

Abstract
Dasabuvir, a nonnucleoside NS5B polymerase inhibitor, is a sensitive substrate of cytochrome P450 (CYP) 2C8 with a potential for drug–drug interaction (DDI) with clopidogrel. A physiologically based pharmacokinetic (PBPK) model was developed for dasabuvir to evaluate the DDI potential with clopidogrel, the acyl‐β‐D glucuronide metabolite of which has been reported as a strong mechanism‐based inhibitor of CYP2C8 based on an interaction with repaglinide. In addition, the PBPK model for clopidogrel and its metabolite were updated with additional in vitro data. Sensitivity analyses using these PBPK models suggested that CYP2C8 inhibition by clopidogrel acyl‐β‐D glucuronide may not be as potent as previously suggested. The dasabuvir and updated clopidogrel PBPK models predict a moderate increase of 1.5–1.9‐fold for Cmax and 1.9–2.8‐fold for AUC of dasabuvir when coadministered with clopidogrel. While the PBPK results suggest there is a potential for DDI between dasabuvir and clopidogrel, the magnitude is not expected to be clinically relevant.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Dasabuvir is a sensitive CYP2C8 substrate. Clopidogrel acyl‐β‐D‐glucuronide was hypothesized to be a strong CYP2C8 mechanism‐based inhibitor and responsible for clinical interaction with repaglinide, thus posing a potential risk of DDI with dasabuvir.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addressed the hypothesized interaction between clopidogrel and dasabuvir via CYP2C8 mechanism‐based inhibition by clopidogrel acyl‐β‐D‐glucuronide.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Based on the PBPK modeling approach supported by additional in vitro hepatic uptake data, the potential DDI between clopidogrel and dasabuvir was evaluated. This study provides PBPK model‐based predictions suggesting moderate potential of DDI between dasabuvir and clopidogrel.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The novel dasabuvir PBPK model along with quantitative understanding of the interaction mechanisms perpetrated by clopidogrel and its acyl‐β‐D‐glucuronide enables better management of clinical DDI with dasabuvir. Clopidogrel and its glucuronide metabolite may be less potent inhibitors of CYP2C8 than previously thought.
Dasabuvir is a nonnucleoside NS5B polymerase inhibitor coadministered with ombitasvir and paritaprevir (given with a low dose of ritonavir) as the three direct‐acting antiviral (3D) regimen for the treatment of chronic HCV genotype 1 infection.1, 2 Dasabuvir is primarily metabolized by cytochrome P450 (CYP) 2C8, and to a lesser extent by CYP3A4.3, 4 Within the 3D regimen, induction and inhibition of CYP3A4 caused by coadministration with low‐dose ritonavir results in a net inhibition of CYP3A4, diminishing this enzyme's contribution to the elimination of dasabuvir. Coadministration of the 3D regimen with gemfibrozil, a strong CYP2C8 mechanism‐based inhibitor (MBI) via its 1‐O‐β glucuronide,5 resulted in 101% and 1,030% increases in dasabuvir Cmax and AUC0‐72, respectively, with a substantial increase in dasabuvir elimination half‐life (t1/2) from ∼5–90 hours, consistent with mechanism‐based or time‐dependent inhibition of hepatic CYP2C8.6 In contrast, coadministration of the 3D regimen with trimethoprim, a weak CYP2C8 competitive inhibitor, increased dasabuvir exposure to a smaller extent (Cmax 15% and AUC 33%) with only a 43% increase in dasabuvir t1/2.7 Based on these results, dasabuvir is considered a sensitive CYP2C8 substrate and gemfibrozil and other strong CYP2C8 inhibitors are contraindicated with dasabuvir.4
Clopidogrel is an extensively metabolized prodrug5 whose clinical drug–drug interaction (DDI) profile as a perpetrator has not been fully characterized. In recent studies, when clopidogrel was coadministered with repaglinide, a significant (3–5‐fold) increase in repaglinide AUC was reported.8, 10 Repaglinide is a known substrate of CYP2C8, CYP3A4, and organic anion transporting polypeptide (OATP) 1B1, and the contribution that each pathway plays in repaglinide disposition has been characterized in in vitro 11, 12, 13 and clinical studies.14, 15 Additional enzymes and transporters that are less well characterized may also be involved in the disposition and DDIs of repaglinide.16 Based on physiologically based pharmacokinetic (PBPK) modeling, the interaction between clopidogrel and repaglinide has been attributed to a strong CYP2C8 MBI by the major circulating metabolite, clopidogrel acyl‐β‐D glucuronide.8 These findings were cited by Stark to raise the possibility for a potentially significant DDI between clopidogrel and dasabuvir via MBI of CYP2C8.18 A PBPK model developed by Tornio et al.8 to fit the clinical DDI data of clopidogrel and repaglinide assumed that clopidogrel acyl‐β‐D glucuronide is actively taken up into hepatocytes, reflecting hepatic unbound concentrations that are 19‐fold higher than plasma. More recent clinical studies with clopidogrel and the sensitive CYP2C8 substrate, pioglitazone, showed only a modest 2‐fold increase in pioglitazone AUC.10, 19 These new observations suggest that clopidogrel acyl‐β‐D glucuronide may not be a strong CYP2C8 inhibitor.
In the present study, a PBPK model for dasabuvir was developed, and a new clopidogrel PBPK model was developed to incorporate additional in vitro data and sensitivity analysis on hepatic uptake and OATP1B1 inhibition of the acyl‐β‐D glucuronide. The new model, verified using clinical DDI observations with other CYP2C8 and OATP1B1 substrates, was used to predict the effect of clopidogrel on dasabuvir pharmacokinetics.
RESULTS
Dasabuvir PBPK model verification
A PBPK model for dasabuvir was developed using a combination of in vitro and clinical data.3, 6, 7 Input parameters are summarized in Supplementary Table 1 and a schematic of the model development approach is shown in Supplementary Figure 1.
The dasabuvir PBPK model was utilized to simulate a single intravenous dose and a single oral dose of dasabuvir. The simulation results were in good agreement with the observed PK profiles of dasabuvir in humans (Figure 1), verifying the baseline PK model of dasabuvir. Based on in vitro 3 and in vivo 6 evidence, clearance of dasabuvir was assumed to be mediated by CYP2C8 (67% of total clearance) and CYP3A4 (33% of total clearance) metabolisms. To confirm the fractional metabolism by CYP3A4 (fmCYP3A4) for dasabuvir elimination, a ketoconazole coadministration trial with dasabuvir (without ritonavir) was simulated.
Figure 1.
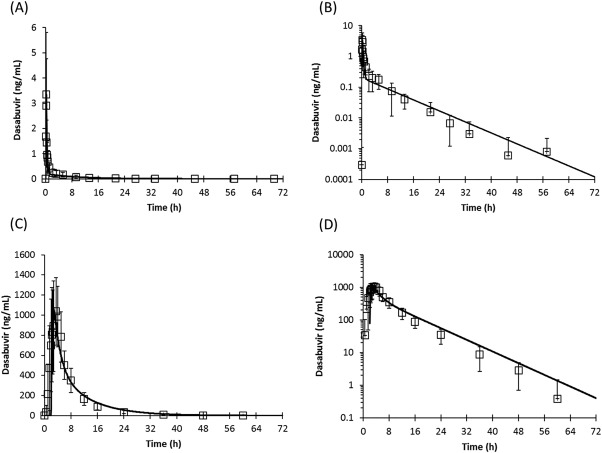
PBPK simulations of the pharmacokinetic profiles of dasabuvir following a single intravenous dose (a,b) or oral dose (c,d) in healthy volunteers. Lines represent simulation results and symbols represent observed data (mean and standard deviation). Pharmacokinetic profiles are shown in linear (a,c) or log‐linear scales (b,d).
The observed interaction with ketoconazole was captured by the dasabuvir PBPK model, and the predicted Cmax and AUC ratios were within 1.1‐fold of the observed data (Table 1), verifying the in vitro observations of an ∼30% contribution of CYP3A4 to dasabuvir metabolism.3
Table 1.
Dasabuvir PBPK model verification and predictions, with predicted vs. observed DDI for Cmax and AUC ratios
| Coadministered drug | Dasabuvir | |||||
|---|---|---|---|---|---|---|
| Cmax ratio | AUC ratio | |||||
| Observed6 | Predictedc | Rpred/obs | Observed6 | Predictedc | Rpred/obs | |
| Ketoconazolea | 1.5 | 1.4 (1.2–2.2) | 0.9 | 1.6 | 1.7 (1.2–3.0) | 1.1 |
| Ritonavir SSb | 0.4 | 0.7 (0.3 –1.4) | 1.6 | 0.5 | 0.5 (0.2 –2.0) | 1.0 |
| Gemfibrozilb | 2.0 | 2.6 (1.4–7.6) | 1.3 | 11 | 10 (2.7–27) | 0.9 |
| Trimethoprimb | 1.2 | 1.6 (1.2–2.2) | 1.3 | 1.3 | 2.0 (1.0–4.5) | 1.5 |
Study designs in PBPK model simulations were matched with each clinical study design. For simulations of the gemfibrozil trial with dasabuvir, the previously validated gemfibrozil PBPK model in Simcyp v14.1 was used, where gemfibrozil 1‐O‐β‐glucuronide metabolite was considered as a CYP2C8 mechanism‐based inhibitor.12 SS, steady state; Rpred/obs, Predicted Cmax or AUC ratio/Observed Cmax or AUC ratio.
Ritonavir not present in this trial simulation to match clinical study design. Dasabuvir 250 mg b.i.d. with ketoconazole 400 mg single dose on day 11.
Ritonavir dosage was 100 mg q.d. with dasabuvir 400 mg b.i.d. to match clinical study design.
Gemfibrozil dosage was 600 mg b.i.d. with dasabuvir 400 mg single dose on day 3 to match clinical study design. Gemfibrozil 1‐O‐glucuronide CYP2C8 KI was optimized from initial value in Simcyp (19 μM) to 4 μM, a 2‐fold lower value than the reported KI of 7.9 μM.13
Trimethoprim dosage was 160 mg b.i.d. with dasabuvir 250 mg single dose on day 3 to match clinical study design.
Predictions are shown as geometric mean and the range (minimum and maximum) is in parentheses.
All simulation results are based on 10 virtual trials of 10 subjects each to account for population variability.
Ritonavir PBPK model accounting for CYP3A4 inhibition and CYP2C8 induction
Ritonavir is coadministered with dasabuvir within the 3D regimen; therefore, a PBPK model was developed to account for ritonavir strong inhibition and weak induction of CYP3A4 and CYP2C8, respectively (Supplementary Table 2).20, 21 The acute and steady‐state effects of ritonavir (100 mg) on CYP3A4 inhibition and induction were verified using clinical data with the probe substrates triazolam and midazolam.20, 22 Simulations resulted in Cmax and AUC ratios within 1.3‐fold of the observed values for triazolam and midazolam (Supplementary Table 3). Ritonavir also induces hepatic CYP2C8 in vitro,23 and at the 100 mg dose results in an in vivo net inhibition of CYP3A49, 17 and induction of CYP2C8, as demonstrated clinically with rosiglitazone.21 Thus, at steady state, weak induction of CYP2C8 by ritonavir could explain the observed decrease in dasabuvir exposure.24 PBPK simulation of ritonavir coadministration with dasabuvir at steady state resulted in Cmax and AUC ratios that were within 1.6‐fold and 1.0‐fold of the clinically observed Cmax and AUC ratios, respectively (Table 1), verifying CYP2C8 induction by ritonavir and its moderate reduction of dasabuvir exposure.
PBPK verification of dasabuvir fmCYP2C8 and simulations with inhibitors
Characterization of the effects of strong and weak CYP2C8 inhibitors on dasabuvir elimination was achieved by simulating trials of dasabuvir (with ritonavir) coadministration with gemfibrozil or trimethoprim, representing strong and weak CYP2C8 inhibitors, respectively.
During initial gemfibrozil simulations with dasabuvir in the presence of ritonavir, the initial value of the CYP2C8 inactivation constant (KI) for gemfibrozil 1‐O‐glucuronide was identified as a sensitive parameter for predicting the observed DDI with dasabuvir. Using the initial value of 19 μM, the predicted AUC0‐72 ratio of dasabuvir was 7.5‐fold compared to the observed 11‐fold. Optimization of the KI value to 4 μM resulted in improved prediction of dasabuvir DDI with gemfibrozil, where the Cmax and AUC0‐72 ratios were within 1.3‐fold and 0.9‐fold, respectively, of the observed clinical results (Table 1). Another sensitive parameter was the first‐order turnover rate constant (kdeg) of CYP2C8, a system parameter in Simcyp. Initial simulations using the CYP2C8 kdeg (0.03 hr‐1) in Simcyp resulted in an underprediction of dasabuvir Cmax and AUC ratios; however, within 2‐fold of the observed results. Optimization of the kdeg from 0.03 to 0.01 hr‐1 improved precision of the model, where the predicted dasabuvir Cmax and AUC0‐72 ratios were within 1.3‐fold of the observed data (data not shown). The predicted concentration–time profile of dasabuvir in the presence of ritonavir and gemfibrozil was also in good agreement with the observed data (Supplementary Figure 2), although part of the terminal phase was underpredicted by the model.
Simulations of the weak competitive CYP2C8 inhibitor trimethoprim with dasabuvir plus ritonavir reasonably predicted the observed interaction within 1.3‐fold and 1.5‐fold of the observed Cmax and AUC ratios, respectively (Table 1).
On the basis of these PBPK verifications steps of dasabuvir DDIs with ketoconazole, ritonavir, gemfibrozil, and trimethoprim, the final dasabuvir model assumed that fractional metabolism by CYP3A4 was 30%, fractional metabolism by CYP2C8 was 70% (in the absence of ritonavir), and that coadministered ritonavir strongly inhibited CYP3A4 and moderately induced CYP2C8‐mediated clearance of dasabuvir. These assumptions and parameters were used for all further dasabuvir simulations and predictions (Supplementary Table 4).
Clopidogrel PBPK model verification
The clopidogrel PBPK model was developed in Simcyp based on published data8 and newly generated in vitro data for hepatocyte uptake, CYP2C8 inactivation, and OATP1B1 inhibition.
To develop a full PBPK model of clopidogrel and its conversion to the acyl‐β‐D glucuronide, and because in vitro uridine diphosphate glucuronosyltransferase (UGT) 2B7 kinetic data for the formation of the acyl‐β‐D glucuronide had not been reported for clopidogrel, a “top‐down” clinical data‐driven approach similar to that used by Tornio et al., with some modifications, was used to estimate the clearance and to model clopidogrel conversion to the acyl‐β‐D glucuronide (Supplementary Tables 5, 6).
Clopidogrel model simulations using this approach resulted in good agreement between the simulated and observed PK profiles of clopidogrel and its acyl‐β‐D glucuronide (Figure 2).
Figure 2.
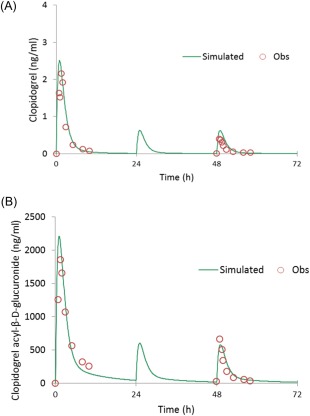
PBPK simulations of the pharmacokinetic profiles of clopidogrel (a) and its acyl‐β‐D glucuronide metabolite (b) following a single 300 mg loading dose on day 1 followed by 75 mg q.d. maintenance doses. Simulated trial design in Simcyp: custom dosing of 300 mg loading dose on day 1 at 8:00 am followed by 75 mg q.d. on days 2 and 3. Observed data were digitized from the Tornio et al. publication.8 Population representative was used for these simulations in Simcyp v. 14.1. [Color figure can be viewed at wileyonlinelibrary.com]
To verify the clopidogrel model, simulations with the sensitive CYP2C8 substrate pioglitazone using a published model in Simcyp,28 and simulations with the dual CYP2C8/OATP1B1 substrate repaglinide from the Simcyp substrate library, were performed and compared to clinical data.8, 10 In addition, our in vitro studies confirmed the CYP2C8 inactivation kinetics parameters, maximum inactivation rate (kinact), and inhibitor concentration needed to cause half of kinact (KI) of 1.14 hr‐1 and 15.0 μM, respectively, for the acyl‐β‐D glucuronide. These values were within 2‐fold of the previously reported values in the literature.8
Simulation of clopidogrel with pioglitazone using the Tornio et al. model assumptions resulted in overprediction (AUC ratio 3‐fold) of the observed 2‐fold AUC ratio of pioglitazone (Supplementary Table 7).
A key assumption in Tornio's model was a 19‐fold hepatic uptake factor for the acyl‐β‐D glucuronide, which was used to fit the observed DDI with repaglinide. Therefore, a sensitivity analysis was performed on this parameter to improve the model prediction of the pioglitazone interaction data. Results of the sensitivity analysis suggested that pioglitazone Cmax and AUC ratios could be accurately predicted by decreasing the hepatic uptake factor of clopidogrel acyl‐β‐D glucuronide from 19‐fold to 5‐fold (Figure 3).
Figure 3.
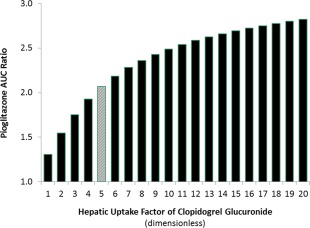
Parameter sensitivity analysis of clopidogrel acyl‐β‐D‐glucuronide hepatic uptake factor and its impact on pioglitazone AUC ratio. Gray bar corresponds to the observed AUC ratio of pioglitazone when coadministered with clopidogrel (Kim et al.).10 Simcyp virtual trial design: clopidogrel 300 mg was administered on day 1 at 8:00 am followed by 75 mg q.d. on days 2 and 3 at the same time. Pioglitazone 15 mg was administered on day 1 at 9:00 am. A population representative was used for the sensitivity analysis in Simcyp v14.1. [Color figure can be viewed at wileyonlinelibrary.com]
Based on these findings, simulations using the updated clopidogrel model (hepatic uptake factor of 5‐fold) and the repaglinide model file from the Simcyp substrate library were performed. As expected, the simulations showed an underprediction of the observed Cmax and AUC ratios of repaglinide (predicted AUC ratio 2‐fold vs. 5‐fold observed), when the hepatic uptake factor was decreased to 5‐fold. In addition, when active hepatic uptake of the acyl‐β‐D glucuronide was tested in vitro using human hepatocytes, uptake of clopidogrel acyl‐β‐D‐glucuronide at 37°C was comparable to that at 4°C (ratio of 0.92 and 0.80 at 0.1 μM and 1 μM, respectively), suggesting no active uptake of clopidogrel acyl‐β‐D glucuronide into hepatocytes (Supplementary Table 8).
Our in vitro OATP1B1 inhibition results were also consistent with previous reports; however, we found that clopidogrel acyl‐β‐D glucuronide inhibited OATP1B1 with an IC50 value of 51 μM, which is less potent than the reported values of 10.9 μM and 33.5 μM.25 A parameter sensitivity analysis on OATP1B1 Ki showed that the increase in repaglinide Cmax and AUC upon coadministration with clopidogrel could be accurately predicted if the acyl‐β‐D‐glucuronide OATP1B1 Ki was reduced from 10.9 to 0.1 μM, and the hepatic uptake factor was fixed to 5‐fold (Figure 4). Under these model assumptions, pioglitazone and repaglinide DDIs with clopidogrel could be predicted accurately (Supplementary Table 7).
Figure 4.
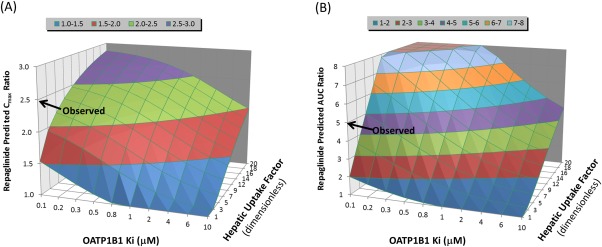
Automated parameter sensitivity analysis demonstrating the impact of hepatic uptake factor and OATP1B1 Ki of clopidogrel acyl‐β‐D‐glucuronide on repaglinide predicted Cmax and AUC ratios. Simulated trial design in Simcyp: clopidogrel was administered as a single 300 mg dose at 8:00 am followed by a single 0.25 mg dose of repaglinide at 9:00 am. Population representative was used for these simulations in Simcyp v. 14.1. Key model parameters of repaglinide: model file from the Simcyp substrate library was used with modification of CYP2C8 fm from 0.64 to 0.89 based on Tornio et al. Key model parameters of clopidogrel acyl‐β‐D glucuronide: hepatic uptake factor ranged from 1–20‐fold. OATP1B1 Ki ranged from 0.1–10 μM.
To verify the optimized OATP1B1 Ki of clopidogrel acyl‐β‐D‐glucuronide, a simulation using the updated clopidogrel model with the selective OATP1B1 substrate pitavastatin was performed using a published model,27 and results were compared with the observed clinical data.10 Simulation results accurately predicted the pitavastatin AUC ratio of 1.1 (Supplementary Table 7). In addition, further verification of the updated clopidogrel acyl‐β‐D‐glucuronide model was performed using another OATP1B1 substrate, rosuvastatin, from the Simcyp substrate library.
When initial simulations using the Tornio et al. model assumptions (19‐fold uptake, OATP1B1 Ki 5.45 μM) were performed with rosuvastatin for comparison, the rosuvastatin AUC ratio was underpredicted (Supplementary Table 7). In contrast, our updated clopidogrel model was able to accurately predict the observed AUC ratio of 1.7‐fold when the optimized OATP1B1 Ki of 0.1 μM and the hepatic uptake factor of 5‐fold were assumed.
On the basis of these PBPK model verification steps of clopidogrel and its glucuronide metabolite DDI with pioglitazone, repaglinide, pitavastatin, and rosuvastatin, the final clopidogrel acyl‐β‐D‐glucuronide model assumed an optimized hepatic uptake factor of 5‐fold, an OATP1B1 Ki of 0.1 μM, and CYP2C8 and CYP3A4 inactivation parameters as previously reported8 without modification for clopidogrel or its acyl‐β‐D‐glucuronide (Supplementary Table 4).
PBPK prediction of clopidogrel impact on dasabuvir using the final model
The final PBPK models of dasabuvir, ritonavir, and clopidogrel with its acyl‐β‐D‐glucuronide were used to predict the magnitude of potential DDI between clopidogrel and dasabuvir. Figure 5 shows the predicted Cmax and AUC ratios of dasabuvir at steady state, following coadministration of a clopidogrel 300 mg loading dose on day 1 and 75 mg q.d. thereafter with dasabuvir 250 mg b.i.d. and ritonavir 100 mg q.d. for 5 days. Utilizing the updated PBPK model assumptions of an active hepatic uptake factor of 5‐fold and MBI of CYP2C8 and CYP3A4 by clopidogrel and its acyl‐β‐D glucuronide, a moderate increase in dasabuvir exposure (Cmax ratio 1.5‐fold and AUC ratio 1.9‐fold) was predicted. Sensitivity analysis to account for a 10‐fold hepatic uptake factor (corresponding to 10‐fold higher hepatic unbound concentrations of clopidogrel acyl‐β‐D glucuronide relative to plasma concentrations), suggested that dasabuvir Cmax and AUC may increase by 1.9‐ and 2.8‐fold, respectively. Even at the worst‐case scenario (glucuronide hepatic concentrations 20‐fold higher than plasma), dasabuvir predicted Cmax and AUC ratios were 2.6‐fold and 4.5‐fold, respectively (Figure 5).
Figure 5.
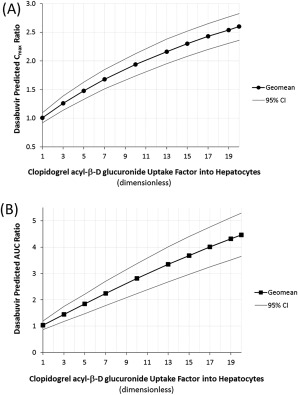
Predicted Cmax and AUC ratios of dasabuvir (with ritonavir) following coadministration with clopidogrel using PBPK modeling. Simulations using the final dasabuvir PBPK model to demonstrate the impact of active uptake factor into hepatocytes on dasabuvir Cmax (a) and AUC (b) ratios. Final model parameters: clopidogrel acyl‐β‐D glucuronide hepatic uptake factor range 1–20‐fold and CYP2C8 inactivation parameters as published in Tornio et al. Trial simulation in Simcyp: dasabuvir dose was 250 mg b.i.d. starting at 9 am for 5 days and a single 250 mg dose on day 6; ritonavir dose was 100 mg q.d. for 6 days at the same time as dasabuvir; clopidogrel dose was 300 mg on day 1 followed by 75 mg q.d. for 5 days starting at 8 am every day. Simulation results were based on 10 virtual trials of 10 subjects each to account for population variability. Results are shown as geometric mean and 95% confidence intervals (CI).
Simulations to mechanistically describe the effects of clopidogrel on dasabuvir in the presence of ritonavir suggested that dasabuvir CYP2C8‐mediated intrinsic clearance (Clint) increased due to induction by ritonavir, while CYP3A4‐mediated Clint was significantly reduced by ritonavir. When clopidogrel was coadministered, dasabuvir CYP2C8‐mediated Clint was predicted to decrease due to MBI by clopidogrel acyl‐β‐D glucuronide; however, the net effect of clopidogrel in the presence of ritonavir was moderate due to the mixed inhibition and induction of dasabuvir hepatic CYP2C8 Clint by clopidogrel and ritonavir, respectively (Figure 6).
Figure 6.
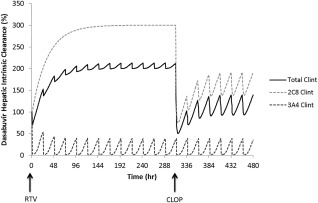
Illustration of the effects of clopidogrel and its acyl‐β‐D glucuronide on dasabuvir CYP2C8 and CYP3A4 mediated intrinsic clearance in the presence of coadministered ritonavir at steady state, using PBPK modeling. Trial simulation in Simcyp: dasabuvir 250 mg b.i.d. was coadministered with ritonavir 100 mg q.d. for 14 days, clopidogrel 300 mg loading dose was coadministered on day 14 followed by 75 mg q.d. maintenance doses for 7 days. RTV: indicates start of ritonavir coadministration; CLOP: indicates start of clopidogrel coadministration.
DISCUSSION
In the presence of ritonavir, dasabuvir is predominantly eliminated via CYP2C8 metabolism and is considered a sensitive CYP2C8 substrate. The high fmCYP2C8 (>0.9) for dasabuvir in the presence of ritonavir has been confirmed clinically; coadministration with gemfibrozil (CYP2C8 and OATP1B1 inhibitor) increased dasabuvir Cmax 2‐fold, AUC0‐72 11‐fold, and the terminal t1/2 from 5 to 90 h.6 In contrast, the weak CYP2C8 inhibitor trimethoprim minimally changed dasabuvir Cmax by 1.15‐fold and AUC by 1.33‐fold, with little impact on the t1/2.7
QTc prolongation by dasabuvir was previously evaluated in healthy subjects, and recently, the risk for QTc prolongation due to DDIs was raised by Stark.18 At concentrations 2‐fold the therapeutic Cmax of dasabuvir, QTc prolongation did not occur to any clinically relevant extent.4 Our PBPK model predicted that the interaction between dasabuvir and a standard clopidogrel dosing regimen would lead to a 1.5–1.9‐fold increase in dasabuvir Cmax, which is within the established QTc safety margin for dasabuvir.
The newly developed PBPK model of dasabuvir as a sensitive substrate of CYP2C8 and a mild substrate of CYP3A4 was verified by comparing the model predictions with clinical data from ketoconazole, ritonavir, gemfibrozil, and trimethoprim DDI studies. All DDIs with dasabuvir were accurately predicted by the model, where the dasabuvir predicted Cmax and AUC ratios were within 1.6‐fold of the clinical data (Table 1). Sensitivity analysis on the CYP2C8 KI of gemfibrozil 1‐O‐glucuronide suggested that a value of 4 μM improved the accuracy of the gemfibrozil DDI prediction. The optimized KI value is within 2‐fold of the reported value of 7.9 μM,13 compared with the initial value in Simcyp of 19 μM. Additional sensitivity analysis suggested that the CYP2C8 hepatic kdeg needed to be lower (0.01 hr‐1) than the initial value in Simcyp to improve the model's accuracy in predicting gemfibrozil DDI with dasabuvir. Although this approach can predict the DDI magnitude between dasabuvir and gemfibrozil with good accuracy, the optimized value was lower than the previously reported kdeg of 0.03 hr‐1 (range of 0.017–0.087 hr‐1)26; therefore, it was not used in the final model assumptions. It should be noted that an extrapolated AUC0‐inf ratio for dasabuvir may be higher than the reported AUC0‐72 ratio of 11‐fold, since the terminal phase of dasabuvir was not fully captured in the gemfibrozil clinical study.6
The updated clopidogrel PBPK model presented here incorporated new in vitro hepatic uptake data, along with sensitivity analyses on hepatic uptake and OATP1B1 inhibition by the acyl‐β‐D glucuronide metabolite. The final clopidogrel model accurately predicted CYP2C8 and OATP1B1‐mediated interactions by simulating DDIs with pioglitazone, repaglinide, pitavastatin, and rosuvastatin (Supplementary Table 7). Similar to dasabuvir, pioglitazone is a sensitive CYP2C8 substrate with minor contribution from CYP3A4; however, clopidogrel increased pioglitazone exposure to a moderate 2‐fold only.10, 19 Pioglitazone simulations using Tornio's clopidogrel model assumptions resulted in overprediction of the clinical data (Supplementary Table 7), and a sensitivity analysis revealed that the hepatic uptake factor of clopidogrel acyl‐β‐D glucuronide could not exceed 5‐fold in order to reproduce the 2‐fold interaction (Figure 2). Although clopidogrel acyl‐β‐D glucuronide concentrations in hepatocytes could be higher than those in plasma, results of the pioglitazone simulation clearly show that the 19‐fold higher liver concentration was overestimated.
Repaglinide is also a sensitive substrate of CYP2C8; however, unlike dasabuvir, it is also a known substrate of OATP1B1 as well as other enzymes and transporters.11, 12, 13, 14, 15, 16, 29, 30 When our updated clopidogrel model was used to simulate the DDI with repaglinide, underprediction of repaglinide's AUC ratio was observed (Supplementary Table 7), suggesting that lowering the hepatic uptake factor allows for accurate prediction of CYP2C8‐mediated pioglitazone DDI but not the CYP2C8/OATP1B1‐mediated repaglinide DDI. This underprediction was resolved by optimizing the OATP1B1 Ki of clopidogrel acyl‐β‐D glucuronide (Figure 4). The combination of optimized hepatic uptake based on pioglitazone DDI data along with OATP1B1 inhibition resulted in accurate prediction of repaglinide DDI with clopidogrel. These results provided an alternative rationale for the clopidogrel interaction with repaglinide, where a relatively more potent inhibition of OATP1B1 by clopidogrel acyl‐β‐D glucuronide combined with MBI of CYP2C8, could also describe accurately the interaction between clopidogrel and repaglinide. Similar to our approach of varying the OATP1B1 Ki value to account for in vitro to in vivo disconnects, the interaction of repaglinide with gemfibrozil and its glucuronide was also rationalized in previous studies using PBPK modeling.12, 13 In those studies, the authors did not increase repaglinide fmCYP2C8 and suggested that OATP1B1 plays a larger role in repaglinide clearance than previously thought. While sensitivity analysis around OATP1B1 inhibition in our model could also capture the DDI with repaglinide in the absence of significant hepatic uptake of the acyl‐β‐D glucuronide (Figure 4), there was a disconnect between the measured in vitro OATP1B1 inhibition potency and the value needed to be assumed in the PBPK model to reproduce the interaction (109‐fold more potent) as part of the sensitivity analysis. In addition, a recent clinical study demonstrated that clopidogrel had a minimal impact on the exposure of the OATP probe substrate, pitavastatin (AUC ratio 1.1), and simvastatin acid (AUC ratio 1.1),10, 31 while in another study, clopidogrel caused a significant increase in rosuvastatin exposure (AUC ratio ∼2.0).32, 33 Even though the in vitro OATP1B1 Ki for clopidogrel acyl‐β‐D glucuronide was altered, the clopidogrel DDI with OATP1B1 substrates was accurately predicted by our model (Supplementary Table 4).
The PBPK modeling analyses supported by the in vitro data described in this report predict that coadministration of clopidogrel with dasabuvir would result in a modest increase in dasabuvir exposure (Figure 5). These predictions are based on verified PBPK models of dasabuvir and coadministered ritonavir, and modification of the clopidogrel PBPK model reported by Tornio et al.8 The new in vitro observation that clopidogrel glucuronide is not actively taken up by human hepatocytes, as was previously assumed, provides justification for reducing the hepatic uptake factor from 19‐fold to a lower value. Although the updated clopidogrel model simultaneously reproduced the clinically observed DDIs, and the final model was used to predict a range for the DDI with dasabuvir, uncertainty regarding the hepatic concentrations of clopidogrel acyl‐β‐D glucuronide and its inhibition potency for OATP1B1 requires further validation. Additional in vitro characterization of the efflux transporters involved in the glucuronide disposition may also help validate the PBPK models. Overall, our additional PBPK analysis of clopidogrel extends the previous findings8 by quantifying the magnitude of CYP2C8 inhibition relative to OATP1B1 inhibition, and suggests that clopidogrel and its metabolite may be considered as moderate CYP2C8 inhibitors.
In conclusion, our PBPK modeling and simulations indicate that the potential DDI between dasabuvir and clopidogrel would be moderate.
METHODS
PBPK modeling
PBPK models were constructed and verified using Simcyp (v. 14.1, Sheffield, UK).34 Model input parameters and their sources are summarized in Supplementary Tables 1, 2, 5, and 6. The default Simcyp virtual Sim‐Healthy Volunteers Population Representative was used for all simulations and sensitivity analyses except for those that included dasabuvir. Dasabuvir simulations were performed using 10 trials with 10 subjects in each trial, age range 20–50 years, and 50% female. The key steps and assumptions of the PBPK modeling are highlighted below, and a diagram that describes the model construction steps and decisions is shown in Supplementary Figure 1.
Dasabuvir model
The model‐building steps used a combination of bottom‐up and top‐down approaches by incorporating physical‐chemical properties, in vitro metabolism data, intravenous pharmacokinetic and absolute oral bioavailability data in humans, and clinical pharmacokinetic data after single and multiple doses of dasabuvir in healthy subjects. Model verification steps used clinical DDI data6 from trials of dasabuvir coadministration with 1) ketoconazole: dasabuvir 400 mg b.i.d. without ritonavir was dosed for 10 days followed by a 400 mg single dose coadministered with ketoconazole 400 mg single dose on day 11, dasabuvir fmCYP2C8 is 0.7 in the absence of ritonavir; 2) ritonavir: dasabuvir 400 mg b.i.d. coadministered with ritonavir 100 mg q.d. to steady state, dasabuvir fmCYP2C8 is ∼1.0 in the presence of ritonavir; 3) gemfibrozil: gemfibrozil 600 mg b.i.d. was dosed for 6 days and dasabuvir (with ritonavir) 400 mg single dose was coadministered on day 3, dasabuvir fmCYP2C8 is ∼1.0 in the presence of ritonavir; and 4) trimethoprim: trimethoprim 160 mg b.i.d. was dosed for 6 days with dasabuvir (with ritonavir) 250 mg single dose coadministered on day 3, dasabuvir fmCYP2C8 is ∼1.0 in the presence of ritonavir. Model applications included simulations of the effects of clopidogrel and its acyl‐β‐D‐glucuronide metabolite on dasabuvir PK parameters following coadministration of both drugs at steady state. Sensitivity analysis simulations were also conducted to predict the DDI magnitude between dasabuvir and clopidogrel under different scenarios (see below under clopidogrel and glucuronide scenarios).
Ritonavir model
The model‐building steps used a combination of bottom‐up and top‐down approaches by incorporating physical‐chemical properties, clinical pharmacokinetic parameters, and DDI results from clinical trials with CYP3A4 probe substrates. Ritonavir PK was assumed to be linear with time; 100 mg q.d. ritonavir dosing results in a net effect of CYP3A4 inhibition (result of reversible and mechanism‐based inhibition and induction)9, 17 and CYP2C8 induction, based on evidence from in vitro and in vivo data.21, 23 Model verification was achieved by simulations of the effects of ritonavir on CYP3A4 using previously validated Simcyp files of the probe substrates midazolam and triazolam.35, 36 Effect of ritonavir on CYP2C8 was verified by simulating a clinical trial with dasabuvir at steady state. Model applications included simulations of DDI trials where ritonavir was coadministered with dasabuvir and a second perpetrator drug.
Clopidogrel and clopidogrel acyl‐β‐D‐glucuronide models
For clopidogrel, all model‐building steps and parameters were the same as those published by Tornio et al.,8 with the following modifications:
Approximately 30% of total clearance was assigned to formation of clopidogrel acyl‐β‐D‐glucuronide metabolite via UGT2B7. About 60% of the remaining clearance was assigned to hepatic and intestinal metabolism via human liver S9 (HLS9 Clint 3000 μL/min/mg‐microsomal protein, HIS9ms Clint 450 μL/min/mg‐microsomal protein).
UGT2B7 CLint estimated (1,000 μL/min/pmol), fu,mic (0.1) to enable formation‐limited metabolite kinetics matching the observed PK profile of the glucuronide metabolite, and to capture dynamic changes of the glucuronide concentrations over time during DDI simulations.
Additional systemic clearance of 15 L/h was estimated to match the reported intravenous clearance of about 110 L/h.37
For clopidogrel acyl‐β‐D‐glucuronide, all model parameters were the same as those published by Tornio et al.,8 with the following modifications:
Direct formation via UGT2B7 metabolism of clopidogrel with fmUGT2B7 of 0.3 as described above.
Distribution using the Single Adjusting Compartment (SAC) model, where clearance into compartment (CLin) was 4.0 L/hr and clearance out from compartment (CLout) was 1.1 L/h.
Active uptake into hepatocytes: factor of 1 to 20 (sensitivity analysis).
The pioglitazone model was taken from Xiao et al.28 and the pitavastatin model was taken from Duan et al.27 with no modifications. Repaglinide and rosuvastatin model files were from the Simcyp v14.1 substrate library. Pioglitazone trial simulations were designed according to a clinical study by Itkonen et al.,19, 31 where clopidogrel 300 mg on day 1 at 8 am followed by 75 mg q.d. for two additional days was coadministered with pioglitazone 15 mg single dose on day 1 at 9 am. Repaglinide trial simulations were designed according to the clinical study by Tornio et al., where clopidogrel 300 mg on day 1 at 8 am followed by 75 mg q.d. for 4 additional days was coadministered with repaglinide 0.25 mg single dose on day 1 at 9 am. Pitavastatin 0.2 mg single dose was coadministered with clopidogrel 300 mg single dose at the same time based on a study by Kim et al.10 Rosuvastatin trial simulations were designed according to the clinical study by Pinheiro et al.,33 where rosuvastatin was given at a daily dose of 40 mg for 1 week and clopidogrel 300 mg was given on day 8, followed by 75 mg daily.
The predictive performance of each model was achieved by comparing the ratio of the predicted exposure change (Cmax ratio and AUC ratio) of each victim drug in the presence of the perpetrator, to the clinically observed Cmax and AUC ratios.
In vitro studies
In vitro hepatic uptake, CYP2C8 time‐dependent inhibition, and OATP1B1 inhibition methods are summarized in the Supplementary Methods.
CONFLICT OF INTEREST/DISCLOSURE
All authors are employees or former employees of AbbVie and may hold AbbVie stock or stock options.
AUTHOR CONTRIBUTIONS
M.S. and D.A.J.B. wrote the article; M.S. designed the research; M.S., W.F., D.A.J.B., and P.B. performed the research; M.S., W.F., D.A.J.B., P.B., and V.F. analyzed the data.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The study was sponsored by AbbVie, Inc. AbbVie contributed to the study design, research, and interpretation of data, and the writing, reviewing, and approving of the publication. The authors thank Dr. Allison Kitten, an employee of AbbVie, for medical writing support. The authors also thank Dr. Rajeev Menon for reviewing the article and for useful discussions.
References
- 1. Feld, J.J. et al Treatment of HCV with ABT‐450/r‐ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 370, 1594–1603 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Ferenci, P. et al ABT‐450/r‐ombitasvir and dasabuvir with or without ribavirin for HCV. N. Engl. J. Med. 370, 1983–1992 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Shebley, M. et al Mechanisms and predictions of drug‐drug interactions of the hepatitis C virus 3‐direct acting antiviral (3D) regimen: paritaprevir/ritonavir, ombitasvir and dasabuvir. Drug Metab. Dispos. (2017). DOI:10.1124/dmd.116.074518 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 4. Viekira Pak (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) [US package insert]. North Chicago, IL; AbbVie, 2016.
- 5. Plavix (clopidogrel) [US package insert]. Bridgewater, NJ; Sanofi Aventis, 2010.
- 6. Menon, R.M. et al Drug‐drug interaction profile of the all‐oral anti‐hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. J. Hepatol. 63, 20–29 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Polepally, A.R. , et al. Drug‐drug interactions between the anti‐hepatitis C virus 3D regimen of ombitasvir, paritaprevir/ritonavir, and dasabuvir and eight commonly used medications in healthy volunteers. Clin. Pharmacokinet. 55, 1003–1014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tornio, A. et al Glucuronidation converts clopidogrel to a strong time‐dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug‐drug interactions. Clin. Pharmacol. Ther. 96, 498–507 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Kim, S.J. et al Clarification of the mechanism of clopidogrel‐mediated drug‐drug interaction in a clinical cassette small‐dose study and its prediction based on in vitro information. Drug Metab. Dispos. DOI: 10.1124/dmd.1116.070276 [Epub ahead of print] (2016). [DOI] [PubMed] [Google Scholar]
- 10. Prandin (repaglinide) [US package insert]. Princeton, NJ; Novo Nordisk, 2011.
- 11. Varma, M.V. , Lai, Y. , Kimoto, E. , Goosen, T.C. , El‐Kattan, A.F. & Kumar, V. Mechanistic modeling to predict the transporter‐ and enzyme‐mediated drug‐drug interactions of repaglinide. Pharm. Res. 30, 1188–1199 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Varma, M.V. , Lin, J. , Bi, Y.A. , Kimoto, E. & Rodrigues, A.D. Quantitative rationalization of gemfibrozil drug interactions: consideration of transporters‐enzyme interplay and the role of circulating metabolite gemfibrozil 1‐O‐beta‐glucuronide. Drug Metab. Dispos. 43, 1108–1118 (2015). [DOI] [PubMed] [Google Scholar]
- 13. Honkalammi, J. , Niemi, M. , Neuvonen, P.J. & Backman, J.T. Dose‐dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab. Dispos. 39, 1977–1986 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Kajosaari, L.I. , Niemi, M. , Neuvonen, M. , Laitila, J. , Neuvonen, P.J. & Backman, J.T. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin. Pharmacol. Ther. 78, 388–399 (2005). [DOI] [PubMed] [Google Scholar]
- 15. Korzekwa, K.R. , Nagar, S. , Tucker, J. , Weiskircher, E.A. , Bhoopathy, S. & Hidalgo, I.J. Models to predict unbound intracellular drug concentrations in the presence of transporters. Drug Metab. Dispos. 40, 865–876 (2012). [DOI] [PubMed] [Google Scholar]
- 16. Stark, J.E. Potential for a significant interaction between clopidogrel and dasabuvir. Clin. Infect. Dis. 61, 134–135 (2015). [DOI] [PubMed] [Google Scholar]
- 17. Itkonen, M.K. , Tornio, A. , Neuvonen, M. , Neuvonen, P.J. , Niemi, M. & Backman, J.T. Clopidogrel markedly increases plasma concentrations of CYP2C8 substrate pioglitazone. Drug Metab. Dispos. 44, 1364–1371 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Kirby, B.J. , Collier, A.C. , Kharasch, E.D. , Whittington, D. , Thummel, K.E. & Unadkat, J.D. Complex drug interactions of HIV protease inhibitors 1: inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab. Dispos. 39, 1070–1078 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sevinsky, H. Effect of atazanavir with and without ritonavir on the pharmacokinetics of the CYP2C8 probe rosiglitazone in healthy subjects. 9th International Workshop on Pharmacology of HIV Therapy, New Orleans, LA, April 7–9, 2008.
- 20. Culm‐Merdek, K.E. et al Effect of extended exposure to grapefruit juice on cytochrome P450 3A activity in humans: comparison with ritonavir. Clin. Pharmacol. Ther. 79, 243–254 (2006). [DOI] [PubMed] [Google Scholar]
- 21. Dixit, V. , Hariparsad, N. , Li, F. , Desai, P. , Thummel, K.E. & Unadkat, J.D. Cytochrome P450 enzymes and transporters induced by anti‐human immunodeficiency virus protease inhibitors in human hepatocytes: implications for predicting clinical drug interactions. Drug Metab. Dispos. 35, 1853–1859 (2007). [DOI] [PubMed] [Google Scholar]
- 22. Mathias, A.A. et al Pharmacokinetics and pharmacodynamics of GS‐9350: a novel pharmacokinetic enhancer without anti‐HIV activity. Clin. Pharmacol. Ther. 87, 322–329 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Morcos, P.N. et al A randomised study of the effect of danoprevir/ritonavir or ritonavir on substrates of cytochrome P450 (CYP) 3A and 2C9 in chronic hepatitis C patients using a drug cocktail. Eur. J. Clin. Pharmacol. 69, 1939–1949 (2013). [DOI] [PubMed] [Google Scholar]
- 24. Menon, R. et al Pharmacokinetics, safety and tolerability following multiple dosing of polymerase inhibitor, ABT‐333 and protease inhibitor, ABT‐450 with ritonavir (Abstract PK_12B) Reviews Antiviral Ther & Infect Dis. 6, 17 (2012). [Google Scholar]
- 25. Xiao et al Physiologically based pharmacokinetics model predicts the lack of inhibition by repaglinide on the metabolism of pioglitazone. Biopharm. Drug Dispos. 36, 603–612 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Tamraz, B. et al OATP1B1‐related drug‐drug and drug‐gene interactions as potential risk factors for cerivastatin‐induced rhabdomyolysis. Pharmacogenet. Genomics. 23, 355–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duan. P , Zhao, P. & Zhang, L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug‐drug interactions (DDIs). Eur. J. Drug Metab. Pharmacokinet. [Epub ahead of print] (2016). [DOI] [PubMed] [Google Scholar]
- 28. Yang, J. et al Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr. Drug Metab. 9, 384–394 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Niemi, M. et al Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin. Pharmacol. Ther. 77, 468–478 (2005). [DOI] [PubMed] [Google Scholar]
- 30. Gan, J. et al Repaglinide‐gemfibrozil drug interaction: inhibition of repaglinide glucuronidation as a potential additional contributing mechanism. Br. J. Clin. Pharmacol. 70, 870–880 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Itkonen, M.K. , Tornio, A. , Neuvonen, M. , Neuvonen, P.J. , Niemi, M. & Backman, J.T. Clopidogrel has no clinically meaningful effect on the pharmacokinetics of the organic anion transporting polypeptide 1B1 and cytochrome P450 3A4 substrate simvastatin. Drug Metab. Dispos. 43, 1655–1660 (2015). [DOI] [PubMed] [Google Scholar]
- 32. Elsby, R. , Martin, P. , Surry, D. , Sharma, P. & Fenner, K. Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug‐drug interaction with rosuvastatin by causing up to a 2‐fold increase in statin exposure. Drug Metab. Dispos. 44, 398–408 (2016). [DOI] [PubMed] [Google Scholar]
- 33. Pinheiro, L.F. et al Pharmacokinetic interactions between clopidogrel and rosuvastatin: effects on vascular protection in subjects with coronary heart disease. Int. J. Cardiol. 158, 125–129 (2012). [DOI] [PubMed] [Google Scholar]
- 34. Jamei, M. , Marciniak, S. , Feng, K. , Barnett, A. , Tucker, G. & Rostami‐Hodjegan, A. The Simcyp population‐based ADME simulator. Expert Opin. Drug Metab. Toxicol. 5, 211–223 (2009). [DOI] [PubMed] [Google Scholar]
- 35. Einolf, H.J. et al Evaluation of various static and dynamic modeling methods to predict clinical CYP3A induction using in vitro CYP3A4 mRNA induction data. Clin. Pharmacol. Ther. 95, 179–188 (2014). [DOI] [PubMed] [Google Scholar]
- 36. Guest, E.J. , Rowland‐Yeo, K. , Rostami‐Hodjegan, A. , Tucker, G.T. , Houston, J.B. & Galetin, A. Assessment of algorithms for predicting drug‐drug interactions via inhibition mechanisms: comparison of dynamic and static models. Br. J. Clin. Pharmacol. 71, 72–87 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cushing, D.J. et al Pharmacokinetics and platelet aggregation inhibitory effects of a novel intravenous formulation of clopidogrel in humans. Clin. Exp. Pharmacol. Physiol. 39, 3–8 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information