

Abstract
In solid‐phase peptide synthesis, the nominal batch size is calculated using the starting resin substitution and the mass of the starting resin. The starting resin substitution constitutes the basis for the calculation of a whole set of important process parameters, such as the number of amino acid derivative equivalents. For Fmoc‐substituted resins, substitution determination is often performed by suspending the Fmoc‐protected starting resin in 20% (v/v) piperidine in DMF to generate the dibenzofulvene–piperidine adduct that is quantified by ultraviolet–visible spectroscopy. The spectrometric measurement is performed at the maximum absorption wavelength of the dibenzofulvene–piperidine adduct, that is, at 301.0 nm. The recorded absorption value, the resin weight and the volume are entered into an equation derived from Lambert–Beer's law, together with the substance‐specific molar absorption coefficient at 301.0 nm, in order to calculate the nominal substitution. To our knowledge, molar absorption coefficients between 7100 l mol−1 cm−1 and 8100 l mol−1 cm−1 have been reported for the dibenzofulvene–piperidine adduct at 301.0 nm. Depending on the applied value, the nominal batch size may differ up to 14%. In this publication, a determination of the molar absorption coefficients at 301.0 and 289.8 nm is reported. Furthermore, proof is given that by measuring the absorption at 289.8 nm the impact of wavelength accuracy is reduced. © 2017 The Authors Journal of Peptide Science published by European Peptide Society and John Wiley & Sons Ltd.
Keywords: SPPS, resin, substitution determination, dibenzolfulvene‐piperidine adduct, Fmoc, absorption coefficient, N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine
Introduction
After introduction of solid‐phase peptide synthesis (SPPS) by R. B. Merrifield 1, the Fmoc group has evolved as the dominating protecting group for temporary amine protection 2, 3, 4, 5, 6, 7, 8. As a consequence, starting resins used in SPPS often contain Fmoc‐protected amine functions. The Fmoc function is either part of the corresponding linker moiety (e.g. Rink amide 9 or Tricyclic amide linker 10 resin) or is introduced by coupling an Fmoc‐protected building block (usually Fmoc‐protected amino acid derivatives) onto a functionalized polymeric support (e.g. polystyrene). The base‐labile Fmoc group is quantitatively cleaved with 20% (v/v) piperidine in DMF, forming the dibenzofulvene–piperidine adduct (Scheme 1). This adduct exhibits two distinct UV absorbance maxima at λ = 301.0 and 289.8 nm. Absorption values measured at either of the absorbance maxima can be used, in combination with the respective molar absorption coefficient, to calculate the substitution of Fmoc‐protected resins 11
Scheme 1.
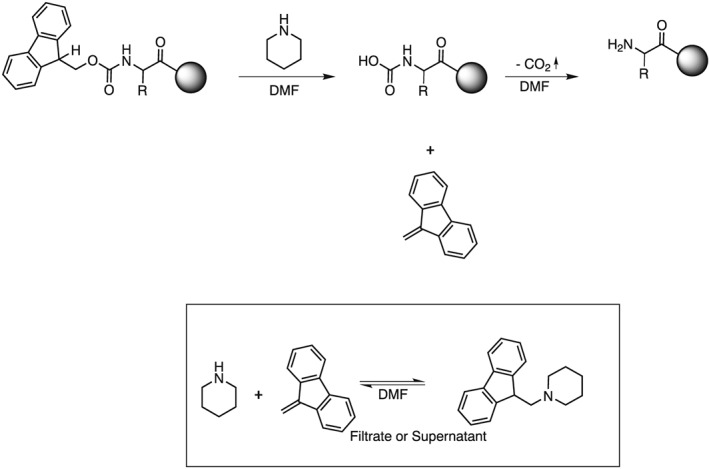
Fmoc cleavage with 20% (v/v) piperidine in DMF and formation of the dibenzofulvene–piperidine adduct.
Obviously, a reliable method for substitution determination is of utmost importance as the determined substitution of a resin defines the SPPS batch size and batch size‐dependent process parameters (e.g. the number of amino acid derivative equivalents, calculated SPPS step yields, etc.). In addition, the resin substitution is an important quality attribute of the starting resin, which has to be considered. For large‐scale peptide manufacturing, this quality attribute strongly impacts the economy of the synthesis and potentially the drug substance quality.
As a result of the distinct UV absorbance of the dibenzofulvene–piperidine adduct, 10–20 mg of resin is a sufficient amount to be used as a sample to determine the substitution in an easy‐to‐perform and robust analytical test method. Predominantly, the resin absorption is measured at 301.0 nm using 20% (v/v) piperidine in DMF as solvent. For these conditions, molar absorption coefficients ranging from 7100 l mol−1 cm−1 12 to 8100 l mol−1 cm−1 13 have been reported. Depending on the applied value, the calculated SPPS batch sizes may differ up to 14% (Figure 1). Evidently this variation applies to any calculated value utilized to determined SPPS batch size. Calculated substitution values that are significantly larger than the actual substitutions may have a substantial financial implication, especially if a synthesis is performed at large scale. For example in a production‐scale SPPS reactor with 1000 l reaction volume, a typical batch size is 35.0 mol. Employing 2.0 eq amino acid building blocks per coupling reaction, a nominal 14% increase due to the selected extinction coefficient (i.e. a nominal batch size of 35.0 mol translates into an actual batch size of 30.1 mol) would result in the unintentional use of additional 9.8 mol of amino acid derivative (i.e. 0.3 eq.) per coupling reaction. In such example, 70.0 mol amino acid derivative (AAD) per coupling cycle would be used instead of 60.2 mol AAD, considering the actual batch size of 30.1 mol. Clearly, such an unintentional use of materials adds unnecessary costs. At an estimated market price of amino acid derivatives, these additional costs are in the range of several 10 000 EUR, and significantly more for longer peptides or in cases where more sophisticated building blocks are required. To be even more specific, taking a peptide such as Bivalirudin (a peptide composed of a linear 20 amino acid chain), that discrepancy would sum up to more than 60 kg of unintentionally used AADs and – assuming a cost of 2 000 EUR per kg amino acid derivative – would result in unintended additional production costs of 120 000 EUR for one batch. In addition and importantly, incorrect substitution values might cause misleading process development data, which in turn might result in erroneous process characterization. As one potential consequence, an adverse impact on the crude peptide quality may result.
Figure 1.
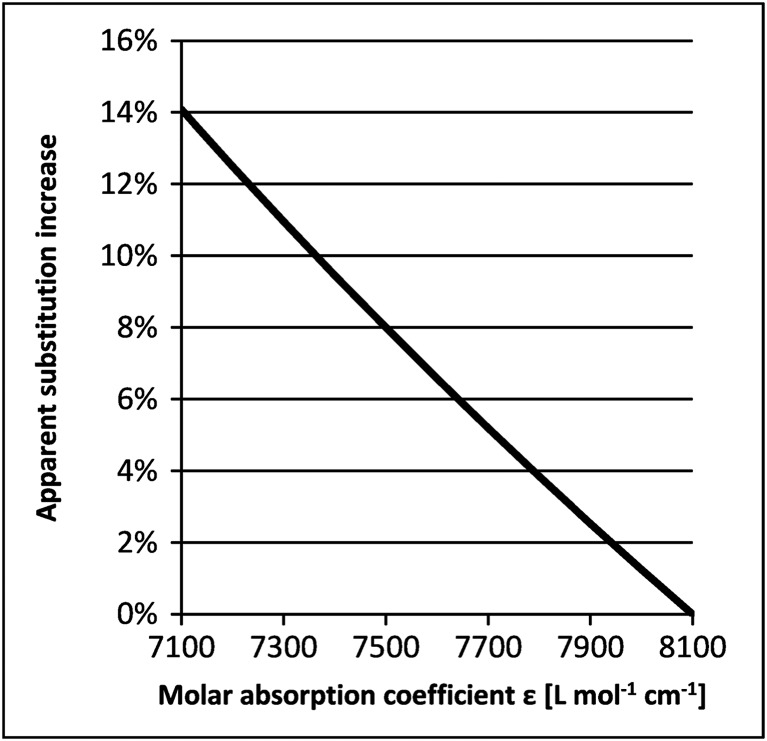
Correlation of the molar absorption coefficient with the determined substitution and substitution‐derived values such as the SPPS batch size according to Lambert–Beer's law. The calculated resin substitution is proportional to ε−1 (ε = molar absorption coefficient). As a consequence, if the molar absorption coefficient decreases by 1% to 99% of the initial value, the obtained substitution increases by 1% of the initial value as (0.99)−1 = 1.01. Accordingly, if the initial molar absorption coefficient is 8100 l mol−1 cm−1 and decreases to a value of 7100 l mol−1 cm−1, this means a decrease of 1000 l mol−1 cm−1, which is 12% of the initial value. As a consequence, the determined substitution with 7100 l mol−1 cm−1 is (0.88)−1‐fold the substitution at 8100 l mol−1 cm−1, which is 1.14‐fold the initial value or 14% more.
Results and Discussion
The UV‐spectroscopic determination of the resin substitution was investigated in detail in the course of a SPPS process development for large‐scale manufacturing including the loading of the resin with the C‐terminal AAD. Applying a value of 7200 l mol−1 cm−1 for the molar absorption coefficient at 301.0 nm, substitution levels above the theoretically expected substitution for quantitative resin loading were calculated. In addition, the substitution values were not in accordance with substitution values that were determined using an orthogonal test method. As an orthogonal test method, the potentiometric titration of the resin suspension in THF/acetic acid with 0.1 M HClO4 after cleavage of the Fmoc group was performed. The titration indicated a lower substitution at the preceding Fmoc stage. A detailed root cause analysis concluded that the molar absorption coefficient (i.e. 7200 l mol−1 cm−1) that was used for the calculation of the substitution was inaccurate. This observation led to further investigations comprising a re‐determination of the molar absorption coefficient. For the first electronic transition at 301.0 nm, an absorption coefficient of 8021 l mol−1 cm−1 was determined, which is close to the previously reported maximum value (see Figure 2 for the absorption spectrum between 270 and 310 nm) 13. The application of this molar absorption coefficient according to Lambert–Beer's law for substitution determination resulted in plausible substitution values. In addition, these substitution values corresponded with the values obtained with the orthogonal titration method for the Fmoc‐deprotected resins (variance within 7%).
Figure 2.
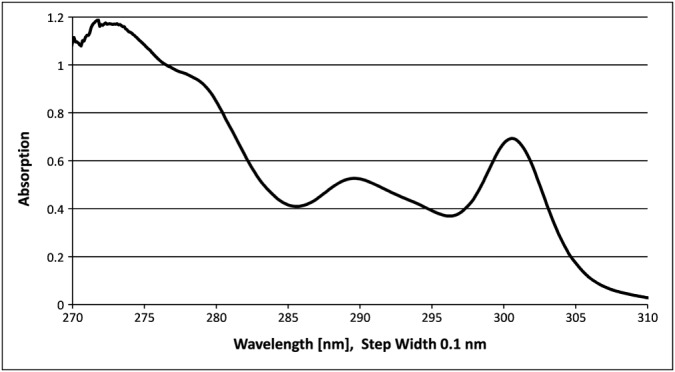
Absorption spectrum of N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine (8.515 · 10−5 mol l−1) in 20% (v/v) piperidine in DMF between 270 and 310 nm.
The determined substitution depended to some degree on the UV spectrometer that was used, which was attributed to different wavelength accuracies and spectral bandwidths. The typical wavelength accuracy of conventional ultraviolet–visible (UV/Vis) spectrometers is approximately ±0.3 nm 14 in the wavelength range used for Fmoc substitution determination, whereas in case of older UV/Vis spectrometers, the wavelength accuracy is lower, for instance ±0.5 nm in case of the HP 8453A (1995) 15 and ±1 nm in case of the HP 8450A (1979) 16. In addition, a small impact of the spectral bandwidth cannot be ruled out either (usually in the range of 1.0–1.5 nm for modern UV/Vis spectrometers 13).
The absorption spectrum of N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine in 20% (v/v) piperidine in DMF (Figure 2) clearly shows that the maximum at 301 nm is a sharp band. For sharp bands, small deviations from the correct maximum wavelength have a significant impact on the determined value for the molar absorption coefficient. That in turn translates in an inaccurate determination of the substitution (Figure 3). To some extent, this impact might also explain the variety of absorption coefficients reported in literature. In general, the adverse impact of equipment wavelength accuracy may be reduced by recording the full UV/Vis spectrum, and reporting the maximum absorption value, independent from the actual wavelength. However this procedure is less feasible for high‐throughput substitution determination, because the read‐out wavelength can no longer be defined in advance and hence automate data collection cannot be easily utilized. Another option to reduce the impact of wavelength accuracy would be to choose a shallower band. Indeed, the absorption spectrum of N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine in 20% (v/v) piperidine in DMF exhibits a shallower band at a wavelength of 289.8 nm. The impact of wavelength accuracy on the determined molar absorption coefficient and in turn on substitution determination is much smaller using the second transition at 289.8 nm. This peculiarity makes substitution determination at 289.8 nm more precise and robust compared with substitution determination at 301.0 nm (Figure 3). Although this advantage comes at the cost of an about 24% lower absorption compared with the transition at 301.0 nm, the reduced intensity is of little importance for the routine substitution determination of Fmoc‐substituted resins. At 289.8 nm, the molar absorption coefficient for the dibenzofulvene–piperidine adduct was determined to be 6089 l mol−1 cm−1 in our laboratories.
Figure 3.
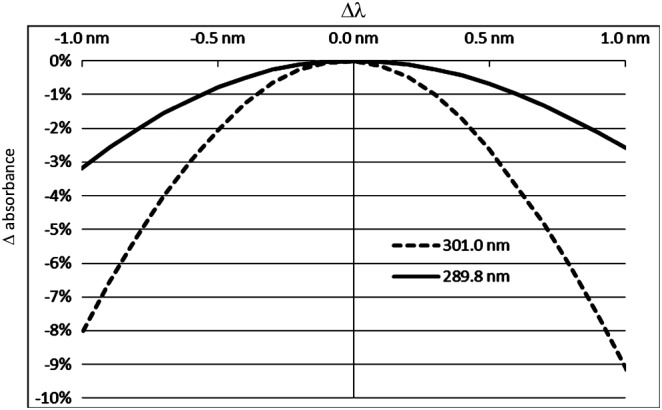
Impact of the wavelength accuracy Δλ on the determined absorption value for the maxima at 301.0 and 289.8 nm, respectively. The corresponding values were taken from the spectrum in Figure 2. The figure covers a Δλ range between −1.0 and +1.0 nm.
The Fmoc substitution may be calculated with the following formula using the maximum at 289.8 nm (for the maximum at 301.0 nm, the calculation is performed analogously):
- SFmoc [mmol g−1] =
where:
- SFmoc =
Fmoc substitution [mmol g−1]
- ε289.8 nm =
Molar absorption coefficient at 289.8 nm: ε289.8 nm = 6089 L mol−1 cm−1
- E289.8 nm =
Absorption of the sample solution at 289.8 nm
- mResin =
Sample weight of the resin [mg]
- 106 mmol mol−1 mg g−1 =
Factor for conversion of mol to mmol and mg−1 to g−1: 1000 [mol to mmol] 1000 [mg−1 to g−1]
- V =
Sample volume in [l] (e.g. 0.1 l)
- l =
Optical path length of the cell in cm (e.g. 1 cm)
- D =
Dilution factor
Using a volume of 0.1 l of 20% (v/v) piperidine in DMF without any dilution steps and a cell with 1 cm optical path length, the formula can be simplified to
- SFmoc [mmol g−1] =
or, neglecting units
- SFmoc [mmol g−1] =
The suitability of this method was demonstrated through recovery experiments with Fmoc‐protected amino acid derivatives (Table 1), as well‐defined model substrates. Only amino acid residues with side chains that do not absorb in the given absorption range were considered to avoid misinterpretation in this part of the study. Recoveries in a range of 99.3% to 102.0% were obtained. For this experiment, the assay‐corrected concentrations of the respective Fmoc‐protected amino acid derivatives were compared with the concentration obtained from absorption determination at a wavelength of 289.8 nm using the previously determined molar absorption coefficient of 6089 l mol−1 cm−1.
Table 1.
Comparison of concentration determination by titrimetric and spectroscopic methods
| Fmoc‐AA‐OH | Conc. 1a | Conc. 2b | Recovery [%] |
|---|---|---|---|
| Fmoc‐His(1‐Trt)‐OH | 1.502 | 1.498 | 99.7 |
| Fmoc‐Phe‐OH | 1.507 | 1.508 | 100.1 |
| Fmoc‐Ala‐OH · H2O | 1.511 | 1.520 | 100.6 |
| Fmoc‐Gly‐OH | 1.514 | 1.517 | 100.2 |
| Fmoc‐Gln(Trt)‐OH | 1.459 | 1.471 | 100.8 |
| Fmoc‐Asp(OtBu)‐OH | 1.523 | 1.513 | 99.3 |
| Fmoc‐Glu(OtBu)‐OH · H2O | 1.517 | 1.513 | 99.7 |
| Fmoc‐Lys(Boc)‐OH | 1.516 | 1.522 | 100.4 |
| Fmoc‐Leu‐OH | 1.525 | 1.526 | 100.1 |
| Fmoc‐Met‐OH | 1.504 | 1.510 | 100.4 |
| Fmoc‐Asn(Trt)‐OH | 1.509 | 1.508 | 99.9 |
| Fmoc‐Ser(tBu)‐OH | 1.505 | 1.514 | 100.5 |
| Fmoc‐Thr(tBu)‐OH | 1.517 | 1.514 | 99.8 |
| Fmoc‐Ile‐OH | 1.495 | 1.508 | 100.9 |
| Fmoc‐Pro‐OH | 1.473 | 1.502 | 102.0 |
| Fmoc‐Val‐OH | 1.537 | 1.540 | 100.2 |
Calculated from sample weight, corrected for assay (by titration) and sample volume, [10−4 mol l−1].
Calculated from the molar absorption coefficient ε289.8 nm = 6089 l mol−1 cm−1 [10−4 mol l−1] and the determined absorption at 289.8 nm.
Formation of the dibenzofulvene–piperidine adduct is considered an equilibrium, that is, the presence of both dibenzofulvene and the dibenzofulvene–piperidine adduct is to be expected in the reaction mixture. With a 10 : 1 ratio of piperidine (0.5 M) and urethane (0.05 M) in deuterated dimethyl sulfoxide, a ratio of 12 : 88 was determined by 1H NMR 17. Compared with these conditions, substitution determination according to this publication is performed with about 20 mg peptide resin in 100 ml 20% piperidine in DMF. Assuming a resin substitution of 2.00 mmol g−1 (i.e. an extremely high resin substitution), a 5000 : 1 ratio of piperidine (2 M) and urethane (0.0004 M) is obtained. Disregarding the different solvent, an even higher excess of the dibenzofulvene–piperidine adduct may be expected because of the 500‐times higher excess of piperidine compared with the example from the literature. These considerations are in accordance to the fact that the impact of the piperidine concentration in DMF in the typically used range of 15% to 25% (v/v) on the determined molar absorption coefficient proved to be negligible, with relative deviations of 0.4% and 0.5%, respectively (Table 2). The impact of the piperidine concentration in DMF on substitution determination is thus equally small. Furthermore, the solutions used for substitution determination proved to be stable for several hours at room temperature, with a maximum relative deviation of 1.3% from the molar absorption coefficient determined after 0 h (Table 3), and excellent linearity was observed during the determination of the molar absorption coefficient (Experimental Part). Hence, even though an equilibrium is expected for the proposed conditions, the procedure itself has been shown to be very robust and reliable.
Table 2.
Influence of the piperidine concentration in DMF on determined substitutions
| Piperidine concentration [%] | Resin [mg]a | Absorption | Substitution [mmol/g] | Relative deviation [%] |
|---|---|---|---|---|
| 15 | 200.0 | 1.1742 | 0.964 | 0.4% |
| 20 | n.a. | n.a. | 0.968b | – |
| 25 | 200.4 | 1.1850 | 0.971 | 0.5% |
Fmoc‐2,4‐dimethoxy‐4′‐(carboxymethyloxy)‐benzhydrylamine linked to Aminomethyl resin
Average value from a sixfold substitution determination.
Table 3.
Stability of analytical solutions: dibenzofulvene‐piperidine adduct concentration of 15.187 · 10−5 mol l−1, determination of the molar absorption coefficient after storage at room temperature
| Time [h] | Molar absorption coefficient [l mol−1 cm−1] | Absolute deviation [l mol−1 cm−1] to 0 h | Relative deviation [%] to 0 h |
|---|---|---|---|
| 0.0 | 6108.4 | – | – |
| 0.5 | 6121.6 | 13.2 | 0.2 |
| 1.0 | 6148.0 | 39.5 | 0.6 |
| 1.5 | 6163.1 | 54.7 | 0.9 |
| 2.0 | 6190.1 | 81.7 | 1.3 |
| 2.5 | 6123.6 | 15.1 | 0.2 |
| 3.0 | 6103.2 | 5.3 | 0.1 |
| 3.5 | 6112.4 | 4.0 | 0.1 |
| 4.0 | 6064.3 | 44.1 | 0.7 |
As an alternative method, a dibenzofulvene‐based substitution determination method has been described in literature 18. The strategy to generate dibenzofulvene with DBU in the absence of a Michael donor avoids the equilibrium in the sample solution. On the other hand, an unquenched, reactive species is present that might have an impact on the results of substitution determination, for example, in case of prolonged holding times. However, for Fmoc‐Gly‐OH as a model substrate, the authors showed good reproducibility of absorption coefficient determination. Furthermore, the authors showed for several different resins good consistency with alternative methods for substitution determination.
Consequently, we were able to successfully perform a method validation according to the ICH guideline 19 using the molar absorption coefficient of 6089 l mol−1 cm−1 at a wavelength of 289.8 nm with 20% (v/v) piperidine in DMF.
Exemplary results of substitution determination for different Fmoc‐protected resins are given in Table 4. Using the maximum absorption of the respective transition from the spectrum and the given molar absorption coefficients, essentially the same substitution values are obtained for the two electronic transitions, irrespective of the exact measured wavelength.
Table 4.
Examples of substitution determination for a variety of polystyrene resins at 289.8 and 301.0 nm
| Entry | Sample weight [mg] | Abs. 289.8 nm | Abs. 301.0 nm | Subst. 289.8 nm [mmol g−1] | Subst. 301.0 nm [mmol g−1] |
|---|---|---|---|---|---|
| 1 | 11.9 | 0.459 | 0.609 | 0.63 | 0.64 |
| 2 | 9.8 | 0.570 | 0.757 | 0.96 | 0.96 |
| 3 | 17.4 | 0.778 | 1.030 | 0.73 | 0.74 |
| 4 | 17.4 | 0.573 | 0.759 | 0.54 | 0.54 |
| 5 | 19.2 | 0.885 | 1.171 | 0.76 | 0.76 |
| 6 | 15.5 | 0.467 | 0.618 | 0.49 | 0.50 |
| 7 | 18.1 | 0.514 | 0.680 | 0.47 | 0.47 |
For the substitution determination, the given amount of resin was treated with 100 ml 20% piperidine in DMF for 20–40 min at room temperature, then the UV absorption spectra between 280 and 320 nm were recorded. The maxima from the spectra corresponding to the respective transitions were taken for substitution determination using 8021 l mol−1 cm−1 (301.0 nm) and 6089 l mol−1 cm−1 (289.8 nm) as the molar absorption coefficients. Resins used: Fmoc‐Tyr(tBu)‐Wang resin (entry 1), Fmoc‐2,4‐dimethoxy‐4′‐(carboxymethyloxy)‐benzhydrylamine linked to Aminomethyl resin (entry 2), Fmoc‐L‐threoninol‐diphenylmethyl ether resin (entry 3), Fmoc‐Ser(tBu)‐Wang resin (entry 4), Fmoc‐met‐Wang resin (entry 5), tricyclic amide linker resin (DL form) (entry 6) and Fmoc‐Phe‐Wang resin (entry 7).
We provide here a rationale for the diversity of previously published molar absorption coefficients for N‐(9H‐fluoren‐9‐ylmethyl)‐piperidine in 20% (v/v) piperidine in DMF at 301.0 nm and demonstrate that UV absorbance determination at the second electronic transition at 289.8 nm is substantially more robust because of enhanced tolerance towards spectrometer wavelength and spectral bandwidth inaccuracies. The reported molar absorption coefficients of N‐(9H‐fluoren‐9‐ylmethyl)‐piperidine in 20% (v/v) piperidine in DMF at 301.0 nm and at the preferred 289.8 nm transition together with the described analytical method enable fast and precise substitution determination of Fmoc‐resins allowing for significant starting material cost reduction and enhanced SPPS process understanding.
Conclusions
For substitution determination of Fmoc‐substituted resins, the cleavage of the Fmoc group with 20% piperidine in DMF is a convenient procedure. Based on our experience regarding substitution determination at 301.0 and 289.8 nm with different UV/Vis spectrometers, we suggest the routine use of the transition at 289.8 nm for which we determined the absorption coefficient. As the band at 289.8 nm is shallower, the impact of wavelength accuracy is reduced, which allows for a more precise and robust substitution determination than in the case of the transition at 301.0 nm that is usually used. This allows for a more accurate substitution determination regardless what type of UV/Vis spectrometer is used. An accurate substitution determination procedure is of utmost importance because the resin substitution is used for the calculation of a whole set of important process parameters such as the batch size and the number of amino acid derivative equivalents, which affects further calculations such as the overall yield.
Experimental Part
Determination of the Molar Absorption Coefficient at 289.8 nm
Solutions at seven different concentration levels (4.735 to 26.477 · 10−5 mol/l) were independently prepared by dissolving different amounts of N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine (M = 263.38 g mol−1, Assay = 99.6%, obtained from Bachem AG, Switzerland) in 20% (v/v) piperidine in DMF (graduated flask, final total volume of 100 ml) and subsequent transferring aliquots of them into other graduated flasks and dilution with 20% (v/v) piperidine in DMF (final total volume of 100 ml). The absorptions at 289.8 nm were determined with a UV spectrometer (UV‐1800, Shimadzu, Quartz cuvette with optical path length = 1 cm) in triplicate. The linearity was assessed by evaluating the coefficient of determination, the slope of the regression line, y‐intercept of the regression line and the residual sum of squares from the mean of a triplicate of each concentration level by linear regression from temperature corrected extinction versus concentration [mol/l]. From the slope of the best‐fit line, a molar absorption coefficient of 6089 l mol−1 cm−1 was obtained. The Pearson coefficient of the regression line was 1.0000.
Determination of the Molar Absorption Coefficient at 301.0 nm
Dissolved in 20% (v/v) piperidine in DMF (graduated flask, final total volume of 100 ml) were 2.5, 6.4, 7.6, 11.1, 21.4, 30.7, 40.6, 52.9 and 60.2 mg N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine (M = 263.38 g mol−1, Assay = 99.6%, obtained from Bachem AG, Switzerland). About 5 ml of the respective obtained solutions were transferred into another graduated flask and diluted with 20% (v/v) piperidine in DMF (final total volume of 100 ml). As a consequence, solutions with a concentration of 4.73, 12.10, 14.37, 20.99, 40.47, 58.06, 76.78, 100.04 and 113.85 nmol ml−1 N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine in 20% (v/v) piperidine in DMF were obtained. The absorption of these solutions against 20 ml piperidine in DMF were determined with a UV spectrometer (UV‐1700, Shimadzu, Quartz cuvette with optical path length = 1 cm). In case of this measurement, the respective absorption maxima as reported by the UV spectrometer were considered. Absorption values of 0.040, 0.095, 0.112, 0.168, 0.324, 0.461, 0.611, 0.804 and 0.913 were obtained. The absorption values were plotted against the corresponding concentrations. From the slope of the best‐fit line, a molar absorption coefficient of 8021 l mol−1 cm−1 was obtained. The Pearson coefficient for this measurement was 0.9999.
Comparison of Concentration Determination by Titrimetric and Spectroscopic Methods
Therefore 235.23 mg Fmoc‐His(1‐Trt)‐OH (Assay 98.9%), 147.19 mg Fmoc‐Phe‐OH (Assay 99.2%), 50.26 mg Fmoc‐Ala‐OH · H2O (Assay 99.0%), 45.32 mg Fmoc‐Gly‐OH (Assay 99.3%), 232.06 mg Fmoc‐Gln(Trt)‐OH (Assay 96.0%), 156.82 mg Fmoc‐Asp(OtBu)‐OH (Assay 99.9%), 168.51 mg Fmoc‐Glu(OtBu)‐OH · H2O (Assay 99.8%), 71.51 mg Fmoc‐Lys(Boc)‐OH (Assay 99.3%), 54.10 mg Fmoc‐Leu‐OH (Assay 99.6%), 141.05 mg Fmoc‐Met‐OH (Assay 99.0%), 226.63 mg Fmoc‐Asn(Trt)‐OH (Assay 99.3%), 145.48 mg Fmoc‐Ser(tBu)‐OH (Assay 99.2%), 150.91 mg Fmoc‐Thr(tBu)‐OH (Assay 99.9%), 53.69 mg Fmoc‐Ile‐OH (Assay 98.4%), 127.96 mg Fmoc‐Pro‐OH (Assay 97.1%), 52.36 mg Fmoc‐Val‐OH (Assay 99.6%) were separately weighed into 50 ml volumetric flasks and dissolved in 20% (v/v) piperidine in DMF under concomitant cleavage of the Fmoc group (all Fmoc amino acid derivatives were obtained from Bachem AG, Switzerland). The flasks were filled to volume with 20% (v/v) piperidine in DMF and shaken vigorously until complete dissolution was achieved. From each 50 ml volumetric flask, 2 ml of the solution were transferred to a separate 100 ml volumetric flask, diluted to volume with 20% (v/v) piperidine in DMF and shaken vigorously. The absorptions of these solutions were measured at 289.8 nm (UV‐1800, Shimadzu, Quartz cuvette with optical path length = 1 cm). Temperature‐corrected absorption values of 0.9118 for Fmoc‐His(1‐Trt)‐OH, 0.9184 for Fmoc‐Phe‐OH, 0.9256 for Fmoc‐Ala‐OH · H2O, 0.9238 for Fmoc‐Gly‐OH, 0.8959 for Fmoc‐Gln(Trt)‐OH, 0.9211 for Fmoc‐Asp(OtBu)‐OH, 0.9212 for Fmoc‐Glu(OtBu)‐OH · H2O, 0.9267 for Fmoc‐Lys(Boc)‐OH, 0.9290 for Fmoc‐Leu‐OH, 0.9192 for Fmoc‐Met‐OH, 0.9181 for Fmoc‐Asn(Trt)‐OH, 0.9216 for Fmoc‐Ser(tBu)‐OH, 0.9220 for Fmoc‐Thr(tBu)‐OH, 0.9181 for Fmoc‐Ile‐OH, 0.9145 for Fmoc‐Pro‐OH and 0.9402 for Fmoc‐Val‐OH were obtained. From the photometric assays, the theoretical initial concentrations were calculated using the molar extinction coefficient of 6089 l mol−1 cm−1. With the photometric assay, the percental recoveries against the assays (by potentiometric titration with tetrabutylammonia hydroxide solution 0.1 mol/l; results were taken from the latest Bachem analytical data sheet of the corresponding Fmoc‐AA‐OH) were calculated.
Influence of the Piperidine Concentration in DMF on Determined Substitutions.
About 200.0 mg Fmoc‐2,4‐dimethoxy‐4′‐(carboxymethyloxy)‐benzhydrylamine linked to Aminomethyl resin (100–200 mesh) were weighed in a 100 ml graduated flask and filled to approximately 60 ml with 15% (v/v) piperidine in DMF. About 200.4 mg Fmoc‐2,4‐dimethoxy‐4′‐(carboxymethyloxy)‐benzhydrylamine linked to Aminomethyl resin (100–200 mesh) were weighed in a 100‐ml graduated flask and filled to approximately 60 ml with 25% (v/v) piperidine in DMF. Both flasks were shaken at room temperature for 20 min (i.e. the cleavage time). Afterwards, the flasks were diluted to volume with 15% (v/v) and 25% (v/v) piperidine in DMF, respectively, and shaken vigorously. Each solution was filtered through a glass funnel with a folded filter (Schleicher&Schuell LS 14 1/2 150 mm). About 10.00 ml of each filtrate was pipetted in a separate 100 ml volumetric flask, diluted to volume with 20% (v/v) piperidine in DMF and shaken vigorously. The absorptions of these solutions were measured at 289.8 nm (UV‐1800, Shimadzu, Quartz cuvette with optical path length = 1 cm).
Stability of Analytical Solutions
About 200.76 mg N‐(9H‐Fluoren‐9‐ylmethyl)‐piperidine (M = 263.38 g mol−1, Assay = 99.6%) were dissolved in 20% (v/v) piperidine in DMF (graduated flask, final total volume 100 ml). About 5 ml of the solution was transferred into another graduated flask and diluted with 20% (v/v) piperidine in DMF (final total volume of 100 ml), producing a final concentration of 15.184 · 10−5 mol/l. The absorption of this solution was measured at 289.8 nm (UV‐1800, Shimadzu, Quartz cuvette with optical path length = 1 cm) after 0.5, 1.0, 2.0 2.5, 3.0, 3.5 and 4.0 h.
Examples of Substitution Determination for a Variety of Resins at 289.8 and 301.0 nm
From 9.8 to 19.2 mg of the analyzed resin were transferred into a 100‐ml graduated flask. The flask was filled to the mark with 20% (v/v) piperidine in DMF. The resin was treated for ≥4 h, then a sample from the supernatant was transferred to a Quartz cuvette and the absorption was measured against 20% (v/v) piperidine in DMF with a UV spectrometer (UV‐1700, Shimadzu, Quartz cuvette with optical path length = 1 cm). For this measurement, the respective absorption maxima as reported by the UV spectrometer were considered. The sample weights, the obtained absorptions at the given maxima for the given wavelengths and the calculated substitution are provided in the accessory information section.
Supporting information
Data S1. Supporting info item
Eissler, S. , Kley, M. , Bächle, D. , Loidl, G. , Meier, T. , and Samson, D. (2017) Substitution determination of Fmoc‐substituted resins at different wavelengths. J. Pept. Sci., 23: 757–762. doi: 10.1002/psc.3021.
References
- 1. Merrifield RB. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963; 85: 2149–2154. [Google Scholar]
- 2. Carpino LA, Han GY. The 9‐Fluorenylmethoxycarbonyl function, a new base‐sensitive amino protecting group. J. Am. Chem. Soc. 1970; 92: 5748–5749. [Google Scholar]
- 3. Carpino LA, Han GY. 9‐Fluorenylmethoxycarbonyl amino‐protecting group. J. Org. Chem. 1972; 37: 3404–3409. https://doi.org/10.1021/jo00795a005. [Google Scholar]
- 4. Atherton E, Fox H, Harkiss D, Logan CJ, Sheppard RC, Williams B, A mild procedure for solid phase peptide synthesis: use of fluorenylmethoxycarbonylamino‐acids . J. Chem. Soc. Chem. Commun. 1978; 537–539. https://doi.org/10.1039/C39780000537. [Google Scholar]
- 5. Chang C‐D, Waki M, Ahmad M, Meienhofer J, Lundell EO, Haug J. Preparation and properties of Nalpha‐9‐fluorenylmethyloxycarbonylamino acids bearing tert.‐butyl side chain protection. Int. J. Pept. Prot. Res. 1980; 15: 59–66. https://doi.org/10.1111/j.1399‐3011.1980.tb02550.x. [DOI] [PubMed] [Google Scholar]
- 6. Atherton E, Logan CJ, Sheppard RC, Peptide synthesis. Part 2. Procedures for solid‐phase synthesis using N ‐fluorenylmethoxycarbonylamino‐acids on polyamide supports. Synthesis of substance P and of acyl carrier protein 65–74 decapeptide . J. Chem. Soc. Perkin Trans. I 1981; 538–546. [Google Scholar]
- 7. Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9‐fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Prot. Res. 1990; 35: 161–214. https://doi.org/10.1111/j.1399‐3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 8. Behrendt R, White P, Offer J. Advances in Fmoc solid‐phase peptide synthesis. J. Pept. Sci. 2016; 22: 4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bernatowicz MS, Daniels SB, Köster H. A comparison of acid labile linkage agents for the synthesis of peptide C‐terminal amides. Tetrahedron Lett. 1989; 30: 4645–4648. https://doi.org/10.1016/S0040‐4039(01)80764‐4. [Google Scholar]
- 10. Ramage R, Irving SL, Mclnnes C. Design of a versatile linker for solid phase peptide synthesis: Synthesis of C‐terminal primary/seconary amides and hydrazides. Tetrahedron Lett. 1993; 34: 6599–6602. https://doi.org/10.1016/0040‐4039(93)88115‐Y. [Google Scholar]
- 11. Meienhofer J, Waki M, Heimer EP, Lambros TJ, Makofske RC, Chang C‐D. Solid phase synthesis without repetitive acidolysis. Int. J. Pept. Protein Res. 1979; 13: 35–41. [PubMed] [Google Scholar]
- 12. Romieu A, Bruckdorfer T, Clavé G, Grandclaude V, Massifa C, Renard P‐Y. N‐Fmoc‐alpha‐sulfo‐beta‐alanine: a versatile building block for the water solubilisation of chromophores and fluorophores by solid‐phase strategy. Org. Biomol. Chem. 2011; 15: 5337–5342. https://doi.org/10.1039/c1ob05730h. [DOI] [PubMed] [Google Scholar]
- 13. Eichler J, Bienert M, Stierandová A, Lebl M. Evaluation of cotton as a carrier for solid‐phase peptide synthesis. Pept. Res. 1991; 4: 296–307. [PubMed] [Google Scholar]
- 14. Instruction Manual System User's Guide UV-1800 Shimadzu Spectrophotometer, 2008, 6.1.1 Hardware Specifications, Page 6–2.
- 15. Agilent 8453 UV‐visible Spectroscopy System ‐ Service Manual ‐ Edition 02/00, 2000, 12.
- 16. Wills BG. Design and performance of a highly integrated parallel access spectrometer. Hewlett‐Packard J. 1980; 2: 3. [Google Scholar]
- 17. Carpino LA, Mansour EME, Knapczyk J. Piperazino‐functionalized silica gel as a deblocking‐scavenger agent for the 9‐Fluoromethyloxycarbonyl amino‐protecting group. J. Org. Chem. 1983; 48: 666–669. [Google Scholar]
- 18. Gude M, Ryf J, White PD. An accurate method for the quantitation of Fmopc‐derivatized solid supports. Lett. Pept. Sci. 2002; 9: 203–206. [Google Scholar]
- 19. ICH Harmonised Tripartite Guideline (Q2(R1)) ‘Validation of Analytical Procedures: Text and Methodology’ (November 2005).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting info item