

Abstract
NDI‐010976, an allosteric inhibitor of acetyl‐coenzyme A carboxylases (ACC) ACC1 and ACC2, reduces hepatic de novo lipogenesis (DNL) and favorably affects steatosis, inflammation, and fibrosis in animal models of fatty liver disease. This study was a randomized, double‐blind, placebo‐controlled, crossover trial evaluating the pharmacodynamic effects of a single oral dose of NDI‐010976 on hepatic DNL in overweight and/or obese but otherwise healthy adult male subjects. Subjects were randomized to receive either NDI‐010976 (20, 50, or 200 mg) or matching placebo in period 1, followed by the alternate treatment in period 2; and hepatic lipogenesis was stimulated with oral fructose administration. Fractional DNL was quantified by infusing a stable isotope tracer, [1‐13C]acetate, and monitoring 13C incorporation into palmitate of circulating very low‐density lipoprotein triglyceride. Single‐dose administration of NDI‐010976 was well tolerated at doses up to and including 200 mg. Fructose administration over a 10‐hour period stimulated hepatic fractional DNL an average of 30.9 ± 6.7% (mean ± standard deviation) above fasting DNL values in placebo‐treated subjects. Subjects administered single doses of NDI‐010976 at 20, 50, or 200 mg had significant inhibition of DNL compared to placebo (mean inhibition relative to placebo was 70%, 85%, and 104%, respectively). An inverse relationship between fractional DNL and NDI‐010976 exposure was observed with >90% inhibition of fractional DNL associated with plasma concentrations of NDI‐010976 >4 ng/mL. Conclusion: ACC inhibition with a single dose of NDI‐010976 is well tolerated and results in a profound dose‐dependent inhibition of hepatic DNL in overweight adult male subjects. Therefore, NDI‐010976 could contribute considerable value to the treatment algorithm of metabolic disorders characterized by dysregulated fatty acid metabolism, including nonalcoholic steatohepatitis. (Hepatology 2017;66:324–334).
Abbreviations
- ACC
acetyl‐CoA carboxylase
- AE
adverse event
- AUC
area under the concentration‐time curve
- AUEC
area under the effect curve
- BMI
body mass index
- Cmax
maximum mean plasma concentration
- CoA
coenzyme A
- DNL
de novo lipogenesis
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OATP
organic anion‐transporting polypeptide
- PD
pharmacodynamic
- PK
pharmacokinetic
- TEAE
treatment‐emergent AE
- TG
triacylglycerol
- VLDL
very‐low‐density lipoprotein
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent hepatic pathology in the Western world, and nonalcoholic steatohepatitis (NASH), the most severe form of NAFLD, is estimated to occur in 10%‐30% of patients with NAFLD.1 Dysregulated fatty acid metabolism, including increased fatty acid synthesis and impaired fatty acid oxidation, has been implicated in the etiology of NAFLD and NASH.2, 3 De novo lipogenesis (DNL), the synthesis of fatty acids such as palmitate from carbohydrates or amino acids, occurs primarily in the liver. The ensuing increase in hepatic steatosis results in formation of complex lipid signaling molecules, leading to lipotoxicity, inflammation, and fibrosis.3 Therefore, inhibition of fatty acid synthesis coupled with stimulation of fatty acid oxidation has the potential to favorably affect a variety of metabolic diseases including NASH. The acetyl‐coenzyme A (CoA) carboxylase (ACC) isozymes ACC1 and ACC2 are critical enzymes in de novo fatty acid synthesis and fatty acid oxidation, respectively. ACC catalyzes the adenosine triphosphate–dependent carboxylation of acetyl‐CoA to form malonyl‐CoA, the rate‐limiting and first committed step in fatty acid synthesis.2, 4, 5 Malonyl‐CoA also acts as an allosteric inhibitor of carnitine palmitoyltransferase, the rate‐limiting enzyme in fatty acid oxidation.2, 4 Due to this unique position in intermediary metabolism,2, 4, 6 pharmacologic inhibition of ACC represents an attractive approach to limiting fatty acid synthesis in lipogenic tissues while simultaneously stimulating fatty acid oxidation in oxidative tissues.2 Mice with targeted mutations that maintain ACC in a constitutively activated state demonstrate histological and clinical signs of NASH, including an elevation in hepatic malonyl‐CoA, enhanced lipogenesis, elevated hepatic triglycerides, insulin resistance, and liver fibrosis.7
NDI‐010976 is an orally available, liver‐targeted, potent, and selective small molecule allosteric inhibitor of ACC being developed for the treatment of metabolic disorders characterized by dysregulated fatty acid metabolism including NASH. NDI‐010976 inhibits ACC by binding to an allosteric site that is not conserved across other human enzymes, resulting in a high degree of specificity for the target.6 NDI‐010976 is a highly potent and selective inhibitor of both ACC1 and ACC2 in biochemical and cellular assays and was designed to be a substrate for hepatic transporter proteins, namely the organic anion‐transporting polypeptide (OATP) transporters, resulting in favorable liver‐directed biodistribution that ensures that the pharmacological effects are focused on the key target tissue for NASH.6 Results from pharmacodynamic (PD) studies in nonclinical models of metabolic disease indicate that NDI‐010976 can reduce de novo fatty acid synthesis and stimulate fatty acid oxidation in the liver, and thus favorably affect de novo lipogenesis, hepatic steatosis, insulin resistance, and body weight/body fat without affecting food consumption or markers of liver function.6 These studies confirm the potential for NDI‐010976 to impact important metabolic endpoints associated with diseases such as NASH.
A reproducible method for assessing hepatic DNL has been developed and validated by measuring the appearance of de novo synthesized palmitate in very‐low‐density lipoproteins (VLDLs) in the plasma in response to oral fructose using [1‐13C]acetate and mass isotopomer distribution analysis.8 The primary objective of this clinical study was to assess the PD effects of a single oral dose of NDI‐010976 on fractional DNL in overweight and/or obese but otherwise healthy adult male subjects. Secondary objectives included assessment of the safety and tolerability of NDI‐010976, determination of pharmacokinetic (PK) parameters, and correlation of the PK and PD effects of NDI‐010976. The results from this study will inform the design and dose selection of NDI‐010976 for use in future clinical studies in subjects with NASH.
Participants and Methods
EXPERIMENTAL DESIGN
This study was a two‐period, two‐treatment crossover, randomized, double‐blind, placebo‐controlled, single‐center study designed to determine the PD activity on fractional DNL of a single oral dose of 20, 50, or 200 mg NDI‐010976 compared to placebo in adult male subjects who were overweight and/or obese but otherwise healthy. Three cohorts of 10 subjects at each dose level (total 30 subjects) were included. All subjects were judged by the principal investigator to be obese or overweight volunteers with no other significant health issues and met all eligibility criteria. Inclusion criteria included age between 18 and 50 years and body mass index (BMI) of 25‐32 kg/m2. Exclusion criteria included intolerance to or malabsorption of fructose, history of clinically significant gastrointestinal disease and/or surgery that would alter absorption or metabolism of study drug, and history of diabetes or other clinically significant medical disease.
Subjects were randomized in period 1 to receive a single oral dose of either NDI‐010976 or matching placebo followed by a minimum of a 5‐day washout period and administration of the opposite study medication during period 2 (Fig. 1). On day –1 of both periods, all subjects received an isocaloric (relative to individual BMI) lunch and dinner and then were fasted for the duration of the study beginning at hour –12. Subjects received an intravenous infusion of [1‐13C]acetate (10 ± 0.25 g in 1,000 mL 0.45% saline solution, GMP grade; Cambridge Isotope Laboratories, Andover, MA) beginning at hour –9, administered at a rate of 50 mL/hour over 19 hours by infusion pump. At hour 0 on day 1 of both period 1 and period 2, subjects received either active NDI‐010976 capsules as a single oral dose (20, 50, or 200 mg) or matching placebo capsules administered with 240 mL of water. Immediately after study medication administration in both periods, subjects began drinking fructose (250 mg fructose/kg body weight) dissolved in tap water mixed with a zero‐calorie flavor agent administered every 30 ± 5 minutes until hour +9.5 for a total of 20 doses.
Figure 1.
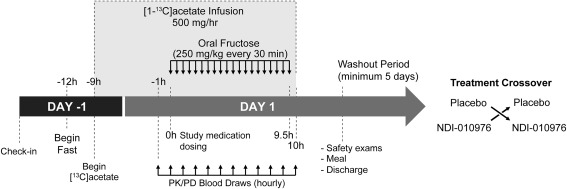
Schematic of study design protocol.
SELECTION OF NDI‐010976 DOSES
This clinical study is the third study of NDI‐010976 in humans including single‐dose and repeat‐dose safety studies. The dose range selected for evaluation in the current study was based on tolerability observed in a single ascending dose clinical safety study in healthy adults in which doses of NDI‐010976 between 30 and 1,000 mg were well tolerated.9 The dose levels of 20, 50, and 200 mg were selected based on a preclinical model of hepatic de novo fatty acid synthesis in Sprague‐Dawley rats to provide assessment of a potential dose–response relationship in humans (Supporting Table S1).6 The highest dose of 200 mg was included to investigate the maximum effect on DNL. The 20 mg dose level was included to explore the lower end of the dose–response curve. The sample size chosen for this study was selected without statistical considerations and was based on typical cohort size for studies of DNL in humans.
BLINDING
This was a double‐blind, randomized, placebo‐controlled, crossover study. The site staff, including the principal investigator and study coordinator, and all subjects were blinded to whether the subjects were receiving active study drug or placebo in each period. The clinic pharmacy staff remained unblinded to ensure proper randomization and dosing. The PD/PK bioanalysis laboratory staff were unblinded, and any data provided by these laboratories to the site staff, principal investigator, study coordinator, and sponsor were provided in a blinded manner until after database lock.
SAFETY
Safety was monitored through physical examinations, vital signs, clinical laboratory tests, and recording of treatment‐emergent adverse events (TEAEs). All reported adverse events (AEs) were coded to a standard set of terms, using the Medical Dictionary for Regulatory Activities, Version 18.0. TEAEs were defined as any AEs that occurred after dosing with study medication (NDI‐010976 or placebo) in period 1. TEAEs that occurred after dosing with study medication in period 1 up to the time of dosing with study medication in period 2 were attributed to the study medication (NDI‐010976 or placebo) that the subject received in period 1. TEAEs that occurred after dosing with study medication in period 2 were attributed to the study medication (NDI‐010976 or placebo) that the subject received in period 2. AEs that started after acetate infusion and before study drug administration in period 1 were counted separately from the study treatments (NDI‐010976 or placebo). The number of subjects experiencing TEAEs and the number of TEAEs were summarized by preferred term for acetate infusion, pooled placebo, each NDI‐010976 treatment, and total NDI‐010976 treatment.
Clinical laboratory assessments (serum chemistry, hematology, and urinalysis) were performed at screening, check‐in (day –1), hour 10 of day 1 of each period, and, if applicable, at early termination visit. Vital signs, including systolic and diastolic blood pressure, pulse rate, respiratory rate, and body temperature, were measured at screening, check‐in (day –1), and hours 0 (pre‐dose), 5.5, and 10.5 on day 1 of each period and, if applicable, at early withdrawal. Baseline was defined as the last predose value, including rechecks, in each period. Safety 12‐lead electrocardiograms were performed at screening.
PHARMACODYNAMICS
Blood samples for the determination of fractional DNL, serum lipids (total cholesterol, high‐density lipoprotein cholesterol, low‐density lipoprotein cholesterol, VLDL, triglycerides, glycerol‐blanked triglycerides, and free glycerol), serum leptin and adiponectin, and blood ketones were collected at protocol‐specified times (Fig. 1). Serum concentrations of lipids and blood ketones by ketometer were determined by the Celerion clinical laboratory (Tempe, AZ). Serum concentrations of leptin and adiponectin were determined using solid‐phase enzyme‐linked immunosorbent assays by Pacific Biomarkers (Seattle, WA). Sample analysis and fractional DNL calculations were performed by KineMed (Emeryville, CA) using gas chromatography‐mass spectrometry and mass isotopomer distribution analysis as described.8, 10, 11 Briefly, at each time point VLDL particles were isolated from plasma by sequential ultracentrifugation, total lipids were extracted from VLDL with chloroform:methanol (2:1), and VLDL‐triacylglycerols (VLDL‐TGs) were then isolated by thin layer chromatography. VLDL‐TG fatty acids were trans‐esterified to fatty acid‐methyl esters for gas chromatography‐mass spectrometry analyses. Fractional DNL represents the fraction of palmitate in VLDL‐TG that was newly synthesized during the period of the [1‐13C]acetate infusion. Fractional DNL was measured after an overnight fast (hour –1 and hour 0) and in response to fructose (from hour +1 until hour +10). Fructose‐induced fractional DNL was determined by calculating the change from fasting baseline DNL at each postdose time point by subtracting individual baseline values from time‐matched active values for each subject. In addition, fractional DNL area under the effect curve (AUEC) was determined using Phoenix WinNonlin, Version 6.3, for both raw values and change from fasting baseline fractional DNL.
PHARMACOKINETICS
Plasma samples for quantitation of NDI‐010976 levels were collected predose and hourly through 10 hours postdose on day 1. Plasma concentrations of NDI‐010976 were determined using validated high‐performance liquid chromatography‐tandem mass spectrometry methods at Covance (West Trenton, NJ). The lower limit of quantitation was 0.5 ng/mL for NDI‐010976 in human plasma. The noncompartmental plasma PK parameters of NDI‐010976 were determined using Phoenix WinNonlin, Version 6.3, and SAS, Version 9.3, including maximum plasma concentrations (Cmax), area under the concentration–time curve (AUC0‐t), and time point of maximum plasma concentration (tmax). Actual sample times were used in the calculations.
PK‐PD ANALYSIS
The PK‐PD relationships of NDI‐010976 were examined using PK estimates of AUC0‐t, Cmax, and C10hours and two PD endpoints including AUEC and the percentage fractional DNL of placebo at 10 hours for each subject. Data were fit using a logarithmic curve fit, and the associated R 2 values were compared to evaluate the strength of the PK–PD correlation (GraphPad Prism 6, Microsoft Excel 2013). The individual PK–PD temporal data for each subject at each sampling time point for each dose was also examined to estimate plasma concentrations at which 70% and 90% inhibition of maximal DNL was observed.
Results
SUBJECT DEMOGRAPHICS AND DISPOSITION
The baseline characteristics and demographics of the study subjects are presented in Table 1, and an enrollment summary is presented in Fig. 2. A total of 32 subjects were enrolled in the study and randomized to study treatment; 30 subjects received one dose of study medication, and 27 subjects completed the study according to protocol. Two subjects in cohort 1 were discontinued on day 1 of period 1 due to an AE prior to dosing (see Safety below) and replaced. Two subjects in cohort 2 were lost to follow‐up in period 2. One subject in cohort 3 withdrew on day –1 of period 2 due to a family emergency. Baseline characteristics of the study population were comparable across cohorts. The majority of subjects (80%) were white, 67% were Hispanic or Latino, and the mean age was 38.5 years (range 26‐49 years). The mean weight was 86.0 kg (range 63.0‐103.2 kg), mean BMI was 28.1 kg/m2 (range 25.7‐31.9 kg/m2), and 30% of subjects were obese (BMI ≥30 kg/m2). None of the subjects were diabetic.
Table 1.
Subject Demographics
| Trait | Category | Number of Subjects (Percent of Subjects) |
|---|---|---|
| Gender | Male | 30 (100%) |
| Race | Black or African American | 3 (10%) |
| White | 24 (80%) | |
| White, American Indian/Alaska Native | 1 (3%) | |
| Asian | 1 (3%) | |
| White, Black or African American | 1 (3%) | |
| Ethnicity | Hispanic or Latino | 20 (67%) |
| Non‐Hispanic or Latino | 10 (33%) | |
| Mean (± standard deviation) | ||
| Age (years) | 38.5 (± 7.6) | |
| Weight (kg) | 86.0 (± 10.0) | |
| Height (cm) | 174.6 (± 8.6) | |
| BMI (kg/m2) | 28.1 (± 1.7) |
Figure 2.
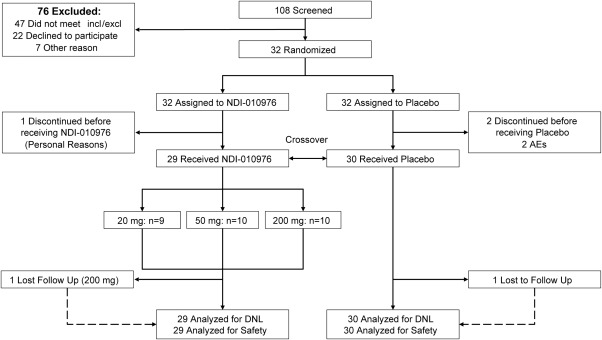
CONSORT flow diagram of patient disposition.
SAFETY
A summary of the safety results is provided in Table 2, and detailed safety results are included in Supporting Table S2. Overall, single oral doses of NDI‐010976 were safe and well tolerated. There were no deaths, serious AEs, or Grade 3 or higher TEAEs in this study. Two subjects were discontinued due to an AE of infusion site extravasation that occurred predose in period 1. There were no clinically important treatment‐related or dose‐related trends in the incidence or severity of TEAEs, clinical laboratory, vital sign, or physical examination assessments in this study. Gastrointestinal disorders including diarrhea and flatulence were the most common TEAEs following both active and placebo treatments, and these may have been related to fructose administration. Diarrhea was the most common, reported by 11 (38%) subjects following NDI‐010976 and 9 (30%) subjects in the pooled placebo population. Flatulence, the next most common, was reported by 5 (17%) subjects following NDI‐010976 and 3 (10%) subjects in the pooled placebo population.
Table 2.
Summary of Safety Observations
| TEAEs | Acetate Infusion | Pooled Placebo |
20 mg NDI‐010976 |
50 mg NDI‐010976 |
200 mg NDI‐010976 |
Total NDI‐010976 |
|---|---|---|---|---|---|---|
| Number of subjects dosed | 30 | 30 | 9 | 10 | 10 | 29 |
|
Number of subjects with TEAEs (% of subjects dosed) |
4 (13%) | 12 (40%) | 3 (33%) | 7 (70%) | 5 (50%) | 15 (52%) |
|
Number of subjects without TEAEs (% of subjects dosed) |
26 (87%) | 18 (60%) | 6 (67%) | 3 (30%) | 5 (50%) | 14 (48%) |
EFFICACY AND BIOMARKERS
The results for fractional DNL demonstrate the successful implementation of the stable isotope methodology to assess the pharmacodynamic effects of NDI‐010976 on fructose‐stimulated hepatic DNL (Fig. 3A). The crossover design used in this study emphasized comparisons within subjects over time rather than comparisons among groups, which mitigates and controls for differences in baseline DNL and other unrecognized intersubject variables. Periodic oral administration of fructose‐stimulated fractional DNL from a fasted baseline of 7.15 ± 4.94% new palmitate at hour 0 (predose) to 38.10 ± 5.66% at hour 10 in the pooled placebo subjects (Supporting Table S3). A comparison of treatment effects across groups for fractional DNL as mean AUEC or fractional DNL at the end of the treatment period (10 hours) is shown in Fig. 3B,C. Analysis of the AUEC for fractional DNL change from fasting baseline (fasting‐adjusted DNL) showed a prominent dose‐dependent inhibition following treatment with NDI‐010976 relative to placebo of approximately 70 ± 6%, 85 ± 3%, and 104 ± 2% (mean ± standard error of the mean) following single‐dose administration of 20, 50, and 200 mg, respectively (Fig. 3B). Following a single dose of 50 mg NDI‐010976, all subjects had >70% inhibition of DNL as determined by fasting‐adjusted DNL AUEC over the 10‐hour study period compared to the matched placebo period, and all subjects administered 200 mg NDI‐010976 had complete or near complete inhibition of DNL. NDI‐010976 demonstrated a prolonged and marked dose‐dependent reduction in fractional DNL at 10 hours postdose when mean reduction of fasting‐adjusted DNL was approximately 41 ± 9%, 53 ± 9%, and 94 ± 4% relative to placebo following 20, 50, and 200 mg NDI‐010976, respectively (Fig. 3C).
Figure 3.
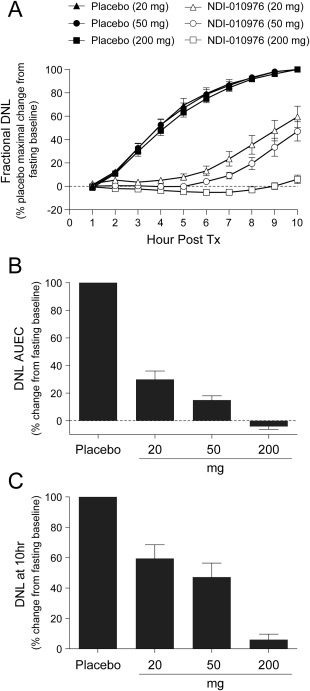
Fractional DNL change from fasting baseline. (A) Time course of fructose‐induced mean fractional DNL following a single dose of placebo or 20, 50, and 200 mg of NDI‐010976. Fasted baseline‐adjusted fractional DNL was normalized as percent of maximal DNL at 10 hours postdose of each individual. (B) Mean (± standard error of the mean) change from fasting baseline‐adjusted fractional DNL by AUEC for NDI‐010976‐treated groups was normalized to matched placebo. (C) Mean (± standard error of the mean) change from fasting baseline‐adjusted fractional DNL at hour 10 end of study for NDI‐010976‐treated groups was normalized to maximal fasting baseline‐adjusted DNL following placebo treatment at 10 hours postdose.
PK AND PK–PD RELATIONSHIPS
Plasma PK concentration time curves and mean PK parameters were estimated from a limited PK sampling time course in this study and are shown in Fig. 4. Mean plasma NDI‐010976 AUC0‐t and observed Cmax values increased with increasing NDI‐010976 dose. The mean observed plasma concentration versus time profiles of NDI‐010976 following a single oral dose at 20, 50, and 200 mg achieved mean maximum plasma concentrations (Cmax) and area under the concentration‐time curve from time 0 to 10 hours (AUC0‐10hours) of 15.5, 36.5, and 222 ng/mL and 39.9, 98.8, and 518 ng*hr/mL, respectively. The median peak plasma concentrations of NDI‐010976 were observed at the earliest sampling time points from 1.0 to 2.0 hours (tmax) following administration of 20 to 200 mg NDI‐010976. Mean concentrations at the last observed time point (10 hours postdose) were low and not dose‐proportional due to high variability at this time point.
Figure 4.
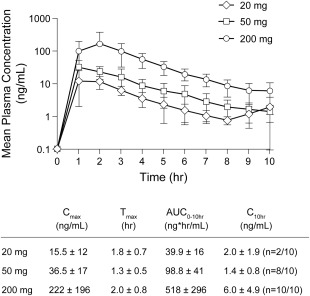
Human plasma PK of NDI‐010976. Mean (± standard deviation) plasma concentration–time profiles of NDI‐010976 in overweight adult male subjects after administration of a single oral dose at 20, 50, and 200 mg (n = 10/group).
As anticipated from the acute, single‐dose nature of the study, no significant NDI‐010976 effects were observed on serum glucose, lipids, leptin, and adiponectin, or blood ketones following a single dose of 20, 50, or 200 mg NDI‐010976 (Supporting Table S4). Based on the scatter plots comparing two pharmacodynamic endpoints for DNL (fasting‐adjusted DNL AUEC and percent maximal DNL at 10 hours) and three measures of plasma NDI‐010976 exposure (concentration at hour 10 [C10hours], AUC0‐t, and Cmax), there was a trend toward decreased fractional DNL with increasing plasma NDI‐010976 exposure (Supporting Fig. S1). The strongest correlation (R 2 = 0.5696) occurred between the percentage change of DNL at 10 hours compared to fasting baseline and C10hours plasma exposure. For DNL AUEC, a similar correlation was observed with plasma exposures of NDI‐010976 determined both by AUC0‐t (R 2 = 0.4548) and C10hours (R 2 = 0.4937). The weakest relationship (R 2 < 0.3) for both DNL PD endpoints was with Cmax exposure of NDI‐010976, likely due in part to the limited PK sampling characterizing early plasma exposures. Further, there was a direct relationship between individual plasma concentrations of NDI‐010976 and inhibition of fructose‐stimulated DNL response across the experimental time course (Supporting Fig. S1D). Plasma concentrations of NDI‐010976 >2.2 ng/mL were associated with at least 70% inhibition of fractional DNL (one data point outlier), while plasma concentrations greater than approximately 4.0 ng/mL yielded >90% inhibition of fractional DNL (two data point outliers) across all dose groups, thus defining minimal drug concentrations required to maintain complete inhibition of the fructose‐stimulated increase in fractional DNL.
Discussion
The objectives of this study were to determine the extent of inhibition of hepatic DNL achievable with a liver‐targeted, allosteric inhibitor of acetyl‐CoA carboxylases, ACC1 and ACC2, and to guide dose selection of NDI‐010976 to be studied in patients with NASH. In overweight and/or obese but otherwise healthy adult male subjects, NDI‐010976 demonstrated a dose‐dependent inhibition of fructose‐stimulated fractional DNL AUEC of approximately 70%, 85%, and 104% relative to placebo following single oral doses of 20, 50, and 200 mg, respectively. Analyses of percentage change of fasting‐adjusted DNL relative to placebo (at hour 10 and AUEC) versus plasma NDI‐010976 exposure (based on plasma NDI‐010976 concentration at hour 10 and AUC0‐t) support an inverse relationship between fractional DNL and plasma NDI‐010976 exposure. The most striking concentration–effect relationship was that fractional DNL was inhibited by at least 70% and 90% when plasma concentrations of NDI‐010976 exceeded 2.2 and 4.0 ng/mL, respectively.
NDI‐010976 was designed to take advantage of chemical properties that enable active transport into the liver through oral administration to achieve high liver drug concentrations while maintaining low systemic exposure, providing for both inhibition of fatty acid synthesis and stimulation of mitochondrial fatty acid oxidation.6 The profound inhibitory effect on DNL at very low plasma concentrations of drug in this clinical study is a reflection of the specific liver‐targeted biodistribution of NDI‐010976. While plasma concentrations of NDI‐010976 required for this pharmacodynamic effect on DNL were low, substantially higher drug levels in liver are expected due to the preferential uptake of NDI‐010976 into hepatocytes through OATP transporters. In preclinical studies, hepatic extraction in cynomolgus monkeys, a species with high homology in amino acid sequence and substrate specificity to human for OATP transporters, was approximately 94%, suggesting the estimated human liver concentrations of NDI‐010976 may be greater than an order of magnitude higher than plasma concentrations12, 13, 14 (also unpublished data). Moreover, the observed PK–PD response relationship of NDI‐010976 in this clinical study was consistent with data in a preclinical model of de novo fatty acid synthesis in Sprague‐Dawley rats (Supporting Tables S1 and S6).
Indeed, further analysis of the 20 mg low‐dose subject data revealed an average of 60 ± 26% (mean ± standard deviation) inhibition of fractional DNL at the earliest measurable fructose‐induced increase in placebo DNL (2 hours postdose), consistent with rapid liver extraction of NDI‐010976. The associated plasma concentrations at 2 hours postdose ranged from 5.7 to 23.5 ng/mL. One subject in this dose group demonstrated the highest plasma Cmax (43.4 ng/mL) and the lowest DNL response (22.6 % inhibition at 2 hours postdose) of all subjects from the 20 mg dose group, consistent with lower hepatic extraction of NDI‐010976 in this individual. Across the three dose groups, subjects with plasma exposure above 2.2 ng/mL were associated with >70% inhibition of maximal DNL (201 of 202 data points), followed by steady recovery of DNL activity at later time points with lower plasma concentrations. Despite variability in the interindividual PK, subjects maintained at least 70% inhibition of hepatic fructose‐stimulated fractional DNL for at least 6 hours following a 20 mg oral dose of NDI‐010976.
There are several mechanisms by which hepatic de novo lipid synthesis leads to an increased hepatic triglyceride content. Increased rate of lipogenesis results from an increased flux of carbons through glycolysis that serve both as the glycerol backbone for TG synthesis and as the fatty acids that are esterified to the glycerol backbone. Moreover, DNL increases malonyl‐CoA levels, which inhibits transport of fatty acids into the mitochondria through negative regulation of carnitine palmitoyltransferase, leading to inhibition of mitochondrial lipid oxidation and increased cytosolic re‐esterification of fatty acids in the liver.15, 16 The result is either an increase in secretion of VLDL‐TG or accumulation of hepatic fat droplets. Palmitate, the primary fatty acid product of DNL, is a saturated fatty acid that has been demonstrated to promote inflammation and endoplasmic reticulum stress.3, 17, 18 The accumulation of malonyl‐CoA also promotes elongation of free fatty acids, enabling the formation of complex lipids important in signaling pathways implicated in inflammation and fibrosis.19 Therefore, elevated DNL, mediated through ACC1 and ACC2, plays an important role in the underlying pathophysiology of NASH.
An important contributor of fat accumulation in NAFLD is the loss of hepatic regulation of the rate of lipogenesis that normally occurs in the transition from the fasted to the fed state. There exists both indirect and direct evidence that elevated lipogenesis provides a significant source of the fatty acids accumulating in the livers of patients with NAFLD.20, 21, 22, 23 Lambert and colleagues reported that fatty acid synthesis through DNL is 3.5‐fold higher in individuals with high liver fat compared to low liver fat, and as a percentage of TG‐palmitate, there was a greater than 2‐fold increase in contribution of VLDL‐TG from DNL in high–liver fat individuals.23 These systemic changes were independently associated with intrahepatic triglyceride levels, demonstrating a positive association between fasting DNL and intrahepatic triglyceride levels in all subjects. The most striking evidence for a role of hepatic DNL in NAFLD is that DNL is not suppressed by fasting in subjects with NAFLD, and fasting lipogenesis is approximately 5‐fold higher in insulin‐resistant compared to insulin‐sensitive individuals.24 Thus, the effect of insulin resistance manifested through increased rate of DNL is not responsive to the nutritional state of the individual. Lastly, the flux of free fatty acids from adipose tissue is not different between high–liver fat and low–liver fat individuals.23
For context in relationship to therapeutic intervention in subjects with NAFLD and specifically NASH, a study evaluating the effect of pioglitazone on DNL in subjects with type 2 diabetes mellitus and hypertriglyceridemia using a similar stable isotope labeling protocol has been reported.11 In that study, pioglitazone reduced hepatic DNL by 40%. The dose of pioglitazone evaluated was similar to that used in the PIVENS trial in subjects with NASH that was associated with significant improvement in steatosis, inflammation, and hepatocellular ballooning, as well as improvements in insulin resistance and liver biochemistry.25 Thus, repeat dosing of pioglitazone at a dose that caused 40% reduction in hepatic DNL translated to evidence of clinical benefit in NASH subjects. However, while glitazones may have favorable effects on hepatic lipogenesis and macrophage inflammatory parameters, the adverse effects associated with glitazone use (adipose fat accumulation, weight gain, cardiovascular effects) limit their use in NASH patients. A single oral dose of 20 mg NDI‐010976 reduced mean fructose‐stimulated fractional DNL by 70%, suggesting that a greater effect may be achieved at steady‐state drug levels with repeat dosing. Longer labeling of the precursor pool may result in a greater effect of NDI‐010976 on fractional DNL than that observed in the present study.26 Future studies will enable determination of the effects of longer duration of inhibition of ACC on DNL and other endpoints of NASH.
Based on the allosteric mechanism of inhibition, NDI‐010976 is anticipated to have a favorable safety profile. NDI‐010976 inhibits ACC through interaction with a binding site that is not conserved across other human enzymes, including carboxylases, resulting in an excellent selectivity profile of NDI‐010976 with reduced off‐target activity.6, 9 The liver‐directed biodistribution of NDI‐010976 ensures that pharmacological effects are focused on the key target tissue for NASH and is anticipated to minimize effects outside of the liver. While this study was not specifically designed to assess the safety of NDI‐010976, a previous clinical study has demonstrated the safety of single‐dose administration in normal healthy subjects up to a dose of 1,000 mg.9 In this current phase 1 clinical study, single oral doses of NDI‐010976 from 20 to 200 mg were well tolerated when administered to overweight and/or obese but otherwise healthy male adult subjects. No clinically important treatment‐related or dose‐related trends in TEAEs, clinical laboratory tests, vital signs, or physical examination findings were observed in this study. In addition, as anticipated from an acute single‐dose study, there were no apparent study drug effects on serum glucose, lipids, leptin or adiponectin, or blood ketones. Full assessment of the safety of prolonged inhibition of ACC by NDI‐010976 will be determined in longer‐term phase 2 clinical studies that are currently ongoing.
Overall, NDI‐010976 is anticipated to add considerable value to the treatment algorithm of NASH, particularly in light of the high unmet medical need, lack of effective existing therapies, and limitations of emerging treatments. First, through the mechanism of inhibiting ACC, NDI‐010976 reduces the de novo formation of long‐chain lipid signaling molecules and significantly decreases hepatic free fatty acids and triglycerides, which are considered the driving force behind lipotoxicity in the liver that leads to hepatocellular injury, inflammation, and fibrosis.3 In addition, inhibition of ACC reduces concentrations of malonyl‐CoA and thereby further reduces free fatty acids by stimulating mitochondrial fatty acid oxidation. Based on nonclinical data showing dramatic reductions in hepatic triglyceride levels in several rodent models, NDI‐010976 is anticipated to have a significant impact on hepatic steatosis in patients with NASH.6
One potential effect of ACC inhibition would be an anticipated increase in cellular acetyl‐CoA due to reduced conversion to malonyl‐CoA and increased mitochondrial beta‐oxidation. The fate of the cytosolic acetyl‐CoA not used for fatty acid synthesis and the fate of the mitochondrial acetyl‐CoA formed through enhanced beta‐oxidation remain to be fully characterized in humans. However, it is clear that the cytosolic acetyl‐CoA that is not converted into fatty acids following inhibition of ACC in cultured cells and experimental animals does not shunt to de novo cholesterol synthesis.2, 6, 27 It is also clear that the acetyl‐CoA formed through enhanced mitochondrial beta‐oxidation is not converted through pyruvate and gluconeogenesis to glucose but is rather converted to ketone bodies, even in the fed state, and that basal metabolic rates are elevated after drug treatment, leading to a greater CO2 production and a correspondingly, albeit smaller, increase in O2 consumption.6, 27, 28 Studies to further explore, and to more completely describe in quantitative terms, the fates of cytosolic and mitochondrial acetyl‐CoA after ACC inhibition in experimental animals are in progress, and the findings from those studies will be used to incorporate similar evaluations into longer‐term clinical studies.
In summary, this clinical study demonstrates that single‐dose treatment with NDI‐010976 dramatically decreases fructose‐stimulated hepatic DNL in overweight and/or obese but otherwise healthy, adult male subjects. Future studies will evaluate the clinical and histologic benefits of NDI‐010976 treatment in subjects with NASH.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29246/suppinfo.
Supporting Information Figure 1.
Supporting Information Tables.
Acknowledgment
We thank Elizabeth Parks, Scott Turner, and Tom Roddy for guidance and helpful suggestions in the design of the DNL study. We also thank Danielle Armas, Clayton Dehn, Toni Grant, Kate Reese, Kristen Westrick, and the outstanding Celerion clinical team for their tireless efforts and superb execution of the clinical study described in this report.
Potential conflict of interest: Dr. Stiede is employed by and owns stock in Nimbus. Dr. Miao is employed by and owns stock in Nimbus. Dr. Blanchette is employed by and owns stock in Nimbus. Dr. Beysen was employed by KineMed. Dr. Harriman owns stock in Nimbus. Dr. Harwood consults and owns stock in Nimbus. Dr. Kapeller is employed by and owns stock in Nimbus. Dr. Westlin is employed by and owns stock in Nimbus.
REFERENCES
- 1. Dyson JK, Anstee QM, McPherson S. Non‐alcoholic fatty liver disease: a practical approach to diagnosis and staging. Frontline Gastroenterol 2014;5:211‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harwood HJ Jr. Treating the metabolic syndrome: acetyl‐CoA carboxylase inhibition. Expert Opin Ther Targets 2005;9:267‐281. [DOI] [PubMed] [Google Scholar]
- 3. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
- 4. Tong L, Harwood HJ Jr. Acetyl‐CoA carboxylases: versatile targets for drug discovery. J Cell Biochem 2006;99:1476‐1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim KH. Regulation of mammalian acetyl‐coenzyme A carboxylase. Annu Rev Nutr 1997;17:77‐99. [DOI] [PubMed] [Google Scholar]
- 6. Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, et al. Acetyl‐CoA carboxylase inhibition by ND‐630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci USA 2016;113:E1796‐E1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinikunnil T, Chen ZP, et al. Single phosphorylation sites in ACC1 and ACC2 regulate lipid homeostasis and the lipid‐sensitizing effects of metformin. Nat Med 2013;19:1649‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hellerstein MK, Neese RA. Mass isotopomer distribution analysis: a technique for measuring biosynthesis and turnover of polymers. Am J Physiol 1992;263:E988‐E1001. [DOI] [PubMed] [Google Scholar]
- 9. Westlin WF, Harriman G, Harwood HJ, Kapeller R, Lennon S, Miao W, et al. NDI‐010976, a potent, liver‐directed, oral inhibitor of acetyl‐CoA carboxylase for non‐alcoholic steatohepatitis: a phase 1 single ascending dose study in healthy volunteers [Abstract]. J Hepatol 2016;2(Suppl.):S501‐S502. [Google Scholar]
- 10. Hellerstein MK. De novo lipogenesis in humans: metabolic and regulatory aspects. Eur J Clin Nutr 1999;53:S53‐S65. [DOI] [PubMed] [Google Scholar]
- 11. Beysen C, Murphy EJ, Nagaraja H, Decaris M, Riiff T, Fong A, et al. A pilot study of the effects of pioglitazone and rosiglitazone on de novo lipogenesis in type 2 diabetes. J Lipid Res 2008;49:2657‐2663. [DOI] [PubMed] [Google Scholar]
- 12. Shen H, Yang Z, Mintier G, Han Y‐H, Chen C, Balimane P, et al. Cynomolgus monkey as a potential model to assess drug interactions involving hepatic organic anion transporting polypeptides: in vitro, in vivo, and in vitro‐to‐in vivo extrapolation. J Pharmacol Exp Ther 2013;344:673‐685. [DOI] [PubMed] [Google Scholar]
- 13. Bleasby K, Castle JC, Roberts CJ, Cheng C, Bailey WJ, Sina JF, et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre‐clinical species: a resource for investigations into drug disposition. Xenobiotica 2006;36:963‐988. [DOI] [PubMed] [Google Scholar]
- 14. Evers R, Chu X‐Y. Role of the murine organic anion‐transporting polypeptide 1b2 (Oatp1b2) in drug disposition and hepatotoxicity. Mol Pharmacol 2008;74:309‐311. [DOI] [PubMed] [Google Scholar]
- 15. McGarry JD. Banting lecture 2001. Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002;51:7‐18. [DOI] [PubMed] [Google Scholar]
- 16. Topping DL, Mayes PA. The immediate effects of insulin and fructose on the metabolism of the perfused liver. Changes in lipoprotein secretion, fatty acid oxidation and esterification, lipogenesis and carbohydrate metabolism. Biochem J 1972;126:295‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid‐induced lipotoxicity. Proc Natl Acad Sci USA 2003;100:3077‐3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 2006;291:E275‐E281. [DOI] [PubMed] [Google Scholar]
- 19. Jump DB, Torres‐Gonzalez M, Olson LK. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem Pharmacol 2011;81:649‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008;134:424‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sevastianova K, Santos A, Kotronen A, Hakkarainen A, Makkonen J, Silander K, et al. Effect of short‐term carbohydrate overfeeding and long‐term weight loss on liver fat in overweight humans. Am J Clin Nutr 2012;96:727‐734. [DOI] [PubMed] [Google Scholar]
- 22. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilke MS, French MA, Goh YK, Ryan EA, Jones PJ, Clandinin MT. Synthesis of specific fatty acids contributes to VLDL‐triacylglycerol composition in humans with and without type 2 diabetes. Diabetologia 2009;52:1628‐1637. [DOI] [PubMed] [Google Scholar]
- 25. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vedala A, Wang W, Neese RA, Christiansen MP, Hellerstein MK. Delayed secretory pathway contributions to VLDL‐triglycerides from plasma NEFA, diet, and de novo lipogenesis in humans. J Lipid Res 2006;47:2562‐2574. [DOI] [PubMed] [Google Scholar]
- 27. Harwood HJ Jr, Petras SF, Shelly LD, Zaccaro LM, Perry DA, Makowski MR, et al. Isozyme‐nonselective N‐substituted bipiperidylcarboxamide acetyl‐CoA carboxylase inhibitors reduce tissue malonyl‐CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem 2003;278:37099‐37111. [DOI] [PubMed] [Google Scholar]
- 28. Griffith DA, Kung DW, Esler WP, Amor PA, Bagley SW, Beysen C, et al. Decreasing the rate of metabolic ketone reduction in the discovery of a clinical acetyl‐CoA carboxylase inhibitor for the treatment of diabetes. J Med Chem 2014;57:10512‐10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29246/suppinfo.
Supporting Information Figure 1.
Supporting Information Tables.