

Abstract
CC‐292, a potent Bruton tyrosine kinase inhibitor, is under development for the treatment of B‐cell malignancies. An analysis was performed to develop a population pharmacokinetic model of CC‐292 and assess the influence of demographics and disease‐related covariates on CC‐292 exposure and to assess the exposure‐response (overall response rate) relationship in patients with chronic lymphocytic leukemia. Population pharmacokinetic analysis was based on a 2‐compartment base model conducted in NONMEM. Categorical exposure‐response analysis was performed using logistic regression in SAS. The population pharmacokinetic analysis results indicated that CC‐292 pharmacokinetic disposition is similar between healthy subjects and patients. CC‐292 showed a larger central compartment volume of distribution than the peripheral compartment volume of distribution (158 L and 72 L, respectively) and a faster clearance than intercompartmental clearance (134 L/h and 18.7 L/h, respectively), indicating that for CC‐292, clearance from blood occurs faster than distribution into deep tissues and organs. CC‐292 clearance is not affected by demographics or baseline clinical lab factors, except for sex. Although sex significantly reduced variation of apparent clearance, the sex effect on apparent clearance is unlikely to be clinically relevant. The exposure‐response analysis suggested that higher drug exposure is linearly correlated with higher overall response rate. A twice‐daily dose regimen showed higher overall response rate as compared to once‐daily dosing, consistent with a threshold concentration of approximately 300 ng/mL, above which the probability of overall response rate significantly increases.
Keywords: CC‐292, population pharmacokinetics, exposure‐response assessment, chronic lymphocytic leukemia, threshold concentration
The importance of B‐cell receptor signaling to the growth and survival of distinct subtypes of B‐cell lymphomas as well as other B‐cell malignancies has been demonstrated,1, 2 and B‐cell receptor signaling modulation has been validated for the treatment of B‐cell malignancies as well as a variety of B‐cell–dependent autoimmune disorders.1, 2 Bruton tyrosine kinase (BTK) is a cytoplasmic, nonreceptor tyrosine kinase that transmits signals from a variety of cell‐surface molecules, including B‐cell receptor and tissue homing receptors.3 BTK is an integral component of the B‐cell receptor signaling complex with expression limited primarily to B lymphocytes, mast cells, monocytes, and osteoclasts4, 5 and plays an important role in the B‐cell signaling pathway linking cell surface B‐cell receptor stimulation to downstream intracellular responses.6, 7, 8 The highly restricted expression pattern of BTK together with the prominent role of BTK in the B‐cell receptor signaling pathway makes it an attractive drug target for the treatment of B‐cell malignancies. It has been reported that numerous B‐cell‐derived malignancies, such as acute lymphoblastic leukemia, chronic lymphocytic leukemia (CLL), non‐Hodgkin lymphoma, mantle‐cell lymphoma, Waldenstrom macroglobulinemia, and multiple myeloma, are dependent on disregulation of BTK kinase activity.8, 9, 10 Ibrutinib, a novel first‐in‐class BTK inhibitor, has been approved by various regulatory agencies for the treatment of mantle‐cell lymphoma, CLL, and Waldenstrom macroglobulinemia.11 More selective and potent BTK inhibitors are currently being investigated clinically.3, 12, 13
CC 292 is a potent, selective, orally administered small‐molecule inhibitor of BTK. CC 292 inhibits BTK activity by binding with high affinity to the adenosine triphosphate binding site of BTK and forming a covalent bond with the target BTK protein, providing rapid, complete, and prolonged inhibition of BTK activity, both in vitro and in vivo.14, 15, 16, 17 In animals, CC‐292 was rapidly absorbed, and the extent of bioavailability ranged from low (11%) in monkeys to moderate (57%) in rats. Mass balance study in rats showed that CC‐292–derived radioactivity was primarily excreted in the bile (∼79% of the dose), with urinary excretion being a minor pathway. CC‐292 is extensively metabolized by CYP3A4 and CYP1A2, and no major metabolite showed inhibition on the BTK activity (data on file).
Pharmacokinetics of CC‐292 have been determined in both healthy subjects (data on file) and patients with relapsed and/or refractory B‐cell malignancies in clinical studies.14 In general, CC‐292 showed similar pharmacokinetic disposition properties in healthy subjects and patients. In healthy subjects, CC‐292 is rapidly absorbed with a time to peak concentration that ranges from 0.34 to 2 hours. A multiple ascending dose study (50, 100, and 200 mg) demonstrates that the drug does not accumulate or dissipate with multiple dosing. After reaching their peak, plasma concentrations decline rapidly with median steady‐state terminal half‐life estimates for CC‐292 ranging from 1.1 to 2.8 hours independent of dose.
Pharmacokinetic results of CC‐292 in patients with relapsed and/or refractory B‐cell malignancies following a CC‐292 once‐daily (QD) dose regimen (125, 250, 400, 625, 750, and 1000 mg) showed that the plasma concentration‐vs‐time profiles were characterized by a rapid absorption phase at all tested dose levels. The mean apparent elimination half‐lives were from 2 hours to 3 hours, similar at all tested dose levels. Area under the plasma concentration‐time curve from time 0 to 24 hours (AUC0‐24) and maximum plasma drug concentration increased in a dose‐proportional manner over the 125 to 1000 mg dose range. Moderate to high between‐subject variability was noted for both AUC0‐24 and peak plasma concentration (coefficient of variation ranging from 37.4% to 82.6%). Pharmacokinetic results of CC‐292 in patients with relapsed and/or refractory B‐cell malignancies following a twice‐daily (BID) dose regimen (375 mg and 500 mg) showed linear pharmacokinetics similar to that observed from QD dose regimens.
Based on cumulative data in healthy subjects and patients with relapsed and/or refractory B‐cell malignancies, analyses were conducted to build a population pharmacokinetic and covariate model that quantitatively describes the disposition of CC‐292 and to characterize the major sources of variability on CC‐292 pharmacokinetic exposure. In addition, a logistic regression model was constructed to quantify the relationship between CC‐292 exposure and antitumor efficacy of CC‐292 from a phase 1b escalating‐dose study.
Methods
Patients and Study Design
The studies were approved by the institutional review boards of the participating centers and conducted according to the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. All subjects gave written informed consent prior to enrollment. CC‐292 was administered orally as a solid dosage form either QD or BID in the 2 studies described below.
AVL‐292‐004 was a phase 1, 2‐part study conducted in healthy adult subjects. Part 1 was a randomized, placebo‐controlled, double‐blind, multiple‐dose study to evaluate the safety, tolerability, and pharmacokinetics of CC‐292. Thirty‐two subjects were enrolled and randomized at a 1:1:1:1 ratio into 4 parallel groups to receive 1 of the 4 treatments (Treatment A, 50 mg; Treatment B, 100 mg; Treatment C, 200 mg; and Treatment D, placebo) as oral doses QD for 7 days. Part 2 of the study was an open‐label, randomized, 2‐period, 2‐way crossover study in 10 subjects, to evaluate the effect of a standard high‐fat breakfast on the pharmacokinetics of a single 200‐mg dose of CC‐292 (Treatment G, a single 200‐mg dose under fasted condition; Treatment H, a single 200‐mg dose under fed condition). In part 1, intensive pharmacokinetic samples were collected in each period at predose, 0.5, 1, 2, 4, 8, 12, and 24 hours on days 1 (parts 1 and 2) and 7 (part 1). In addition, sparse pharmacokinetic samples were collected at predose (trough concentration) on days 3, 4, 5, and 6.
AVL‐292‐003 was a phase 1b, escalating‐dose study of CC‐292 as monotherapy in subjects with relapsed and/or refractory B‐cell non‐Hodgkin lymphoma, CLL, and Waldenstrom macroglobulinemia.14 The study was conducted in 2 parts. In part 1, 61 subjects with B‐cell non‐Hodgkin lymphoma, CLL/small lymphocytic lymphoma (SLL), or Waldenstrom macroglobulinemia were enrolled into a series of escalating‐dose cohorts: 125 mg QD (n = 3), 250 mg QD (n = 3), 400 mg QD (n = 6), 625 mg QD (n = 6), 750 mg QD (n = 6), 1000 mg QD (n = 13), 375 mg BID (n = 12), and 500 mg BID (n = 12). The dose escalation in part 1 followed a 3 + 3 study design. Part 2 of the study enrolled 52 subjects with relapsed and/or refractory CLL/SLL alone into 2 doses/regimens: 750 mg QD (n = 25) and 500 mg BID (n = 27). A total of 113 patients received continual dosing with CC‐292 in 28‐day cycles at doses ranging from 125 mg to 1000 mg QD and 375 mg and 500 mg BID, continuing into dose expansion cohorts of 750 mg QD and a preliminary recommended phase 2 dose expansion cohort of 500 mg BID. A secondary objective was to characterize preliminary antitumor efficacy of CC‐292 against relapsed and/or refractory B‐non‐Hodgkin lymphoma, CLL, and Waldenstrom macroglobulinemia. Intensive pharmacokinetic samples were collected at predose, 0.5, 1, 2, 4, 6, and 8 hours on cycle 1 day 1 and cycle 1 day15. Sparse pharmacokinetic samples were collected on cycle 1 day 2 (predose), cycle 1 day 8 (predose), cycle 1 day 16 (predose and 1 hour postdose), cycle 1 day 22 (predose), cycle 2 day 1 (predose, 1 and 4 hours postdose), and cycle 3 day 1 (predose).
Pharmacokinetic data from AVL‐292‐004 (part 1) and AVL‐292‐003 studies were combined in the population pharmacokinetic analysis. Response assessment for CLL/SLL patients (according to the updated International Workshop on Chronic Lymphocytic Leukemia) from AVL‐292‐003 study was employed in the exposure‐response analysis.
Bioanalytical Methodology
Validated liquid chromatography‐tandem mass spectrometry methods with lower limits of quantification of ≤0.5 ng/mL were used to determine CC‐292 concentrations in human plasma samples. Plasma samples were spiked with stable labeled CC‐292 (as an internal standard) processed by liquid‐liquid extraction and analyzed using reversed‐phase high‐performance liquid chromatographywith electrospray mass spectrometry detection. Peak separation was achieved using high‐performance liquid chromatography with a gradient of organic solvent and aqueous mobile phases.
Population Pharmacokinetic Model Building
Population pharmacokinetic analysis of the concentration‐time data of CC‐292 was performed using the nonlinear mixed‐effect modeling program (NONMEM version 7.2; Icon Development Solutions, Ellicott City, Maryland) with first‐order conditional estimation with the interaction option throughout population pharmacokinetic data analysis. CC‐292 concentration data were ln‐transformed. The S‐Plus (version 8.2, TIBCO Software Inc, Somerville, Massachusetts) and R‐based model building aid Perl‐Speaks‐NONMEM (PsN, version 3.5.3, by Kajsa Harling and Andrew Hooker, Department of Pharmacy, Uppsala University, Uppsala, Sweden) postprocessing software were used for graphic data processing. NONMEM was installed on Windows XP with the Intel Visual FORTRAN Compiler (version 9.1). Comparison of base models was based on the objective function value and goodness‐of‐fit criteria. A value of P < .001, representing a decrease in objective function value >10.83, was considered statistically significant. Selection criteria during the model development process were based on goodness‐of‐fit plots, changes in objective function value, residual distributions, parameter estimates, and their relative standard error values.
Population pharmacokinetic model building started with a 1‐compartment model and tested 2‐ and 3‐compartment base pharmacokinetic models. On the basis of visual data plots and prespecified data‐fitting criteria, CC‐292 concentration‐time data were best described by a 2‐compartment base pharmacokinetic model with the first‐order absorption rate constant, absorption lag time, apparent clearance (CL/F), apparent central compartment volume of distribution (V2/F), apparent intercompartmental clearance between central and peripheral compartments (Q/F), and peripheral volume of distribution (V3/F).
Assuming a log‐normal distribution for interindividual variability in pharmacokinetic parameters, the interindividual variability was modeled as follows:
| (1) |
where P is the typical value of the parameter in the population, Pi is the value of the parameter for the i‐th individual, and ηi is a random interindividual effect in the parameter for the i‐th subject with a mean of 0 and variance ω2 (ie, η ∼ N[0, ω2]).
Intraindividual or residual variability was modeled as follows:
| (2) |
where Cmij is the model‐predicted j‐th concentration in the i‐th subject, Cij is the observed j‐th concentration in the i‐th subject, and εij is the random residual effect for the j‐th concentration in the i‐th subject with a mean of 0 and variance of σ2. Given that the studies conducted in healthy subjects are well controlled vs patient studies, assumption of the same residual variability for all individuals may result in biased parameter estimates. To reduce this possible bias, residual variability was modeled separately for healthy subjects and patients with relapsed and/or refractory B‐cell malignancies.
Covariate Analysis
Demographics and disease covariates were tested for their correlation with all pharmacokinetic parameters of the 2‐compartment model, including age, body weight, body surface area, sex, race, hepatic function markers (total bilirubin, albumin, aspartate aminotransferase, or other markers as appropriate), renal function markers (creatinine clearance [CLcr] estimated by Cockcroft‐Gault formula),18, 19, 20 and status of health (healthy subjects vs patients). Covariates were initially selected by graphic inspection and biological plausibility. Further testing of potential covariates was performed by a 3‐stage approach for the selection of covariates.
First, covariates identified by graphic analysis were introduced into the base model individually for univariate analysis. In the second step (forward selection [P < .05]), the covariate with the highest significance by univariate analysis was included first, and other significant covariates from univariate analysis were included in rank order of their significance. In the third step (backward elimination [P < .005]), covariates were removed from the full model obtained from forward selection, in sequence, until there were no further insignificant covariates remaining.
The stepwise covariate model building tool of PsN was used for development of the CC‐292 covariate model, which implemented forward selection and backward elimination of covariates for the CC‐292 population pharmacokinetic model. There is a fixed set of pharmacokinetic parameter covariate relations defined in the stepwise covariate modeling; predefined shapes for the parameter‐covariate relations for continuous covariates for CC‐292 covariate model development include the following:
Linear equation
| (3) |
and Power equation
| (4) |
where P is the typical value of a pharmacokinetic parameter in the population after adjusting for values of covariates of individual subjects, θ is the typical value of the pharmacokinetic parameter, θcov is the coefficient for the effect of the covariate, COVi is the covariate value for individual subjects, and COVm is the median value of covariates in the study population.
Categorical covariates included in the CC‐292 covariate model development were as follows:
| (5) |
where Zind,k is an indicator variable representing 1 from of a binary covariate, and θcov is the coefficient for the effect of the covariate.
Model Evaluation
Model evaluation was performed using traditional visual predictive check script in Perl speaks NONMEM (PsN, version 3.5.3, by Kajsa Harling and Andrew Hooker, Department of Pharmacy, Uppsala University, Uppsala, Sweden), which provided an evaluation of model assumption and population parameter estimates by comparing model predictions with observations. The ability of the final population pharmacokinetic model to describe the observed concentration data was evaluated by simulating 1000 data sets with the same doses, dosing schedules, and sampling times as in the original data set and by performing visual predictive checks.
Stability of the final pharmacokinetic parameter estimates and 90%CIs was evaluated using the nonparametric bootstrap approach. The final model was fit to each of the 1000 bootstrap data sets, and all the model parameters were estimated for each data set; 96% of the 1000 bootstrap runs were successful. Median and nonparametric 90%CIs (5th‐95th percentiles) for each of the 1000 estimates were calculated for each parameter.
Exposure‐Response Analysis
Exposure‐response analysis (logistic regression) was performed using SAS (version 9.2; SAS Institute, Cary, North Carolina). CC‐292 is an irreversible inhibitor that binds to the adenosine triphosphate site of BTK and forms a covalent bond with cysteine 481 near the active site.14 Although the plasma half‐life is short, its pharmacokinetic effect is durable because CC‐292 is an irreversible BTK inhibitor. The exposure‐response analysis assumed the drug exposure may be related to its efficacy.
Intensive pharmacokinetic samples were collected for all patients from the AVL‐292‐003 study. The exposure metric logarithm‐transformed AUC at steady state (AUCss) was calculated by noncompartmental analyses. The efficacy end point in CLL/SLL patients from the AVL‐292‐003 study was overall response rate (ORR), defined as the proportion of subjects achieving a best overall response of complete response (disappearance of all evidence of disease) (CR); CR with incomplete bone marrow recovery (CRi), or partial response (PR) based on updated International Workshop response criteria for patients with CLL/SLL.
The covariates tested in the univariate and multivariate logistic regression analyses included the following: age, body surface area, sex, race, dose regimen (QD or BID), CLcr, total bilirubin, albumin, alanine aminotransferase, and aspartate aminotransferase.
Initially, a univariate analysis to assess the relationship of CC‐292 exposure parameters with the response was conducted. Logistic regression model was used for binary end points to describe the relationship between systemic CC‐292 exposure and probability of the event (responder vs nonresponder). The probability that the event occurred as a function of independent variables was described as follows:
| (6) |
where Pi is the probability of the event in the i‐th patient and α is the baseline Ln (odds) of the event. The β1…βn are adjusted odds ratios characterizing the dependence of the Ln (odds) on 1 or more covariates, X1…Xn.
The estimate of slope (for the logistic regression model) and associated P (from likelihood ratio test) were used to determine if an exposure metric was statistically significantly correlated with an increase in response rate.
End points exhibiting a statistically significant relationship to CC‐292 exposure were further evaluated with the multivariate logistic regression model to assess the effects of covariates in conjunction with CC‐292 exposure on response. Statistically significant covariate effects were identified in the multivariate logistic regression analysis by the use of an automated search procedure. Then, a stepwise forward/backward elimination algorithm was used to add covariates to the model in stages.
Threshold Analysis
A threshold analysis21 was conducted to explore whether there was a CC‐292 concentration level above which a correlation with clinical response could be detected. Briefly, the cumulative duration above a threshold concentration at the steady‐state exposure was computed based on approximately 100 concentration cutoff values, ranging from 50 ng/mL to 500 ng/mL with an increment of 5 ng/mL for each patient. Univariate logistic regression was performed for the duration above each concentration cutoff level vs the clinical response of ORR for approximately 100 data sets to test the statistical significance in predicting the probability of response. Model evaluation and diagnostics including goodness of fit of a likelihood ratio test, Wald test, validation of predicted values using receiver operating characteristic curve from each logistic regression were conducted. The duration above the cutoff value that provides the best statistically significant correlation with the clinical response of ORR and the best goodness of fit will be identified as the threshold concentration.
Results
Data Included in the Analyses and Demographics
A total of 145 subjects with 3156 observations from AVL‐292‐004 and AVL‐292‐003 studies were included in the final population pharmacokinetic analysis data set. Baseline characteristics are summarized in Table 1. Twenty‐five subjects (17.2%) had renal impairment (CLcr 30 mL/min to 60 mL/min), and no subjects had severe renal impairment (CLcr < 30 mL/min).
Table 1.
Demographic and Baseline Characteristics of the Pharmacokinetic Patient Population
| Variable | Combined | AVL‐292‐004 | AVL‐292‐003 |
|---|---|---|---|
| Demographics | |||
| Sex, n (%) | |||
| Male | 87 (60%) | 23 (72%) | 64 (57%) |
| Female | 58 (40%) | 9 (28%) | 49 (43%) |
| Race, n (%) | |||
| Asian | 2 (1.4%) | 0 (0%) | 2 (2%) |
| Black | 20 (13.8%) | 14 (44%) | 6 (5%) |
| Other | 2 (1.4%) | 0 (0%) | 2 (2%) |
| White | 121 (83.4%) | 18 (56%) | 103 (91%) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 15 (10.3%) | 9 (28%) | 6 (5%) |
| Not Hispanic or Latino | 130 (89.7%) | 23 (72%) | 107 (95%) |
| Patient characteristics, median (range) | |||
| Age, year | 62.0 (20.0, 89.0) | 40.5 (20.0, 65.0) | 66.0 (29.0, 89.0) |
| Body weight, kg | 79.5 (49.9, 128.4) | 80.0 (55.1, 108.7) | 79.4 (49.9, 128.4) |
| Height, cm | 170.0 (149.0, 200.0) | 171.4 (153.0, 187.2) | 169.4 (149.0, 200.0) |
| Body mass index, kg/m2 | 27.0 (19.0, 41.0) | 27.5 (20.5, 32.9) | 26.8 (19.0, 41.0) |
| Hepatic function, median (range) | |||
| Albumin, g/dL | 4.3 (2.6, 5.0) | 4.2 (3.5, 4.8) | 4.3 (2.6, 5.0) |
| Alkaline phosphatase, IU/L | 77.0 (37.0, 294.0) | 62.0 (39.0, 103.0) | 82.0 (37.0, 294.0) |
| Alanine aminotransferase IU/L | 19.0 (4.0, 87.0) | 20.0 (12.0, 36.0) | 18.0 (4.0, 87.0) |
| Aspartate aminotransferase, IU/L | 22.0 (8.0, 70.0) | 23.0 (16.0, 30.0) | 22.0 (8.0, 70.0) |
| Total bilirubin, mg/dL | 0.4 (0.1–1.3) | 0.6 (0.4–1.3) | 0.3 (0.1–1.2) |
| Lactase dehydrogenase IU/L | 190.0 (95.0, 2000.0) | 133.0 (98.0, 173.0) | 229.0 (95.0, 2000.0) |
| Total protein, g/dL | 6.7 (4.6, 9.9) | 7.5 (6.5, 8.1) | 6.5 (4.6, 9.9) |
| Renal function, median (range) | |||
| Serum creatinine, mg/dL | 0.93 (0.45, 1.65) | 0.93 (0.60, 1.34) | 0.94 (0.45, 1.65) |
| Creatinine clearance, mL/min | 86.3 (31.0, 193.3) | 112.0 (70.7, 157.8) | 77.9 (31.0, 193.3) |
Base Pharmacokinetic Model Characterization
Visual examination of dose‐normalized concentration‐vs‐time profiles (Figure 1) showed similar pharmacokinetic profiles in patients with relapsed and/or refractory B‐cell malignancies vs healthy normal subjects. Concentration‐time data of CC‐292 were best described by a 2‐compartment model, which was preferred over a 1‐ or 3‐compartment model. Therefore, a 2‐compartment model containing the same Q/F, CL/F, V2/F, and V3/F values was tested and identified as the base pharmacokinetic model. Introducing an absorption lag time significantly improved the model fit according to goodness‐of‐fit and statistical criteria. Considering the inherently better quality of pharmacokinetic data collected from well‐controlled studies in healthy subjects compared with those from patient studies, a different residual error model was tested and preferred over the model with the same residual error model.
Figure 1.
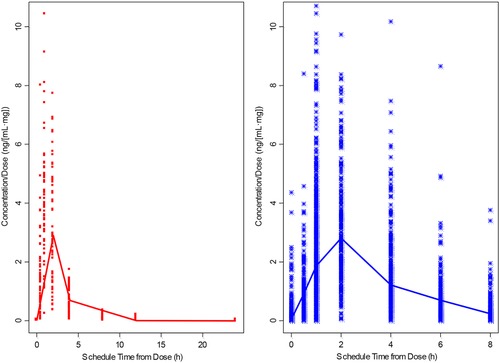
Individual dose‐normalized CC‐292 concentration (from both single‐ and multiple‐dose PK)‐vs‐time profiles: healthy normal subjects (left) vs patients (right) with relapsed and/or refractory B‐cell malignancies. PK indicates pharmacokinetics.
This 2‐compartment model well described CC‐292 pharmacokinetics in both healthy subjects and patients with relapsed and/or refractory B‐cell malignancies and was selected as the final base population pharmacokinetic model. Final population variable estimates are presented in Table 2. The population pharmacokinetic analysis revealed that healthy subjects and patients with relapsed and/or refractory B‐cell malignancies have a larger central compartment volume of distribution than the peripheral compartment volume of distribution (158 L and 72 L, respectively) and a faster elimination clearance value than intercompartmental clearance (134 L and 18.7 L/h, respectively).
Table 2.
Population Pharmacokinetic Parameters for the Final Population Pharmacokinetic Model of CC‐292
| Pharmacokinetic Parameter | Estimate | Bootstrap Estimate (95%CI) | Shrinkage (%) |
|---|---|---|---|
| ka, h−1 | 0.974 | 0.97 (0.86‐1.11) | |
| V2/F, L | 158.0 | 156.4 (130.2‐187.8) | 8.54 |
| V3/F, L | 72.0 | 71.7 (61.4‐83.5) | |
| Q/F, L/h | 18.7 | 18.4 (15.3‐21.7) | |
| CL/F, L/h | 134.0 | 131.9 (109.0‐166.6) | 3.63 |
| Alag1, h | 0.427 | 0.428 (0.414‐0.439) | |
| Effect of sex on CL/F | 0.26 | 0.26 (0.09‐0.45) | |
| Effect of age on V2/F | 0.946 | 0.946 (0.592‐1.355) | |
| Interindividual variability | |||
| ω2 V2/F | 0.755 | 0.750 (0.526‐1.010) | |
| ωV2/F: ωCL/F | 0.328 | 0.321 (0.198‐0.461) | |
| ω2 CL/F | 0.317 | 0.306 (0.202‐0.454) | |
| Residual variability | |||
| HNP | 0.234 | 0.230 (0.180‐0.288) | 6.49 |
| Patients | 0.659 | 0.654 (0.537‐0.708) | 4.21 |
Alag1, absorption lag time; CL/F, apparent clearance; HNP, healthy normal study subject; ka, first‐order absorption rate constant; Q/F, apparent intercompartmental clearance between central and peripheral compartments; V2/F, apparent central compartment volume of distribution; V3/F, apparent peripheral compartment volume of distribution.
Covariate Analysis
The majority of the subjects in the population pharmacokinetic data set were white (83.4%); therefore, all nonwhite patients were grouped as 1 population for the covariate analysis of race. Exploratory graphic analysis demonstrated that female study subjects appeared to have a lower CL/F than male subjects, and there was a correlation between the age and the central volume of distribution.
All proposed covariates were included in covariate model development using the stepwise covariate model‐building tool of PsN. The output of the stepwise covariate model‐building log file indicated that inclusion of sex into CL/F and age into V2/F in the forward selection step significantly improved the model fit. Both linear and power equations were tested for the V2/F vs age relationships. A power equation between V2/F and age significantly improved the model fit by decreasing objective function value from −2758 to −2722.
By graphic analysis, although body weight and albumin appeared to be positively correlated with CL/F, and age appeared to be negatively correlated with CL/F, these correlations were not statistically significant in the forward selection step. The final model was identified through a backward elimination process. No covariates identified from the forward selection step were removed from the full model.
The final covariate model at the population level was described as follows:
and
Typical values of CC‐292 CL/F and V2/F for male subjects with a median age of 62 years were 134 L/h and 158 L, respectively. The final population pharmacokinetic model suggested that female subjects had 26% lower CL/F compared to male subjects, and V2/F increases with increasing age. Despite the statistically significant effect of sex on CL/F and the effect of age on V2/F, the contribution of sex and age to interindividual variability was marginal. Separating female from male reduced the interindividual variability for CL/F from 58% in the base model to 56% in the final model, and including age reduced interindividual variability for V2/F from 88% in the base model to 86%. Therefore, neither of these covariates (sex on CL/F and age on V2/F) was deemed to be clinically relevant for CC‐292 exposure.
Model Evaluation
The final population pharmacokinetic model was subjected to a bootstrap resampling stability test to assess its robustness. Bootstrap analyses (N = 1000) were performed using the final population pharmacokinetic model, with 959 (95.9%) successfully minimized. As shown in Table 2, median values of the parameters obtained from bootstrap replications were similar to the original NONMEM estimates. The relative difference between the final model estimate and the bootstrap median was ≤15% for the fixed‐effect parameters and ≤4% for the random‐effect parameters, suggesting that the final model is robust and stable.
The results from goodness‐of‐fit plots suggested that there was good agreement in the time course and central tendency between distributions of observed and simulated data, with no obvious bias (data not shown). The results of the visual predictive check evaluation are presented in Figure 2. Approximately 90% of the observed concentration data were well contained within the 90% prediction intervals. The 5th, 50th, and 95th percentiles of the observed concentration data at each time point were generally contained within the respective 95%CI (shaded area, pink for the 50th percentile and blue for 5th and 95th percentiles) of the simulated data. There was a good agreement in the time course and central tendency between distributions of observed and simulated data, with no obvious bias. Overall, the estimated interindividual variability adequately described observed variability in CC‐292 concentrations. As a result, CC‐292 concentrations in the logarithmic range of –0.67 to 9.152 (ie, 0.51 ng/mL to 9440 ng/mL) were well characterized by the final population pharmacokinetic model.
Figure 2.
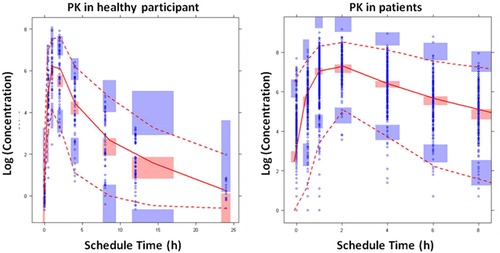
Visual predictive checks for the time profiles of CC‐292 concentrations (all dose levels). Blue circles represent observed data. Lines represent the 5th (dashed), 50th (solid), and 95th (dashed) percentiles of the observed data. Shaded areas represent nonparametric 95% confidence intervals about the 5th (blue), 50th (pink), and 95th (blue) percentiles for the corresponding model‐predicted percentiles.
Exposure Response Assessment and Threshold Analysis for CLL/SLL Patients
A summary of best overall response parameters by dose cohort and baseline diagnosis is provided for the Efficacy population in Table 3. For subjects with a baseline diagnosis of CLL/SLL, the ORR was 50.0% (41 subjects with a best response of PR and no subjects with CR). Stable disease was the best response in 40 subjects (48.8%). One subject with CLL/SLL in the 750 mg QD cohort had a best response of progression disease. The response rates were higher in the higher dose cohorts, suggesting a dose response (although the number of subjects in the lower dose cohorts was too small to evaluate properly). The exposure‐response relationship from a total of 73 subjects with a baseline diagnosis of CLL/SLL treated with 750 mg QD, 1000 mg QD, 375 mg BID, and 500 mg BID dose regimens was assessed using a logistic regression model.
Table 3.
Summary of Best Overall Response for Subjects With a Baseline Diagnosis of CLL/SLL
| AVL‐292 Dosing Cohorts | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | 125 mg QD n (%) | 250 mg QD n (%) | 400 mg QD n (%) | 625 mg QD n (%) | 750 mg QD n (%) | 1000 mg QD n (%) | 375 mg BID n (%) | 500 mg BID n (%) | Total n (%) |
| Baseline diagnosis: CLL/SLL | |||||||||
| n | 3 | 1 | 4 | 1 | 29 | 7 | 6 | 31 | 82 |
| Best overall response | |||||||||
| Complete response | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Partial response | 1 (33.3) | 0 | 1 (25.0) | 0 | 12 (41.4) | 4 (57.1) | 4 (66.7) | 19 (61.3) | 41 (50.0) |
| Stable disease | 2 (66.7) | 1 (100) | 3 (75.0) | 1 (100) | 16 (55.2) | 3 (42.9) | 2 (33.3) | 12 (38.7) | 40 (48.8) |
| Progression disease | 0 | 0 | 0 | 0 | 1 (3.4) | 0 | 0 | 0 | 1 (1.2) |
| Overall response rate | 1 (33.3) | 0 | 1 (25.0) | 0 | 12 (41.4) | 4 (57.1) | 4 (66.7) | 19 (61.3) | 41 (50.0) |
CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma.
In the univariate analysis a statistically significant relationship between increase in ORR and the AUCss was identified, as determined by logistic regression (Table 4 and Figure 3). The mean ORR in the first and fourth quartiles of average AUC (3875 to 20 533 ng·h/mL) was 35% and 50.2%, respectively. The higher AUC in patients with CLL generally corresponds to a higher ORR in the AVL‐292‐003 study. In the univariate analysis only CC‐292 steady‐state exposure (AUC) was statistically significant. In addition, no other covariate was identified as a significant predictor of clinical response in multivariate logistic regression analyses. Because QD and BID dose regimens showed different response rates in exploratory analysis, and dosing schedule (QD and BID) was identified as a marginally statistically significant predictor of clinical response due to the limited number of patients enrolled in each dose regimen, logistic regressions were conducted separately for each dose regimen. Of note, the slope of the ORR achieved by the BID dose regimens is higher than that achieved by the QD dose regimens, indicating that at the same total daily doses and CC‐292 exposures, the BID regimen provided better clinical response. As a result, total plasma exposure to CC‐292 as measured by AUCss was not the best predictor of the clinical response. In an attempt to explain the relationship between the clinical response and the dose regimen (QD vs BID), it was hypothesized that the clinical response could be related to the time that plasma CC‐292 concentrations were at or above a certain threshold concentration. This duration would therefore be a function of dose, dose schedule, and the disposition of CC‐292. The results of threshold analysis identified a threshold concentration of approximately 300 ng/mL, and the duration above this threshold was best correlated with the clinical response and was a more sensitive exposure metrics than AUCss. The duration/coverage above the threshold concentration of 300 ng/mL from 750 mg QD vs 375 mg BID were compared in Figure 4. For the same total daily dose, 375 mg BID provided ∼10.4‐hour coverage of the threshold concentration at and above 300 ng/mL, as compared to 6.6 hours by 750 mg QD, driving a better clinical response than that by the QD regimen of 750 mg.
Table 4.
Logistic Regression Parameters for the Final Exposure Response Model of CC‐292
| Estimate | Lower 90%CI | Upper 90%CI | P > Chi Squared | |
| Intercept | –6.31 | –11.03 | –1.59 | .028 |
| Log(AUCss) | 0.66 | 0.14 | 1.17 | .035 |
Figure 3.
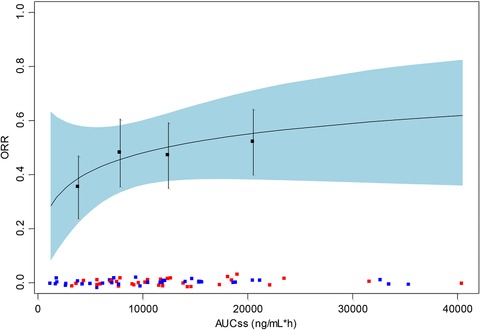
Logistic regression of the probability of overall response vs AUCss. Black line indicates the predicted response. Light blue ribbon indicates 95% confidence interval of predicted response. Black solid squares and vertical error bars indicate mean and 95% confidence interval for the observed response rate within each quartile of exposure. Individual exposure values are shown as colored squares (red squares represent responders and blue squares represent nonresponders). AUCss indicates area under the concentration‐time curve at steady state.
Figure 4.
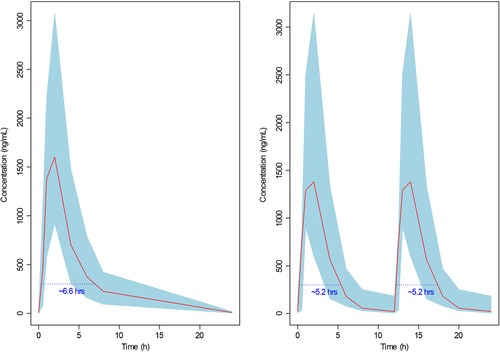
Duration above the threshold concentration from the same total daily dose: 750 mg QD (left) vs 375 mg BID (right). Solid red lines represent the mean of observed concentration data. Shaded areas represent 90% confidence intervals of observed concentration data. Dotted blue lines represent the threshold concentration of 300 ng/mL. Blue numbers represent the duration above the threshold concentration. QD indicates once daily; BID, twice daily.
Discussion
The final population pharmacokinetic model of CC‐292 provided an adequate description of CC‐292 concentration‐time data and associated variability from both healthy subjects at a dose range from 50 mg to 200 mg and patients with relapsed and/or refractory B‐cell malignancies at a dose range from 125 mg to 1000 mg from both QD and BID regimens.
The present analysis indicated that the drug is rapidly absorbed with a model predicted absorption rate constant of 0.97 h−1, which was estimated with good precision because there were a sufficient number of observations in the absorption phase from both healthy subjects and patients. CC‐292 exhibits approximately linear, time‐independent pharmacokinetics. Following oral administration, CC‐292 undergoes biphasic disposition. Healthy subjects and patients showed comparable pharmacokinetic properties demonstrated by similar dose‐normalized plasma concentration profiles (Figure 1).
In the population pharmacokinetic analysis Q/F was found to be 7.2‐fold lower than CL/F, and V3/F was 2.2‐fold smaller than V2/F, suggesting that CC‐292 clears more rapidly from the blood than it distributes to tissues/organs. Based on the final population pharmacokinetic parameters, the plasma elimination clearance is ∼7‐fold faster than the intercompartmental clearance (134 L/h and 18.7 L/h, respectively), and the central volume of distribution is more than 2‐fold larger than the peripheral volume of distribution (158 L and 72 L, respectively), resulting in a faster elimination rate (0.85 h−1) than distribution rate (0.11 h−1) of CC‐292.
In the covariate analysis the most influential covariates in the disposition of CC‐292 were sex and age. Statistically significant effects of sex on CL/F and age on V2/F were found in improving the model fitting. Although body weight and albumin levels appeared to be positively correlated with CL/F in the graphic analyses (correlation coefficient = 0.192), these correlations did not reach statistical significance in the following covariate model assessment. Creatinine clearance, a marker associated with renal function, was not identified as a statistically significant covariate of CC‐292 clearance. In the current population pharmacokinetic data set, there are an adequate number of patients with impaired renal function (25 subjects with impaired renal function), suggesting that the nonsignificant association between renal function marker (CLcr) and apparent clearance (CL/F) is applicable for a wide range of CLcr observed (ie, 31.0 mL/min to 193.3 mL/min). The finding is consistent with minimal renal elimination of unchanged CC‐292 and its predominant metabolite (data on file).
None of the hepatic function markers (albumin, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, total bilirubin levels) shows correlation with CL/F. However, all subjects included in this analysis showed normal levels of hepatic function markers. Specifically, the number of patients with elevated total bilirubin findings in this analysis was small (only 3% of patients had total bilirubin 1.5‐fold higher than the upper limit of normal). Therefore, the relationship between the hepatic function and the CL/F should be interpreted with caution.
Race (white vs nonwhite) was found to have no effect on CC‐292 pharmacokinetics. The population pharmacokinetic data set consisted of a majority of whites and only 2 Asians, 20 blacks, and 2 subjects of other races. Thus, the lack of effect of race on CC‐292 pharmacokinetics was limited to the white vs nonwhite population and consisted of 121 and 24 subjects, respectively, in the population pharmacokinetic data set.
The analysis indicated that the systemic clearance of the drug was only significantly correlated to sex according to the criteria defined for covariate analysis. Sex‐related pharmacokinetic disparities have been reported for other drugs. There are many potential reasons for sex differences in CC‐292 pharmacokinetics, such as differences in gastric pH, which is higher in females, lower hepatic blood flow and consequently lower hepatic metabolic capacity,22, 23 which could partly explain the findings. However, the resulting differences in drug exposures between female and male subjects were small (below 26%) relative to the observed overall variability in the pharmacokinetics of CC‐292. Therefore, this finding is not deemed to be clinically relevant and requires no dosage adjustment.
The contribution of sex to the interindividual variability of CL/F and the contribution of age to the interindividual variability of V2/F were marginal, reducing the interindividual variability from 58% in the base model to 56% in the final model, and from 88% in the base model to 86% in the final model for CL/F and V2/F, respectively. Therefore, none of these covariates (sex on CL/F, age on V2/F) appears to be clinically relevant, which is consistent with the clinical data; ie, there is no difference of ORR between male and female patients (48.7% and 46.4% for male and female patients, respectively).
The exposure‐response analysis for the patients with chronic lymphocytic leukemia showed that the higher AUC observed in patients with chronic lymphocytic leukemia generally corresponds to a higher overall response rate. Interestingly, the BID dose regimen provided a higher overall response rate than the QD dose regimen under the same total daily dose of 750 mg or 1000 mg. The threshold analysis has been previously conducted to explore whether there was a drug concentration level above which exposure correlated with a clinical event (clinical responses or toxicity).21, 24 The current threshold analysis identified a relatively high threshold concentration of approximately 300 ng/mL (Figure 4), exposure above which is well correlated with the clinical response and is consistent with the findings from the population pharmacokinetic analysis, which showed that CC‐292 has a relatively poor distribution property; ie, the CC‐292 clearance rate is much faster than its distribution rate. Therefore, maintaining CC‐292 concentration above this threshold may help to drive CC‐292 from blood/plasma into the desired tissues/organs.
In conclusion, the population pharmacokinetic data described here suggest that systemic CC‐292 exposure is comparable between healthy subjects and patients. CC‐292 showed the pharmacokinetic property that it is more readily cleared from the blood than distributed into tissues/organs. CC‐292 clearance is not complicated by demographics or baseline factors, other than sex. Furthermore, the finding that sex influences CL/F is unlikely to be clinically relevant, given that only 26% reduction in clearance was observed in female subjects. The exposure‐response analysis suggested that the higher drug exposure is correlated with higher overall response rate, and the BID dose regimen showed a higher overall response rate than the QD dose regimen, which may be due to the relatively poor distribution property of CC‐292.
Disclosures
Yan Li, Francisco Ramírez‐Valle, Yongjun Xue, Judith I. Ventura, Olivier Gouedard, Jay Mei, Kenichi Takeshita, Maria Palmisano and Simon Zhou are employees of and hold equity ownership in Celgene Corporation.
References
- 1. Gururajan M, Jennings CD, Bondada S. Cutting edge: constitutive B cell receptor signaling is critical for basal growth of B lymphoma. J Immunol. 2006;176(10):5715–5719. [DOI] [PubMed] [Google Scholar]
- 2. Kuppers R. Somatic hypermutation and B cell receptor selection in normal and transformed human B cells. Ann N Y Acad Sci. 2003;987:173–179. [DOI] [PubMed] [Google Scholar]
- 3. Burger JA. Bruton's tyrosine kinase (BTK) inhibitors in clinical trials. Curr Hematol Malig Rep. 2014;9(1):44–49. [DOI] [PubMed] [Google Scholar]
- 4. Smith CI, Baskin B, Humire‐Greiff P, et al. Expression of Bruton's agammaglobulinemia tyrosine kinase gene, BTK, is selectively down‐regulated in T lymphocytes and plasma cells. J Immunol. 1994;152(2):557–565. [PubMed] [Google Scholar]
- 5. Mohamed AJ, Yu L, Backesjo CM, et al. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228(1):58–73. [DOI] [PubMed] [Google Scholar]
- 6. Lewis CM, Broussard C, Czar MJ, Schwartzberg PL. Tec kinases: modulators of lymphocyte signaling and development. Curr Opin Immunol. 2001;13(3):317–325. [DOI] [PubMed] [Google Scholar]
- 7. Zhao X, Huang W, Wang Y, et al. Discovery of novel Bruton's tyrosine kinase (BTK) inhibitors bearing a pyrrolo[2,3‐d]pyrimidine scaffold. Bioorg Med Chem. 2015;23(4):891–901. [DOI] [PubMed] [Google Scholar]
- 8. Zhao D, Huang S, Qu M, et al. Structural optimization of diphenylpyrimidine derivatives (DPPYs) as potent Bruton's tyrosine kinase (BTK) inhibitors with improved activity toward B leukemia cell lines. Eur J Med Chem. 2017;126:444–455. [DOI] [PubMed] [Google Scholar]
- 9. Rickert RC. New insights into pre‐BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013;13(8):578–591. [DOI] [PubMed] [Google Scholar]
- 10. Davis RE, Ngo VN, Lenz G, et al. Chronic active B‐cell‐receptor signalling in diffuse large B‐cell lymphoma. Nature. 2010;463(7277):88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. IMBRUVICA (ibrutinib) [package insert]. Sunnyvale, CA: Pharmacyclics LLC; 2016. [Google Scholar]
- 12. Wu J, Zhang M, Liu D. Bruton tyrosine kinase inhibitor ONO/GS‐4059: from bench to bedside. Oncotarget. 2017;8(4):7201–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akinleye A, Chen Y, Mukhi N, Song Y, Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol. 2013;6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown JR, Harb WA, Hill BT, et al. Phase I study of single‐agent CC‐292, a highly selective Bruton's tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2016;101(7):e295–e298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eda H, Santo L, Cirstea DD, et al. A novel Bruton's tyrosine kinase inhibitor CC‐292 in combination with the proteasome inhibitor carfilzomib impacts the bone microenvironment in a multiple myeloma model with resultant antimyeloma activity. Leukemia. 2014;28(9):1892–1901. [DOI] [PubMed] [Google Scholar]
- 16. Arnason JE, Brown JR. B cell receptor pathway in chronic lymphocytic leukemia: specific role of CC‐292. Immunotargets Ther. 2014;3:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC‐292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–228. [DOI] [PubMed] [Google Scholar]
- 18. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. [DOI] [PubMed] [Google Scholar]
- 19. Taylor GO, Bamgboye EA, Oyediran AB, Longe O. Serum creatinine and prediction formulae for creatinine clearance. Afr J Med Med Sci. 1982;11(4):175–181. [PubMed] [Google Scholar]
- 20. Konishi K, Saruta T, Abe S, Wada T, Ozawa Y, Kato E. Prediction of creatinine clearance from the serum creatinine concentration. Nihon Jinzo Gakkai Shi. 1984;26(9):1195–1203. [PubMed] [Google Scholar]
- 21. Chen N, Li Y, Ye Y, Palmisano M, Chopra R, Zhou S. Pharmacokinetics and pharmacodynamics of nab‐paclitaxel in patients with solid tumors: disposition kinetics and pharmacology distinct from solvent‐based paclitaxel. J Clin Pharmacol. 2014;54(10):1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanaka E. Gender‐related differences in pharmacokinetics and their clinical significance. J Clin Pharm Ther. 1999;24(5):339–346. [DOI] [PubMed] [Google Scholar]
- 23. Kekki M, Samloff I, Ihamaki T, Varis K, Siuala M. Age‐ and sex‐related behaviour of gastric acid secretion at the population level. Scand J Gastroenterol. 1982;17(6):737–743. [DOI] [PubMed] [Google Scholar]
- 24. Gianni L, Kearns CM, Giani A, et al. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol. 1995;13(1):180–190. [DOI] [PubMed] [Google Scholar]