated with gain‐of‐function mutations in MDA5, this is not the case with T1D. Instead, we propose that viral infection and subsequent IFN signaling induce MDA5 activity. The low incidence of T1D necessitates that this antiviral response must be accompanied by additional defects in immune regulation. Importantly, gain‐of‐function mutations in MDA5 have been identified in healthy people and the development of autoimmune pathology in mice that overexpress Mda5 or IFNs is dependent upon the genetic background (Stewart et al, 1993; Crampton et al, 2012). Accordingly, MDA5‐dependent activity and IFN signaling are not sufficient to induce pathophysiology and additional conditions are required in order to break immune tolerance.
Gene association studies have identified a number of other potential T1D candidate genes. Although not shown to be causal and by no means comprehensive, the current genes that have been identified to correlate with T1D implicate the patient's leukocyte function. This is characterized by polymorphisms in the human leukocyte antigen (HLA) variants DQA1, DQB1, and DRB1; C‐C chemokine receptor type 5 (CCR5); cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4); hepatic nuclear factor 1α (HNF1A); interleukin 2 receptor subunit α (IL2RA); and protein tyrosine phosphatase, non‐receptor type 22 (PTPN22).
In aggregate, the data suggest that the minor alleles of T1D‐related IFIH1 SNPs diminish the innate immune response to RV infection, resulting in an impaired ability to limit RV production compared to individuals with the major allele. This, with the strong selective pressure from infectious virus, likely explains the rarity of these polymorphisms in the population. However, the more robust innate immune response in individuals with the major IFIH1 allele, with the tissue‐specific activity of MDA5 combined with additional uncertain defects in immune regulation, predisposes them to progress to autoimmune destruction of their β‐cells following viral infection. In this light, polymorphism within IFIH1 that decreases the function of MDA5 might be retained in the population, as they protect against more injurious immune responses. Together, these data reassert the concept that it is not viral infection per se, but MDA5‐induced cell death, IFN signaling, and ensuing inflammation that prime the individual's lymphocyte function to promote autoimmune pathology in T1D.
Materials and Methods
Ethics statement
All procedures were conducted in accordance with protocols approved by the Monash University Animal Welfare Committee (approval number MMCA/2007/43) under relevant institutional guidelines, the Prevention of Cruelty to Animals Act 1986 and associated regulations, and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Mice
Mice were sedated with intraperitoneal injection of Avertin (tribromoethanol), 12.5 mg/ml in water, 4 mg/10 g of body weight. Animals were euthanized by cervical dislocation or using a carbon dioxide inhalation chamber. C57BL/6 mice were obtained from Monash Animal Services, and Ifih1 −/− mice were obtained from Dr Marco Colonna (Gitlin et al, 2006). Mice were bred and housed homogeneously in the Monash Medical Centre Animal Facility under conventional conditions. Mice aged 5 weeks were orally inoculated with trypsin‐activated RV (1 × 106 fluorescent cell‐forming units (FCFU) per 10 g body weight) and mock RV (TSC buffer) for 5 days using a peripheral venous catheter (BD Insyte™ 381233, 20 G × 1.00 in, 1.1 mm × 25 mm). Mice were treated with 30 μl of 1.33% NaHCO3 solution 10 min before virus inoculation, to reduce stomach acidity. After 5 days, tissues for biological fluid and RNA extraction were isolated and snap‐frozen in liquid nitrogen and then stored at −80°C. The tissue for histology was fixed in 10% formalin at 4°C overnight and 70% ethanol at 4°C for up to 1 week before processing.
Cells
MEFs were generated from age‐matched Ifih1 −/− and WT mice as described previously (Xu, 2005). Immortalized Ifnar1 −/− MEFs were obtained from Professor Paul Hertzog (Hudson Institute of Medical Research). MEFs, HEK293 (ATCC), HEK293FT (Invitrogen), and MA104 were grown in DMEM with 10% FBS at 37°C, with 5% CO2, and were passaged every 2–5 days under sterile conditions. Peritoneal macrophages were isolated as described previously (Zhang et al, 2008) and were grown in RPMI with 10% FBS at a density of 2 × 105 cells/well in 24‐well plates for 72 h before treatments. Cells were transfected using FuGENE 6 (Roche) transfection reagent as recommended.
Virus
RV was activated in DMEM containing 10 μg/ml porcine trypsin at 37°C for 30 min and was added to serum‐free confluent cells at a final concentration of 105 FCFU/ml. Virus was titrated, and then, the remaining supernatant was inactivated by treatment with UV for 30 min with agitation. RV titers were determined by performing twofold serial dilutions of harvested virus in DMEM containing 1 μg/ml porcine trypsin in a 96‐well plate. After incubation for 18 h at 37°C, the supernatants were aspirated and the cells were fixed with 80% (v/v) acetone, the acetone removed, and anti‐SA11 polyclonal antiserum added, diluted 1:400 with PBS, for 1 h at 37°C, followed by three washes and incubation with 30 μl goat anti‐rabbit antibody (Alexa® Fluor 594) diluted 1:1,000 in PBS at 37°C/5% CO2 for 1 h. Fluorescent cells were counted and virus titers represented as FCFU/ml.
Lentivirus production
The IFIH1 open reading frame (ORF) was cloned into the lentiviral vector pLV‐GFP as an XbaI‐BamHI fragment to generate pLV‐Ifih1‐GFP. The SNPs (T946, V923, and X627) were generated by site‐directed mutation using Pfu turbo (Agilent) with the oligonucleotides: E627X‐TGCGTATACTCATCTTTAAACTTTCTATAATGAAGAG, A946T‐GTAAGAGAAAACAAAACACTGCAAAAGAAGTGT, and I923V‐GGGAAGATATCCATGTAGTTGAGAAAATGCATCAC. Recombinant lentivirus was packaged using the ViraPower™ Packaging Mix (Invitrogen) as directed. Transfected HEK293FT cells were incubated overnight, and then, the medium was supplemented with 2 mM l‐glutamine, 1 mM sodium pyruvate, and 1× MEM non‐essential amino acids (Invitrogen). Cell supernatants were harvested 72 h later, centrifuged at 1,500 g for 15 min at 4°C, and filtered through a sterile 0.45‐μm PVDF filter. Viral stocks were stored at −80°C. Lentivirus was titrated on HEK293 cells over a 10‐fold serial dilution, and then after 72 h, 50–100 positive cells per field were counted and the viral titer was expressed as FCFU/ml.
Fluorescence assays
The IFIH1 and MAVS ORFs were cloned into pcDNA3‐V1/V2 (as AccIII‐XbaI fragments) to produce amino‐terminal split‐Venus‐tagged proteins, respectively. The WT IFIH1 sequence was first mutated to alter an XbaI restriction site. HEK293 cells were seeded into a 24‐well culture plate, transfected with 40 ng/well of each construct, and imaged after 24–48 h with an Olympus IX70 microscope and U‐RFL‐T burner. Images were captured using the SPOT‐RT3 camera and software (Diagnostic Instruments Inc.) and analyzed with ImageJ (http://imagej.nih.gov/ij). Alternatively, cells were grown on coverslips and then stained with MitoTracker and Hoechst after 48 h and processed as described below.
Immune detection
Primary antibodies used for Western blots were as follows: mouse anti‐Mda5 (Axxora, ALX‐210‐352), anti‐IκBα (Cell Signaling, 9242), mouse anti‐Il‐1β (Abcam, ab9722), anti‐β‐actin (Abcam, ab8226), mouse anti‐P56 (Dr Ganes C Sen, Cleveland Clinic, USA), and anti‐GFP (SIGMA). The proteins on the membranes were detected using an Odyssey imaging system (LI‐COR Biosciences) and quantitated using ImageJ (Schneider et al, 2012).
Reagents used for ELISA were as follows: rat anti‐mouse Ifnβ monoclonal antibody 7F‐D3 (Abcam, ab24324), rabbit anti‐mouse Ifnβ polyclonal antibody (PBL Biomedicals, 32400) and goat anti‐rabbit IgG‐HRP (Santa Cruz, E2908), rat anti‐mouse Il‐6 (BD Pharmingen, 554400), biotin rat anti‐mouse Il‐6 (BD Pharmingen, 554402), HRP‐streptavidin conjugate (Invitrogen, 43‐4323) and recombinant murine Il‐6 standards (BD Pharmingen, 554582), and a recombinant murine Ifnβ standard (Professor Paul Hertzog). Tnfα and Il‐1β were measured using kits (BD OptEIA™, 558534 and 559603).
For microscopy, cells were seeded on sterile coverslips (Menzel‐Glaser, 12 × 12 mm, #1.5, Thermo Fisher Scientific, CSC1215GP), infected with RV, then fixed with 10% formalin for 30 min after washing with PBS, permeabilized with 0.1% Triton X‐100 in PBS for 5 min, and then blocked with Assay Diluent (BD OptEIA) for 30 min before detection. Infected cells were probed with polyclonal anti‐SA11 (Donker et al, 2011), goat anti‐rabbit antibody (Alexa® Fluor 594, Invitrogen), phalloidin (Biotium), and Hoechst (Invitrogen). IRF3 was detected using anti‐IRF3 rabbit polyclonal antibody (FL‐425, Santa Cruz Biotech) and goat anti‐rabbit (Alexa® Fluor 488, Invitrogen). Coverslips were mounted with Mowiol (13% Mowiol, 33% glycerol, and 20% sodium azide, pH 8.5) and recorded using a DeltaVision imaging system (Applied Precision). Three‐dimensional pictures of the cells were taken using SoftwoRx and the numbers and intensities of RV particles and the numbers of cells in each image were counted using Imaris from Bitplane, and the total viral particle intensities per cell were calculated and analyzed using Prism 6 (GraphPad).
qRT–PCR
Total RNA was extracted with TRIzol (Invitrogen), and cDNA was synthesized using a First Strand SuperScript™ II RT kit (Invitrogen). qRT‐PCR was performed using SYBR GreenER™ qPCR SuperMix (Invitrogen) in iCycler® PCR Instrument (Bio‐Rad Laboratories), using the following primers: Ifih1‐F GCCCAGAAGACAACACAGAATCAGACA, Ifih1‐R TGCAGTTCTGGCTCGGGGGA, Ifnb1‐F TCCGAGCAGAGACTTCAGGAA, Ifnb1‐R TGCAACCACCACTCATTCTGAG, Il6‐F GAGGATACCACTCCCAACAGACC, Il6‐R AAGTGCATCATCGTTGTTCATACA, Tnf‐F GAAAAGCAAGCAGCCAACCA, Tnf‐R CGGATCATGCTTTCTGTGCTC, Il1b‐F CCTGCTGGTGTGTGACGTTCC, Il1b‐R TCCTTTGAGGCCCAAGGCCACA, Rn18s‐F GTAACCCGTTGAACCCCATT, and Rn18s‐R CCATCCAATCGGTAGTAGCG.
Luciferase assay
Cells co‐transfected with gene‐expressing constructs and the firefly and Renilla luciferase plasmids and then infected with RV were processed following the protocol for the Dual‐Luciferase® Reporter Assay System (Promega) and quantitated using a FLUOstar Optima imager (BMG Labtech). Firefly luciferase expression was normalized by the levels of Renilla luciferase activity.
Assays of cell death
Cell survival was assessed by directly counting cell number and similarly by staining cells with crystal violet, then quantitating the intensity of this stain. Additionally, the relative uptake of annexin V, 7‐AAD, and the nuclear YO‐PRO stain was assessed to positively identify dying cells. The relative uptake of these different fluorescent stains can distinguish different modes of cell death, although alternative modes of cell death were not determined in these experiments.
Assessment with annexin V and 7‐AAD was performed by trypsinizing cells, then washing with cold cation‐free PBS (Gibco®), and resuspending in PBS with 1 mM EDTA (pH 7) and 1% serum at a density of 1 × 106 to 2 × 107 cells/ml for a minimum volume of 0.5 ml, passed through the strainer of 12 × 75 mm polypropylene tubes (BD Falcon™), and then, GFP‐expressing cells were isolated by Cell Sorter (MoFlo™ XDP, Beckman Coulter) and then cultured in medium supplemented with 100 U/ml penicillin and 100 g/ml streptomycin (SIGMA). For the analysis of cell death, cells were resuspended in 1× Binding Buffer at a concentration of 1 × 106 cells/ml in polystyrene tubes (Techno Plas) and incubated with PE annexin V and 7‐AAD (BD Pharmingen™) for 15 min at room temperature in the dark, and then, the reaction was terminated by additional 1× Binding Buffer. RV‐containing samples were fixed with 2% paraformaldehyde for 20–30 min on ice after staining, and the cells were analyzed using a BD FACSCanto II Analyzer and the data were analyzed using FlowJo. Additionally, cells were fixed with 10% formalin before addition of crystal violet solution (Sigma, C3886‐25G, 0.1% in 2% ethanol) and then incubated for 30 min at room temperature. After three PBS rinses and air‐drying in a fume hood, 100% methanol was added to the plate to solubilize the stain. The absorbance of the dissolved cell stain was read at 540 nm on a FLUOstar Optima imager (BMG Labtech).
Alternatively, HEK293 cells were transfected with 25 ng MAVS, 120 ng MDA5, or 100 ng of a constitutively active IRF3 (IRF3‐5D)‐expressing construct and 100 ng of either the IκBα, or dominant‐negative constructs of IRF3 (IRF3‐ΔN), IKKε, or TBK1 constructs in a 24‐well plate, and then passaged to a 96‐well plate 24 h later before being treated with 2 μg/ml pIC and FuGENE for 10 h after a further 24 h. YO‐PRO (Thermal Fisher Scientific) and Hoechst dyes were added to the medium, and the accumulation of the fluorescent YO‐PRO signal in the cell nucleus was quantitated using a Cellomics bioimager (Thermal Fisher Scientific).
Author contributions
Conceptualization: AJS; Methodology: YD, AJS; Investigation: YD, HCHY, CDK, AJS; Writing—original draft: YD, AJS; Visualization: YD, AJS; Funding acquisition: BRGW, AJS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We are grateful to Dr Marco Colonna for the Ifih1 −/− mouse; Dr Daniel Cowley for purified RV; Dr Ganes Sen for anti‐P56 antibody; Dr Paul Hertzog for Ifnar1 −/− MEFs and Ifnar1‐neutralizing antibody; Dr Nicole De Weerd for recombinant Ifnβ and Ifn1α; Drs Curt Hovath, John Hiscott, and Ashley Mansell for expression constructs (MDA5, LGP2, MAVS, IRF3‐5D, and dominant‐negative IKKε, IRF3, and TBK1); and Drs Kirstin Elgass and Sarah Creed for conducting the Cellomics analysis. We also thank Dr Aneta Strzelecki for technical support, Dr Frances Cribbin for editing this manuscript, and Drs Len Harrison and Margo Honeyman for their advice. This research was supported by grants from the National Health and Medical Research Council of Australia (Grantor ID: 491094 (BRGW) and 1043398 (AJS, BRGW); https://www.nhmrc.gov.au), a Monash University‐China Scholarship Council Doctoral Scholarship (YD; http://www.monash.edu), and the Victorian Government's Operational Infrastructure Support Program (http://www.vic.gov.au). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The EMBO Journal (2017) 36: 2742–2757
References
- Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ (2012) Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol 86: 8161–8170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, Hartmann G (2009) Proapoptotic signaling induced by RIG‐I and MDA‐5 results in type I interferon‐independent apoptosis in human melanoma cells. J Clin Invest 119: 2399–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broquet AH, Hirata Y, McAllister CS, Kagnoff MF (2011) RIG‐I/MDA5/MAVS are required to signal a protective IFN response in rotavirus‐infected intestinal epithelium. J Immunol 186: 1618–1626 [DOI] [PubMed] [Google Scholar]
- Bruns AM, Leser GP, Lamb RA, Horvath CM (2014) The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5‐RNA interaction and filament assembly. Mol Cell 55: 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER (2013) Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PLoS ONE 8: e59701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampton SP, Deane JA, Feigenbaum L, Bolland S (2012) Ifih1 gene dose effect reveals MDA5‐mediated chronic type I IFN gene signature, viral resistance, and accelerated autoimmunity. J Immunol 188: 1451–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R et al (2006) PD‐1 expression on HIV‐specific T cells is associated with T‐cell exhaustion and disease progression. Nature 443: 350–354 [DOI] [PubMed] [Google Scholar]
- Di Fiore IJ, Holloway G, Coulson BS (2015) Innate immune responses to rotavirus infection in macrophages depend on MAVS but involve neither the NLRP3 inflammasome nor JNK and p38 signaling pathways. Virus Res 208: 89–97 [DOI] [PubMed] [Google Scholar]
- Donker NC, Foley M, Tamvakis DC, Bishop R, Kirkwood CD (2011) Identification of an antibody‐binding epitope on the rotavirus A non‐structural protein NSP2 using phage display analysis. J Gen Virol 92: 2374–2382 [DOI] [PubMed] [Google Scholar]
- Dotta F, Censini S, van Halteren AGS, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Prato SD, Elliott JF, Covacci A, Rappuoli R, Roep BO, Marchetti P (2007) Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent‐onset type 1 diabetic patients. Proc Natl Acad Sci USA 104: 5115–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyavar Shetty R, Velu V, Titanji K, Bosinger SE, Freeman GJ, Silvestri G, Amara RR (2012) PD‐1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest 122: 1712–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli A, Santer DM, O'Shea D, Barakat K, Syedbasha M, Vollmer M, Baluch A, Bhat R, Groenendyk J, Joyce MA, Lisboa LF, Thomas BS, Battegay M, Khanna N, Mueller T, Tyrrell DL, Houghton M, Humar A, Kumar D (2014) IL‐28B is a key regulator of B‐ and T‐cell vaccine responses against influenza. PLoS Pathog 10: e1004556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enevold C, Kjaer L, Nielsen CH, Voss A, Jacobsen RS, Hermansen ML, Redder L, Oturai AB, Jensen PE, Bendtzen K, Jacobsen S (2014) Genetic polymorphisms of dsRNA ligating pattern recognition receptors TLR3, MDA5, and RIG‐I. Association with systemic lupus erythematosus and clinical phenotypes. Rheumatol Int 34: 1401–1408 [DOI] [PubMed] [Google Scholar]
- Feng N, Kim B, Fenaux M, Nguyen H, Vo P, Omary MB, Greenberg HB (2008) Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J Virol 82: 7578–7590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M (2006) Essential role of mda‐5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 103: 8459–8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, Sullivan J, Phillips R, Pape GR, Klenerman P (2001) Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol 75: 5550–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A, Hummel O, Lee YA, Paskas S, Rintisch C, Saar K, Cooper J, Buchan R, Gray EE, Cyster JG, Erdmann J et al (2010) A trans‐acting locus regulates an anti‐viral expression network and type 1 diabetes risk. Nature 467: 460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hober D, Sauter P (2010) Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol 6: 279–289 [DOI] [PubMed] [Google Scholar]
- Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, Couper JJ, Tait BD, Colman PG, Harrison LC (2000) Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 49: 1319–1324 [DOI] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I (1995) A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA 92: 11284–11288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Fisher PB (1993) Use of a sensitive and efficient subtraction hybridization protocol for the identification of genes differentially regulated during the induction of differentiation in human melanoma cells. Mol Cell Differ 1: 285–299 [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB (2002) mda‐5: an interferon‐inducible putative RNA helicase with double‐stranded RNA‐dependent ATPase activity and melanoma growth‐suppressive properties. Proc Natl Acad Sci USA 99: 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C‐S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 441: 101–105 [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincez PJ, Shanina I, Horwitz MS (2015) Reduced expression of the MDA5 Gene IFIH1 prevents autoimmune diabetes. Diabetes 64: 2184–2193 [DOI] [PubMed] [Google Scholar]
- Lind K, Richardson SJ, Leete P, Morgan NG, Korsgren O, Flodstrom‐Tullberg M (2013) Induction of an antiviral state and attenuated coxsackievirus replication in type III interferon‐treated primary human pancreatic islets. J Virol 87: 7646–7654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M Jr, Silverman RH (2007) Small self‐RNA generated by RNase L amplifies antiviral innate immunity. Nature 448: 816–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi I, Whitmire JK (2014) IFN‐lambda exerts opposing effects on T cell responses depending on the chronicity of the virus infection. J Immunol 192: 3596–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op de Beeck AO, Eizirik DL (2016) Viral infections in type 1 diabetes mellitus–why the beta cells? Nat Rev Endocrinol 12: 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschläger N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, Ruland J (2010) Recognition of RNA virus by RIG‐I results in activation of CARD9 and inflammasome signaling for interleukin 1Β production. Nat Immunol 11: 63–69 [DOI] [PubMed] [Google Scholar]
- Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW (2011) IFN‐lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci USA 108: 7944–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader‐Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L et al (2014) Gain‐of‐function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46: 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 52: 1143–1151 [DOI] [PubMed] [Google Scholar]
- Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, Nurnberg P, Hohne W, Crow YJ, Feigenbaum A, Hennekam RC (2015) A specific IFIH1 gain‐of‐function mutation causes Singleton‐Merten syndrome. Am J Hum Genet 96: 275–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, Pruijssers AJ, Dermody TS, Garcia‐Sastre A, Greenberg HB (2011) The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS‐1, RIG‐I, MDA‐5, and IRF3. J Virol 85: 3717–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo‐Arthur C, Carrero JA, White JM, Hertzog PJ, Schreiber RD (2006) Blocking monoclonal antibodies specific for mouse IFN‐alpha/beta receptor subunit 1 (IFNAR‐1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res 26: 804–819 [DOI] [PubMed] [Google Scholar]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu‐Tirgoviste C, Widmer B, Dunger DB, Savage DA, Walker NM, Clayton DG, Todd JA (2006) A genome‐wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon‐induced helicase (IFIH1) region. Nat Genet 38: 617–619 [DOI] [PubMed] [Google Scholar]
- Stewart TA, Hultgren B, Huang X, Pitts‐Meek S, Hully J, MacLachlan NJ (1993) Induction of type I diabetes by interferon‐alpha in transgenic mice. Science 260: 1942–1946 [DOI] [PubMed] [Google Scholar]
- Sun SC, Ganchi PA, Ballard DW, Greene WC (1993) NF‐κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science 259: 1912–1915 [DOI] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G et al (2007) Robust associations of four new chromosome regions from genome‐wide analyses of type 1 diabetes. Nat Genet 39: 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control C (2007) Genome‐wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Werf N, Kroese FGM, Rozing J, Hillebrands JL (2007) Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev 23: 169–183 [DOI] [PubMed] [Google Scholar]
- Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S (2013) Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152: 276–289 [DOI] [PubMed] [Google Scholar]
- Xu J (2005) Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol Chapter 28: Unit 28 21 [DOI] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM (2008) The isolation and characterization of murine macrophages. Curr Protoc Immunol 111: 14.1.1–14.1.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File