

Abstract
The ethylene-forming enzyme (EFE) from Pseudomonas syringae pv. phaseolicola PK2 is a member of the mononuclear non-heme Fe(II)- and 2-oxoglutarate (2OG)-dependent oxygenase superfamily. EFE converts 2OG into ethylene plus three CO2 molecules while also catalyzing the C5 hydroxylation of L-arginine (L-Arg) driven by the oxidative decarboxylation of 2OG to form succinate and CO2. Here we report eleven X-ray crystal structures of EFE that provide insight into the mechanisms of these two reactions. Binding of 2OG in the absence of L-Arg resulted in predominantly monodentate metal coordination, distinct from the typical bidentate metal-binding species observed in other family members. Subsequent addition of L-Arg resulted in compression of the active site, a conformational change of the carboxylate side chain metal ligand to allow for hydrogen bonding with the substrate, and creation of a twisted peptide bond involving this carboxylate and the following tyrosine residue. A reconfiguration of 2OG achieves bidentate metal coordination. The dioxygen binding site is located on the metal face opposite to that facing L-Arg, thus requiring reorientation of the generated ferryl species to catalyze L-Arg hydroxylation. Notably, a phenylalanyl side chain pointing towards the metal may hinder such a ferryl flip and promote ethylene formation. Extensive site-directed mutagenesis studies supported the importance of this phenylalanine and confirmed the essential residues used for substrate binding and catalysis. The structural and functional characterization described here suggests that conversion of 2OG to ethylene, atypical among Fe(II)/2OG oxygenases, is facilitated by the binding of L-Arg which leads to an altered positioning of the carboxylate metal ligand, a resulting twisted peptide bond, and the off-line geometry for dioxygen coordination.
Graphical Abstract
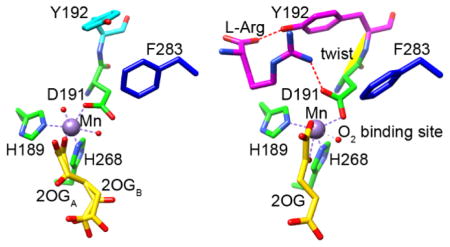
INTRODUCTION
Ethylene, H2C=CH2, is an important building block in the production of plastics, fibers, foams, soaps, detergents, antifreeze, etc.1 This hydrocarbon is produced worldwide at levels that exceed all other synthetic organic chemicals, with its production expected to increase at a compound annual growth rate of ~11.26% from 2016–2025.2 Ethylene is primarily obtained from thermal cracking of natural gas and petroleum,3 but this method of production unfortunately releases into the atmosphere large quantities of CO2 and other greenhouse gases with consequent negative environmental impacts.4 Therefore, technologies are required that produce ethylene from renewable resources. Examples of these alternative or “green” methods include production of ethylene from biomass5–6 and the use of genetically engineered microorganisms including yeast,7–8 Escherichia coli,9–10 and cyanobacteria11–16 that express the efe gene encoding an ethylene-forming enzyme (EFE). The use of transgenic organisms with this gene offers a promising sustainable method for the commercial production of ethylene.
EFE is associated with selected plant pathovars such as the bacteria Pseudomonas syringae and Ralstonia solanacearum and the fungus Penicillium digitatum.17–18 The archetype EFE, from P. syringae pv. phaseolicola PK2, catalyzes two reactions in approximately a 1:2 ratio: (1) the oxidative decarboxylation of 2-oxoglutarate (2OG) coupled to C5 hydroxylation of L-arginine (L-Arg), with the hydroxy-L-Arg intermediate decomposing to guanidine and L-Δ-1-pyrroline-5-carboxylate (P5C), and (2) the decomposition of 2OG to ethylene and three molecules of CO2 (Scheme 1).19–20 The first reaction is typical of those catalyzed by a large family of mononuclear non-heme Fe(II)- and 2OG-dependent oxygenases21–24 and may be the ancestral activity of this enzyme, whereas the second (major) reaction is unique to EFE.
Scheme 1.

Reactions catalyzed by EFE
To clarify how this unique member of the Fe(II)/2OG-dependent oxygenases carries out its dual transformations, we crystallized EFE apoenzyme and ten enzyme complexes with metals and substrates or substrate analogues. We highlight the important differences between our results and three just reported structures (EFE•Mn•bis-Trispropane (BTP), EFE•Mn•2OG, and EFE•Fe•N-oxalylglycine (NOG)•L-Arg), especially those related to ethylene formation.25 In addition, we examine the properties of a range of EFE variants and compare our findings with results of prior mutagenesis efforts.25–27 Together, our structural, mutagenesis, and biochemical analyses provide new insights into the unique ethylene-forming capability of EFE compared to other Fe(II)/2OG oxygenases and they provide a blueprint for engineering EFE to enhance 2OG transformation ethylene without carrying out the wasteful hydroxylation of L-Arg.
RESULTS
Overall Structure of EFE
We solved eleven crystal structures of EFE: the EFE apoprotein, EFE•Ni, and EFE•Mn with 2OG, 2OG•L-Arg, 2-oxoadipate (2OA)•L-Arg, 2OG•Nγ-hydroxy-L-Arg (HO-L-Arg), 2OG•L-argininamide (L-Arg-amide), L-Arg, tartrate, malate, and malate•L-Arg, with EFE•Mn•2OG•L-Arg-amide exhibiting the best resolution at 1.14 Å (Figure 1, Tables S1–S3). Ten structures contained a single EFE molecule per asymmetric unit, consistent with the monomeric species observed in our earlier size-exclusion chromatography/multi-angle light scattering analysis,20 whereas EFE•Mn•2OG contained two EFE molecules per unit cell.
Figure 1.

Active site views of 11 EFE structures. Carbon atoms of the metal-chelating residues (H189, D191 and H268) are shown in green; 2OG, 2OA, malate, or tartrate are depicted in yellow; L-Arg, HO-L-Arg, or L-Arg-amide are illustrated in magenta; Mn and Ni atoms are shown as purple and green spheres. The 2FO - FC maps for metal atoms and ligands are shown as blue meshes at 1 σ. The anomalous maps for metal atoms are shown as magenta meshes at 5 σ. Metal chelation is indicated by dashed lines. Bound water molecules are shown as red spheres. (A) EFE apoprotein. (B) EFE•Ni. (C) EFE•Mn•2OG. (D) EFE•Mn•tartrate. (E) EFE•Mn•malate. (F) EFE•Mn•2OA•L-Arg. (G) EFE•Mn•2OG•L-Arg. (H) EFE•Mn•2OG•HO-L-Arg. (I) EFE•Mn•2OG•L-Arg-amide. (J) EFE•Mn•malate•L-Arg. (K) EFE•Mn•L-Arg.
EFE contains a double-stranded β-helix (DSBH, also known as the jellyroll or cupin fold)28 core typically found in members of the Fe(II)/2OG-dependent oxygenases. Nine β-strands, six from the major β-sheet and three from the minor β-sheet, form the DSBH that is surrounded and stabilized by 10 α-helices (Figure S1A). The eleven EFE structures exhibited the same overall fold with only slight differences observed (Figure S1B). In particular, residues 80–93 move significantly towards the active site when metal and substrate are bound, acting like a “lid” that covers or shields the active site from the solvent.
2OG Adopts Two Modes of Monodentate Metal Binding in the Absence of L-Arg
The 2-histidine-1-carboxylate metal-binding site of EFE is located in a cavity formed by the major and minor β-sheets of the DSBH fold with the H189, D191, and H268 ligands coming from β-8, the loop connecting β-8 to β-9, and β-13 (Figure S1). Compared to the structure of the apoprotein (Figure 1A), Ni (and Mn) binding led to conformational changes of the metal-chelating residues (Figure 1B). In particular, the side chain of H268 flips up and participates in metal binding with the pre-positioned H189 and D191.
In the EFE•Mn•2OG structure, the metal ion is hexa-coordinated and electron density was observed near the metal center corresponding to 2OG (Figure 1C). In contrast to the usual bidentate metal-binding mode of 2OG found in other enzymes of this family,28–29 this substrate was bound monodentate to Mn in EFE lacking L-Arg and was observed in two inverted conformations (Figure 2A). In the 2OGA conformation, the substrate bound Mn with its C1 carboxyl group; in the 2OGB conformation, it bound the metal using its C5 carboxyl group. Monodentate binding to Mn also was observed for the shorter dicarboxylates, tartrate and malate, including for the EFE•Mn•malate•L-Arg complex (Figure 1, D–E and J).
Figure 2.

Active site comparisons of EFE•Mn•2OG and EFE•Mn•2OG•L-Arg. Carbon atoms of the metal-chelating residues are shown in green; 2OG and stabilizing residues are depicted in yellow; L-Arg and its stabilizing residues are illustrated in magenta (these residues are cyan when L-Arg is absent); and water and Mn are shown as red and purple spheres, respectively. The 2FO - FC maps for metal atoms and ligands are shown as blue meshes at 1 σ. The anomalous maps for metal atoms are shown as magenta meshes at 5 σ. Metal chelation is indicated by dashed lines colored the same as the metal, hydrogen bonds are indicated by red dashed lines. (A) EFE•Mn•2OG (1.85 Å). (B) EFE•Mn•2OG•L-Arg (1.43 Å).
L-Arg Binding Leads to Bidentate Metal Coordination by 2OG
In the presence of L-Arg, 2OG shifts from monodentate coordination of the Mn site to the typical chelate binding mode (Figures 1G and 2B). Similarly, the L-Arg analogues L-Arg-amide and HO-L-Arg also facilitate bidentate Mn chelation by 2OG (Figure 1, H and I). 2OA, an alternative 2-keto acid substrate greater in size by one methylene unit compared to 2OG, also bound the metallocenter in a chelate mode when L-Arg was present (Figure 1F).
In all cases of bound 2OG, the carboxylate distal to the metal was stabilized by a salt bridge with R277 and the carboxylate coordinating the metal was stabilized by hydrogen bonds with R171 (Figure 2). Efforts to produce the R277A variant led to the formation of inclusion bodies and we did not study this protein further. Furthermore, while the R171A variant enzyme was soluble, it produced no detectable ethylene and only a trace of P5C under the standard assay conditions (Figure 3A and Table S4). These results indicate the critical nature of these two arginine residues for interacting with 2OG.
Figure 3.

Activity comparisons of WT and selected EFE variants. Ethylene and P5C production by variants of (A) selected hydrophilic residues near the active site along with their location (orange) in the EFE•Mn•2OG•Arg structure, (B) selected hydrophobic residues and their locations (dark blue), and (C) additional EFE variants and their locations (purple). Carbon atoms of metal ligands are shown in white, 2OG in yellow, and L-Arg in magenta. Mn atoms are purple spheres. Assay conditions: 2 mL reaction consisting of 0.5 mM 2OG, 0.5 mM L-Arg, 0.2 mM Fe(II), 0.4 mM L-ascorbic acid, and 1 μM EFE in 25 mM HEPES, pH 7.5, incubated at 25 °C for 80 min and terminated with HCl. WT enzyme generates ~2 ethylene per P5C, and comparisons are shown as percentage of the WT values. Error bars represent the standard errors of at least two independent enzyme preparations. Variants marked with * were found in inclusion bodies.
In contrast to its carboxylate-binding interactions, 2OG binds to a pocket lined by hydrophobic residues (L173, F175, L206, V270, A279, and F283 (Figure S2A); and the nearby V196, A198, and A281 (Figure 3B)). Replacing F283 by tryptophan, tyrosine, arginine, alanine, and valine led to the near elimination of ethylene production (Figure 3B, Table S4), but several of these variant proteins continued to produce significant levels of P5C. Single substitutions of V196 and A281 to arginine resulted in enzymes producing negligible levels of products. By contrast, the A281V variant produced low levels of products in comparison to the WT enzyme. The V196F variant was insoluble and found in inclusion bodies. Notably, the A198V variant produced large amounts of P5C but very little ethylene. These results highlight the importance of maintaining a hydrophobic environment surrounding the 2OG-binding pocket, with strict limitations on the size of the hydrophobic residue. These residues may function in 2OG stabilization, binding of dioxygen, or release of the small nonpolar product ethylene.
L-Arg-Binding Introduces Significant Conformational Changes in the Active Site
L-Arg was observed to bind near, but did not coordinate, the metal center in EFE•Mn•2OG•L-Arg (Figure 1G) as well as in EFE•Mn•malate•L-Arg, EFE•Mn•L-Arg-amide, EFE•Mn•HO-L-Arg, EFE•Mn•2OA•L-Arg, and EFE•Mn•L-Arg (Figure 1, F, H–K). In all structures with bound L-Arg and HO-L-Arg a single conformation was observed, with only the L-Arg-amide ligand showed two conformations differing slightly in the position of the C5 and NE (Figure 1I). The binding of L-Arg led to several notable structural changes at or near the active site: (i) 2OG shifts to bidentate coordination of the metal and defines a dioxygen binding site (Figures 1G, 2B, and 4A), (ii) D191 alters its orientation to engage in hydrogen bonding interactions with the NH1 of L-Arg (Figures 2B and 4B), (iii) significant positional changes were noted for several residues, leading to a twisted peptide bond between D191 and Y192 (Figure 4, B and C), and (iv) helix α-8 is brought closer to the metal center making the active site more compact (Figure S1B).
Figure 4.
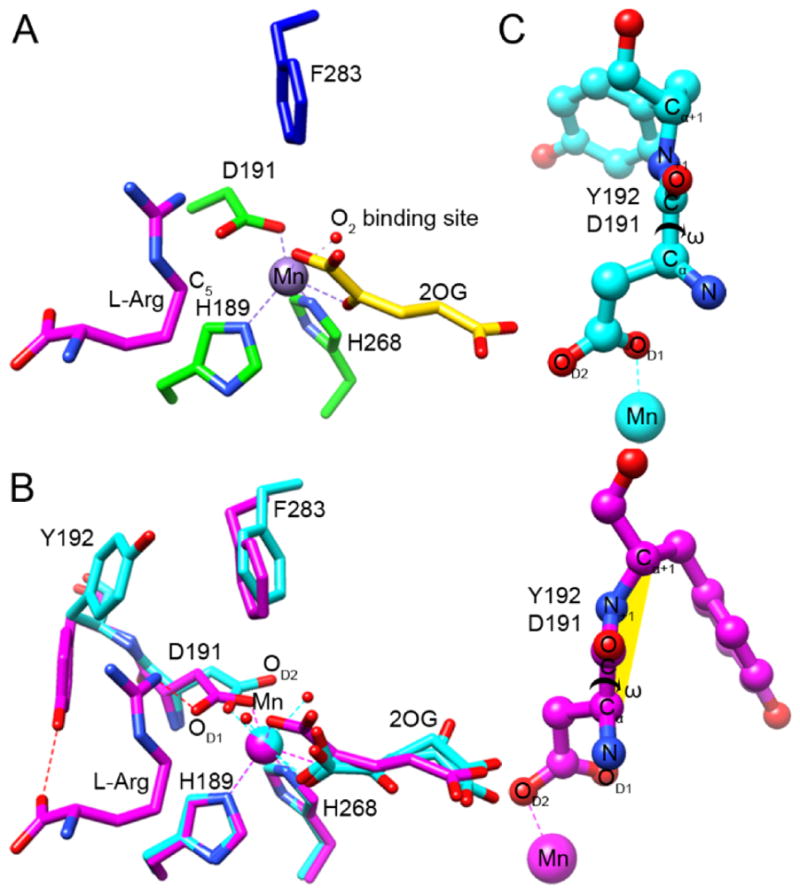
Identification of the O2 binding site and a twisted peptide bond in EFE•Mn•2OG•L-Arg. (A) The C1-carboxylate oxygen of 2OG (yellow carbons) binds approximately trans to the distal H189 and the C2-keto oxygen binds opposite D191, thus defining a dioxygen-binding site in the “off-line” configuration that points away from the C5 position of the L-Arg substrate (magenta carbons). The 2-histidine-1-carboxylate metal-binding motif is shown with green carbons, and the nearby F283 residue is shown in blue. The dioxygen-binding site is illustrated by a water molecule (red sphere) seen in our EFE•Mn•2OG•HO-L-Arg structure. (B) Conformational changes occurring upon L-Arg binding. Comparison of the active sites of EFE•Mn•2OG in cyan and EFE•Mn•2OG•L-Arg in magenta, based on an active site alignment. Metal chelation is shown by dashed lines, a key hydrogen bond between NH1 of L-Arg and OD1 of D191 is indicated by a red dashed line. Alteration of the D191 orientation leads to a switch in metal chelation from OD1 to OD2. (C) Ball and stick representations of the D191-Y192 peptide bond before and after upon L-Arg binding, showing the dihedral peptide angle ω (Cα, C, N+1, Cα+1). The twisted peptide is highlighted in yellow.
For the structures containing both 2OG (or 2OA) and L-Arg (or L-Arg-amide or HO-L-Arg), the C1 carboxyl and C2 keto groups of the 2-oxo acid coordinate the metal in a bidentate manner and are nearly in the same plane as H268 and D191 (the distal histidine and carboxylate ligands). This configuration defines the open coordination site for dioxygen binding opposite H189 as shown for EFE•Mn•2OG•L-Arg (Figure 4A). Indeed, the highest resolution structures containing HO-L-Arg (1.17 Å) and L-Arg-amide (1.14 Å) revealed a water molecule bound at this site. This configuration has been termed “off-line”30 because a ferryl species formed with this same orientation would not point towards the oxygenase substrate; i.e., the C5 of L-Arg which becomes hydroxylated by EFE. Rather, to abstract the pro-S-hydrogen from this carbon atom of the substrate, located only ~4.2 Å from the metal center, the newly created ferryl species would need to “flip” to a very different position, a notion that has been proposed for other off-line members of this enzyme family.30–31 The repositioning of F283 upon binding of L-Arg (Figure 4B) may have implications regarding this ferryl flip. This residue adopts a position pointing at the metal center (~4.6 Å away) and may hinder the flipping of the ferryl species (Figure 4A). Substitution by the larger residues tyrosine, tryptophan, and arginine essentially abolished ethylene-forming activities (Figure 3B, Table S4), probably due to diminished dioxygen binding, while greatly decreasing P5C formation. Conversely, substitution with the smaller residues valine and alanine allowed for significant levels of L-Arg hydroxylation in the absence of ethylene formation.
L-Arg binding induces a repositioning of the metal ligand D191, allowing it to form a hydrogen bond with the newly introduced substrate (Figures 1G, 2B, and 4B). Accompanying the shift in D191 conformation, Y192 also undergoes a large change in position to hydrogen bond with L-Arg (Figure 4B). These changes create a twisted peptide bond between D191 and Y192 at the enzyme active site (Figure 4C). A peptide is annotated as twisted when one of its dihedral angles deviate over 30° from its ideal value (in this case from 180°).32 For the six L-Arg and L-Arg analog bound structures, the ω torsion angle (D191-Cα, D191-C, Y192-N, Y192-C) is 146.8° on average compared to the 5 unbound structures with an angle of 168.1° (Table S5). We substituted Y192 with arginine, phenylalanine, or tryptophan, leading to reduced product formation in the latter two cases (Figure 3A, Table S4). These results suggest Y192 is important, but not essential, to catalysis and it most likely both helps position L-Arg in the correct orientation for the hydroxylation step and directs the 2OG conversion to ethylene.
In addition to the hydrogen bonding with D191 and Y192, L-Arg binds in a hydrophilic pocket with stabilization achieved by interactions with E84, V85, T86, R316, and Van der Waals interactions with C317 (Figures 3A, S2B, and S2C). E84 forms hydrogen bonds with both the NE and amide nitrogens of L-Arg. Substitution of this residue with alanine leads to near elimination of ethylene and P5C production (Figure 3A, Table S4). Both T86 and the carbonyl oxygen of V85 form hydrogen bonds to the amide nitrogen. Hydrogen bonding with R316 stabilizes the substrate carboxylate, and this residue undergoes a reorientation when L-Arg binds (Figure S2C). We substituted the neighboring residue, C317, with serine or asparagine leading to relatively minor changes in activity (Figure 3A, Table S4). We conclude that this residue is not critical to catalysis.
Non-Active Site Regions
We examined the effects on EFE activities of a series of additional variants that involved residues more distant from the active site (Figure 3C and Table S4). S81 is located in a region that undergoes major rearrangement upon binding of the metal ion and substrates (Figure S1B). Both the S81R and S81Y variants exhibited substantial reductions in product formation, likely due to increased instability of this region. Because E285 hydrogen bonds with Y306 in helix α-8, we investigated the E285Q and E285A variants. Both species lost nearly all ethylene-forming activity, while retaining some P5C production. Residues Y306 and F310 form an aromatic network with F314 and Y318 (all located in the α-8 helix) along with Y192 and F283. Substitution of Y306 with alanine or phenylalanine decreased the level of all products. Similarly, elimination of aromatic interactions by substituting F310 with arginine proved detrimental for EFE activity; however, substitution with the aromatic residue tryptophan resulted in retention of substantial activities. The large reduction in catalysis by EFE when these residues were changed to non-aromatic side chains is likely the result of destabilization throughout the helix that prevents it from moving closer to the active site when L-Arg binds, as needed to make it ready for dioxygen binding. No activity changes were expected for the L73K, L73R, R184A, and E213A variants with substitutions positioned distant to the active site, and only modest activity losses were observed (Figure 3C, Table S4). Surprisingly, the E215A and E213A/E215A variants exhibited substantial reductions in EFE activities despite the distance of E215 from the active site (Figure 3C, Table S4); we have no explanation for this sensitivity. In contrast, the Δ338–350 truncation variant was highly active, consistent with the non-essential role of the carboxyl terminus of EFE from P. syringae pv. phaseolicola PK2 which is extended compared to a related sequence for a known ethylene-forming enzyme that is missing this region (Figure S3).
DISCUSSION
EFE exhibits the typical DSBH fold and 2-histidine-1-carboxylate metal-binding motif found in other members of the Fe(II)/2OG oxygenase family, yet it uniquely catalyzes the transformation of 2OG into ethylene. Below, we compare our findings derived from 11 EFE structures to those associated with three just-published EFE structures,25 especially in terms of understanding how the enzyme catalyzes its two reactions. In addition, we place the previously published mutagenesis studies into a structural context.
Comparisons to Prior EFE Structures
Zhang et al. reported a model for an EFE•Mn•2OG structure (PDB ID 5MOF, 1.45 Å)25 that exhibits substantial differences from our model (PDB ID 5V2X, 1.85 Å). In 5MOF, 2OG has bidentate binding to Mn at 70% occupancy with the sixth coordination site being water at 100% occupancy. The C5 carboxyl group weakly interacts with R277, with only O4 forming a hydrogen bond. At the same position, a chloride ion (modeled at 30% occupancy) strongly interacts with R277. In addition, they model several water molecules at 30% occupancy around 2OG and at the positions of the metal-chelating O2 and O5 atoms (Figure S4B). In 5V2X, we interpreted the map with 2OG binding as a monodentate ligand to Mn, with two water molecules coordinated at 100% occupancy. We chose two conformations (~50%+~30%) for 2OG, either with the C1 or C5 carboxyl group facing R277, in each case with both carboxylate oxygen atoms involved in a salt bridge (Figure 2A and S4A). Both interpretations are consistent that 2OG does not bind tightly to the active site in the absence of L-Arg or an L-Arg analog. We further argue that the monodentate binding is more plausible for the structure lacking L-Arg because the electron density for the bidentate linkage is not strong in our case and forcing a bidentate model would lead to an overbuilt situation. Furthermore, our observed strong interaction of 2OG with R277 appears to be a better interpretation than including a chloride ion at this position. In both EFE•Mn•2OG structures, the D191 coordination is not switched to introduce the twisted peptide bond, consistent with our other monodentate-bound structures that lack L-Arg, EFE•Mn•tartrate (Figure 1D) and EFE•Mn•malic acid (Figure 1E), which also contain water molecules at the fifth and sixth coordination sites. Also, our structural interpretation of predominantly monodentate coordination is compatible with our previously reported anaerobic UV-visible difference spectra of the Fe(II)-containing enzyme.20 In those studies, a low intensity (~28 M−1 cm−1) metal-to-ligand charge-transfer (MLCT) transition at 515 nm was observed for the EFE•Fe(II)•2OG sample, but the addition of L-Arg led to intensification of the MLCT transition at 510 nm (~79 M−1 cm−1) to a value that is more typical of related enzymes. The MLCT transition is known to be associated with chelate binding of the metal in other members of this enzyme family,20 so the sample free of L-Arg exhibits a perturbed binding mode for 2OG. Our model of the EFE•Mn•2OG complex reveals the nature of this perturbation as a mixed ligand state with primarily monodentate coordination.
Zhang et al. additionally presented the EFE•Fe•NOG•L-Arg structure (NOG is an inhibitor of the enzyme; PDB ID 5LUN, 1.08 Å)25 that matched many features of our EFE•Mn•2OG•L-Arg (and several other) structures. At this high resolution, the authors reported two conformations (A:B in a 50:50 ratio, Figure S5) with two models of D191 coordination and their effects on the active site. Interestingly, although the authors did not mention it in their report, a similar twisted peptide bond between D191 and Y192 is identified in their conformation A, with ω being 146.6°, while conformation B had an angle of 152.5° (average of four chains in the unit cell Table S5). In their conformation A, the side chain of D191 forms a hydrogen bond with L-Arg via OD1 and chelates the metal via OD2 (Figure S5A). NOGA forms a salt bridge with R277 while also binding bidentate to iron. This conformation has C5 of L-Arg pointed toward the metal and matches very well with our EFE•Mn•2OG•L-Arg structure. In contrast, their conformation B has D191 chelating iron similar to our structures that lack L-Arg or its analogs (Figure S5B). Furthermore, in conformation B the L-Arg shifts towards the metal (but with the C5 carbon pointed away) and NOG, still with bidentate coordination, now interacts weakly with R277 using a single hydrogen bond. The authors concluded that conformation B facilitates ethylene production. Significantly, we have not seen this second conformation of L-Arg (or L-Arg analog) in any structure we solved so far. For EFE•Mn•2OG•L-Arg-amide we modeled two slightly different L-Arg-amide conformations (Figure 1I); however, neither matches the conformation of 5LUN and we observed no effects on the rest of the active site.
There are at least two possible explanations for why Zhang et al. observed the second L-Arg conformation in 5LUN.25 First, the metal might need to be Fe for the second conformation to appear; however, this hypothesis is unlikely because L-Arg does not directly interact with the metal. Furthermore, all other observations seen in our Mn structures appear to match those of 5LUN, including the two conformations of D191. More likely, the substitution of 2OG by NOG in 5LUN could influence the binding of L-Arg and make this second conformation possible. Our finding of clear coupling between 2OG binding and L-Arg binding adds support to this notion.
Another point of discrepancy between the Zhang et al. studies25 and our findings centers on the intrinsic conformation of NOG/2OG. They emphasize the atypical situation found in their 5LUN structure in which the plane formed by the C5/O3/O4 carboxylate atoms differs significantly from the plane formed by the C3(or N in NOG)/C4/C5 carbon atoms (Figure S5B).25 In contrast, these groups of atoms are coplanar in our EFE•Mn•2OG•L-Arg structure (Figure 4A and S5A), as well as those for EFE•Mn•2OG•L-Arg-amide and EFE•Mn•2OG•HO-L-Arg (Figure 1, H and I). The crystal structures of several other enzyme family members have NOG bound in nearly coplanar orientations (e.g., PDB IDs 3O2G, 3HQR, 4NM6),33–34 so the identity of the ligand does not account for these observed differences. One possible explanation is that the polar amide group in NOG is not compatible with the unusual hydrophobic environment of the 2OG binding site in EFE, so that NOG has to adopt a distorted and distinct conformation.
Mechanism of Ethylene formation
A notable structural aspect of EFE centers on the apparent dioxygen binding site in the EFE•Mn•2OG•L-Arg, EFE•Mn•2OA•L-Arg, EFE•Mn•2OG•L-Arg-amide, and EFE•Mn•2OG•HO-L-Arg crystal structures (Figures 4, A and B, and S6B). In these complexes, C5 of L-Arg is located close to the metal as needed for performing the typical steps of substrate hydroxylation. On the other hand, the open sixth coordination site for dioxygen binding is on the opposite face of the metal from this substrate, indicated by a water molecule in the EFE•Mn•2OG•L-Arg-amide and EFE•Mn•2OG•HO-L-Arg structures (Figures 4 and S6B). Several Fe(II)/2OG oxygenases exhibit a similar off-line configuration30 (Figure S6A), and the reaction mechanisms for those enzymes have been suggested to involve a ferryl flip in which the initially generated ferryl group (with the oxygen atom pointed away from substrate) undergoes a conformational change that allows the ferryl oxygen to approach the substrate.31 The extent of shift required for EFE to hydroxylate L-Arg (~130° based on the positions of the waters in the L-Arg-amide and HO-L-Arg structures) appears to exceed that of most other representatives (Figure S6A). Furthermore, L-Arg binding causes the F283 side chain to shift position to approach the metal (Figure 4B); thus, F283 may partially hinder the flipping of the ferryl group. In addition, the distinctly altered position of D191 compared to other off-line enzymes (Figure S6C) may reduce the tendency of the ferryl group to flip by reducing the space available on the opposite side of ferryl group. The generation of a ferryl group that is poorly positioned to react with substrate L-Arg could reasonably facilitate the aberrant degradation of 2OG, thus leading to ethylene formation.
The occurrence of the twisted peptide bond at the active site of EFE appears to be unique, not only within related enzymes but also in general. Deviations greater than 20° from planarity are considered very rare features. Whereas twisted peptide bonds are not generally associated with active sites and catalysis,35 the fact that the D191-Y192 peptide bond is severely twisted only upon L-Arg binding suggests it may play a significant role in the reaction. We postulate that such a strong structural constraint imposed by L-Arg (or L-Arg analog) binding could alter the energy state of the enzyme-substrate complex, facilitating ethylene formation through an unknown mechanism, as the other non-ethylene producing members within the same enzyme family lack this feature.
In addition to the extreme off-line orientation of the dioxygen binding site for L-Arg hydroxylation and the unusual twisted peptide bond at the active site, the highly hydrophobic environment of 2OG could stimulate the decarboxylative fragmentation of this substrate into ethylene. Zhang et al. propose that an EFE intermediate with succinate bound to a ferryl group decomposes to produce ethylene by a Grob-type fragmentation.36 The authors supported their hypothesis by using quantum calculations of carboxylate model compounds with good leaving groups at the C3 position.25 Such fragmentation of succinate would release the formate dianion, a very poor leaving group; however, simultaneous electron transfer from the formate dianion to the ferryl group would form carbon dioxide and Fe(II)-OH. In contrast, we had proposed earlier that a cyclic peroxide-Fe(IV) intermediate formed during the EFE reaction would decarboxylate and fragment to release ethylene while generating an oxalate-bound ferryl species.20 The latter species could decompose further by several mechanisms including (A) decarboxylation with formation of a formyl radical anion bound to Fe(III)-OH and subsequent release of a second CO2, (B) decarboxylation with formation of bicarbonate bound to Fe(II), or (C) formation of bicarbonate while generating a formyl radical anion bound to Fe(III) with subsequent release of CO2 (Scheme 2). Notably, these reactions are reminiscent of steps proposed in the decomposition of oxalate by oxalate decarboxylase and oxalate oxidase.37–38 Because of the multiple pathways available for oxalate metabolism and the precedent for this type of chemistry, we continue to favor an ethylene-forming mechanism that includes formation of transiently-bound oxalate at the metallocenter.
Scheme 2.

Potential mechanisms for decomposing a putative oxalate intermediate of EFE
Structural Context for Previously Described EFE variants
Our site-directed mutagenesis efforts confirmed the importance of many residues that bind substrates and affect catalysis, with our product measurements including not just ethylene, but also P5C. An alignment of EFE sequences from eight strains known to produce ethylene (Figure S3)17, 27 reveals extensive conservation of these residues, with identity noted in many cases. Zhang et al. also reported the loss or diminishment of ethylene formation in several variants affecting substrate-binding residues.25 Two mutational studies of P. syringae pv. phaseolicola PK2 efe were reported prior to elucidation of the structure. Nagahama et al. substituted glutamine for each of the ten histidine residues of EFE and studied the ethylene-producing activities of recombinant E. coli cells producing these proteins.26 In addition, Johansson et al. created recombinant yeast cells producing an assortment of EFE variants and studied their ethylene production activities.27 We depict the locations of these prior substitutions in the EFE structure in Figure S7 and summarize their ethylene-forming activities in Table S6. Substitution of the universally-conserved metal-binding ligands (H189, D191, and H268) by alanine eliminates activity,27 as does the substitution of H189 by glutamine.26 In contrast, the H168Q variant retains a trace of activity, as has been described for similar substitutions in other family members.39 Curiously, among the residues distant from the active site, the H233A variant was fully active27 while the H233Q protein was inactive,26 and substantial activity losses were reported for the H116Q, H168Q, H169Q, H284Q, and H309Q variants. The reasons for these great diminishments in activity are unclear.
CONCLUSIONS
Our structural and mutational studies of EFE provide keen insights into the catalytic mechanisms of this enzyme. Based on our earlier biochemical and spectroscopic studies and new insights from the present structural and mutagenesis investigations, we propose an updated version of the dual-pathway EFE reaction mechanism (Scheme 3). In this revised model, the EFE-catalyzed metabolism of L-Arg (blue pathway) appears to take place via the prototypical hydroxylation mechanism; i.e., a ferryl intermediate is generated by the oxidative decarboxylation of 2OG, the ferryl flips its orientation in order to abstract a hydrogen atom from the nearby C5 position, and hydroxyl radical rebound leads to an unstable hydroxylated intermediate that decomposes to guanidine and P5C. Three novel aspects of this mechanism are (1) the predominant monodentate coordination of 2OG to the metal in the absence of L-Arg, (2) a shift in the orientation of the D191 metal-binding ligand upon binding of L-Arg accompanied by formation of a twisted peptide bond at the active site, and (3) the drastically off-line direction of the ferryl group with F283 and D191 hindering the flip needed for reaction with the L-Arg substrate. These features along with the hydrophobic environment of 2OG may be important for catalysis of the atypical ethylene-forming reaction carried out by this enzyme (red pathway).
Scheme 3. Two-pathway reaction mechanism of EFE with insights provided by the enzyme structuresa.

aEFE•Fe(II) binds 2OG predominantly in two monodentate conformations, with a small amount of chelate coordination to the metal. The nearby binding of L-Arg leads to a change in orientation of D191 (shown in cyan) as it forms a hydrogen bond with this substrate and the 2OG switches to bidentate coordination. Dioxygen binds to the metallocenter opposite of L-Arg to form the Fe(III)-superoxo species that transforms to a cyclic peroxide-Fe(IV) intermediate. The mechanism diverges at this intermediate, with the predominant pathway invoking a decarboxylative fragmentation to release ethylene with oxalic acid transiently bound to the ferryl species. Subsequent reactivity of this intermediate yields CO2 and bicarbonate or two molecules of CO2 (Scheme 2) and the recycled EFE•Fe(II). The second pathway depicts oxidative decarboxylation of 2OG to yield succinate and a ferryl species pointing away from the L-Arg substrate. A ferryl flip repositions the ferryl group to point toward C5 of L-Arg, with F283 (and the repositioned D191) partly hindering this process. Hydrogen atom abstraction, hydroxyl radical rebound, elimination of guanidine from 5-hydroxy L-Arg, and ring formation complete this cycle.
We previously showed that the two reactions can be separated by the use of substrate analogues.20 For example, 2OA allows for hydroxylation of L-Arg, but it cannot generate ethylene. Similarly, HO-L-Arg can stimulate 2OG-dependent formation of ethylene, but it does not undergo hydroxylation. Many variants involving residues that interact with 2OG or L-Arg lead to substantial reduction or loss of activity, but thus far no substitution has led to the exclusive production of ethylene. Of interest, however, several variants (most notably A198V) were highly capable of L-Arg hydroxylation yet had severely repressed ethylene-forming ability. The findings presented here provide new insights into the properties of this unique enzyme and offer the potential for creating a version of EFE that exhibits high-level synthesis of ethylene in the absence of the wasteful L-Arg hydroxylation reaction.
Supplementary Material
Acknowledgments
This work was initiated with support by the National Institutes of Health (Grant GM063584 to R.P.H.). We thank Nicholas Henning for assistance with some assays and creation of some variants, and Tuo Zhang for help with collection of crystallographic data. We thank the beamline scientists at LS-CAT at APS for assistance during data collections.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: xxx.
Experimental procedures, eight supplementary figures, and eight supplementary tables (PDF) along with X-ray crystallographic data for eleven EFE structures (CIF). The reported coordinates and structure factors have been deposited in the PDB as apoprotein (PDB ID 5V2U); EFE•Ni, PDB ID 5V2V); Mn•2OG (PDB ID 5V2X); Mn•2OG•L-Arg (PDB ID 5V2Y); Mn•2OA•L-Arg (PDB ID 5V2Z); Mn•2OG•HO-L-Arg (PDB ID 5VKA); Mn•2OG•L-Arg-amide (PDB ID 5VKB); Mn•malate•Arg (PDB ID 5V34); Mn•Arg (PDB ID 5V31); Mn•tartrate (PDB ID 5V2T) and Mn•malate (PDB ID 5V32)
References
- 1.Chenier PJ. Survey of Industrial Chemistry. Springer US; Boston, MA: 2002. pp. 143–162. [Google Scholar]
- 2.Global Ethylene Market By End Use, By Region, Competition Forecast and Opportunities, 2011 – 2025. PR Newswire. 2017 [Google Scholar]
- 3.Chenier PJ. Survey of Industrial Chemistry. Springer US; Boston, MA: 2002. pp. 117–141. [Google Scholar]
- 4.Ghanta M, Fahey D, Subramaniam B. Appl Petrochem Res. 2014;4:167–179. [Google Scholar]
- 5.Lundgren A, Hjertberg T. Surfactants from Renewable Resources. John Wiley & Sons, Ltd; Chichester, U.K: 2010. pp. 109–126. [Google Scholar]
- 6.Dijkmans T, Pyl SP, Reyniers MF, Abhari R, Van Geem KM, Marin GB. Green Chem. 2013;15:3064–3076. [Google Scholar]
- 7.Johansson N, Persson KO, Quehl P, Norbeck J, Larsson C. FEMS Yeast Res. 2014;14:1110–1118. doi: 10.1111/1567-1364.12208. [DOI] [PubMed] [Google Scholar]
- 8.Johansson N, Quehl P, Norbeck J, Larsson C. Microb Cell Fact. 2013;12:1–7. doi: 10.1186/1475-2859-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan D, Lenz P, Hwa T. Appl Environ Microbiol. 2011;77:6763–6771. doi: 10.1128/AEM.05257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch S, Eckert C, Yu J, Gill R, Maness PC. Biotechnol Biofuels. 2016;9:1–10. doi: 10.1186/s13068-015-0413-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu T, Xie X, Li Z, Tan X, Lu X. Green Chem. 2015;17:421–434. [Google Scholar]
- 12.Markham JN, Tao L, Davis R, Voulis N, Angenent LT, Ungerer J, Yu J. Green Chem. 2016;18:6266–6281. [Google Scholar]
- 13.Guerrero F, Carbonell V, Cossu M, Correddu D, Jones PR. PLoS ONE. 2012;7:e50470. doi: 10.1371/journal.pone.0050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong W, Morgan JA, Ungerer J, Wang B, Maness PC, Yu J. Nature Plants. 2015;1:15053. [Google Scholar]
- 15.Zavřel T, Knoop H, Steuer R, Jones PR, Červený J, Trtílek M. Bioresource Technol. 2016;202:142–151. doi: 10.1016/j.biortech.2015.11.062. [DOI] [PubMed] [Google Scholar]
- 16.Veetil VP, Angermayr SA, Hellingwerf KJ. Microb Cell Fact. 2017;16:34. doi: 10.1186/s12934-017-0645-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weingart H, Völksch B, Ullrich MS. Phytopath. 1999;89:360–365. doi: 10.1094/PHYTO.1999.89.5.360. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda H, Kitajima H, Fujii T, Tazaki M, Ogawa T. FEMS Microbiol Lett. 1989;59:1–5. doi: 10.1111/j.1574-6968.1989.tb03428.x. [DOI] [PubMed] [Google Scholar]
- 19.Fukuda H, Ogawa T, Tazaki M, Nagahama K, Fujiil T, Tanase S, Morino Y. Biochem Biophys Res Commun. 1992;188:483–489. doi: 10.1016/0006-291x(92)91081-z. [DOI] [PubMed] [Google Scholar]
- 20.Martinez S, Hausinger RP. Biochemistry. 2016;55:5989–5999. doi: 10.1021/acs.biochem.6b00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez S, Hausinger RP. J Biol Chem. 2015;290:20702–20711. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hausinger RP. In: 2-Oxoglutarate-Dependent Oxygenases. Schofield CJ, Hausinger RP, editors. The Royal Society of Chemistry; Cambridge, UK: 2015. pp. 1–58. [Google Scholar]
- 23.Kal S, Que L. J Biol Inorg Chem. 2017;22:339–365. doi: 10.1007/s00775-016-1431-2. [DOI] [PubMed] [Google Scholar]
- 24.Wu LF, Meng S, Tang GL. Biochim Biophys Acta. 2016;1864:453–470. doi: 10.1016/j.bbapap.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Smart TJ, Choi H, Hardy F, Lohans CT, Abboud MI, Richardson MSW, Paton RS, McDonough MA, Schofield CJ. Proc Natl Acad Sci USA. 2017;114:4667–4672. doi: 10.1073/pnas.1617760114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagahama K, Yoshino K, Matsuoka M, Tanase S, Ogawa T, Fukuda H. J Ferment Bioeng. 1998;85:255–258. [Google Scholar]
- 27.Johansson N, Persson KO, Larsson C, Norbeck J. BMC Biochem. 2014;15:1–8. doi: 10.1186/1471-2091-15-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aik WS, Chowdhury R, Clifton IJ, Hopkinson RJ, Leissing T, McDonough MA, Nowak R, Schofield CJ, Walport LJ. In: 2-Oxoglutarate-Dependent Oxygenases. Schofield CJ, Hausinger RP, editors. The Royal Society of Chemistry; Cambridge, U.K: 2015. pp. 59–94. [Google Scholar]
- 29.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. J Inorg Biochem. 2006;100:644–669. doi: 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 30.Hausinger RP. Crit Rev Biochem Molec Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Ren J-s, Harlos K, McKinnon CH, Clifton IJ, Schofield CJ. FEBS Lett. 2002;517:7–12. doi: 10.1016/s0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall B, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Crystallogr. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhury R, McDonough MA, Mecinovíc J, Loenarz C, Flashman E, Hewitson KS, Domene C, Schofield CJ. Structure. 2009;17:981–989. doi: 10.1016/j.str.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 35.Berkholz DS, Driggers CM, Shapovatov MV, Dunbrack RL, Jr, Karplus PA. Proc Natl Acad Sci USA. 2012;109:449–453. doi: 10.1073/pnas.1107115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grob CA, Baumann W. Helv Chim Acta. 1955;38:594–610. [Google Scholar]
- 37.Molt RW, Jr, Lecher AM, Clark T, Bartlett RJ, Richards NGJ. J Am Chem Soc. 2015;137:3248–3252. doi: 10.1021/ja510666r. [DOI] [PubMed] [Google Scholar]
- 38.Zhu W, Easthon LM, Reinhardt LA, Tu C, Cohen SE, Silverman DN, Allen KN, Richards NG. Biochemistry. 2016;55:163–173. doi: 10.1021/acs.biochem.6b00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grzyska PK, Müller TA, Campbell MG, Hausinger RP. J Inorg Biochem. 2007;101:797–808. doi: 10.1016/j.jinorgbio.2007.01.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.