

Abstract
Tenapanor (RDX5791, AZD1722) is an inhibitor of sodium/hydrogen exchanger isoform 3 in development for the treatment of constipation‐predominant irritable bowel syndrome and the treatment of hyperphosphatemia in patients with chronic kidney disease on dialysis. We aimed to investigate whether tenapanor inhibits or induces cytochrome P450s (CYPs). In vitro experiments assessing the potential of tenapanor to affect various CYPs indicated that it could inhibit CYP3A4/5 (IC50 0.4‐0.7 μM). An open‐label, phase 1 clinical study (NCT02140268) evaluated the pharmacokinetics of the CYP3A4 substrate midazolam when administered with and without tenapanor. Healthy volunteers received a single oral dose of midazolam 7.5 mg on day 1 followed by tenapanor 15 mg twice daily on days 2 to 15, with an additional single 7.5‐mg midazolam dose coadministered on day 15. Midazolam exposure was similar whether it was administered alone or with tenapanor (geometric least‐squares mean ratio [90%CI] for [midazolam + tenapanor]/midazolam: area under the concentration‐time curve, 107% [101% to 113%]; Cmax 104% [89.6% to 122%]). Findings were similar for metabolites of midazolam. These results indicate that tenapanor 15 mg twice daily does not have a clinically relevant impact on CYP3A4 in humans and suggest that tenapanor can be coadministered with CYP3A4‐metabolized drugs without affecting their exposure.
Keywords: cytochrome P‐450 enzyme system, cytochrome P‐450 CYP3A, drug interactions, midazolam, tenapanor
Tenapanor (RDX5791, AZD1722) is a first‐in‐class small‐molecule inhibitor of sodium/hydrogen (Na+/H+) exchanger isoform 3 (NHE3), a transporter located in the apical membrane of enterocytes that plays an important role in sodium absorption from the gastrointestinal tract.1, 2, 3 Following oral administration of tenapanor, inhibition of gastrointestinal NHE3 reduces intestinal sodium uptake in both rats and healthy humans.4, 5 Furthermore, preclinical and early clinical studies have shown that tenapanor also reduces intestinal phosphate uptake.5, 6, 7 Tenapanor is currently being evaluated for the treatment of constipation‐predominant irritable bowel syndrome (IBS‐C) (ClinicalTrials.gov identifiers NCT01923428,8 NCT026218929 and NCT0268613810), and the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD) on dialysis (ClinicalTrials.gov identifiers NCT0208153411 and NCT0267599812).
As part of the drug development process, it is important to evaluate potential drug‐drug interactions caused by inhibition or induction of cytochrome P450 enzymes (CYPs) by the investigational agent. Tenapanor has been shown to have minimal systemic availability in all clinical studies to date, including in healthy volunteers and patients with CKD stage 5D treated with dialysis.4, 5, 13 After oral administration of a single dose of tenapanor 180 mg or tenapanor 15‐90 mg twice daily to healthy Japanese volunteers for 7 days, serum levels of tenapanor were below the lower limit of quantification (0.5 ng/mL) in 567 of 570 postdose samples.5 Furthermore, in a study of tenapanor treatment in patients with CKD stage 5D on dialysis, tenapanor was below the limit of quantification in 387 of 390 serum samples.13
Most CYPs are found in the liver, so tenapanor would not be expected to interact with them. Some CYPs, however, including CYP3A4, are also present in cells of the gut wall.14, 15 Therefore, minimally systemic drugs with limited ability to traverse cell membranes have the potential to cause drug‐drug interactions via intestinal CYPs. This study evaluated whether tenapanor affects CYP‐mediated metabolism in vitro and in vivo in healthy volunteers. The inhibition or induction of various CYPs by tenapanor was assessed in human liver microsomes and in the human HepaRG cell line, respectively. The results of these assays justified a clinical study in healthy volunteers in which the potential of tenapanor to inhibit or induce CYP3A4 was investigated by evaluation of the pharmacokinetics of the CYP3A4 model substrate midazolam16, 17 when administered with and without tenapanor.
Methods
In Vitro Assessment of CYP Inhibition by Tenapanor
The potential of tenapanor to inhibit various CYPs was assessed by studying its interactions with CYP model substrates.
Reversible inhibition was assessed in triplicate by coincubation of tenapanor at 6 concentrations (0.1, 0.3, 1.0, 3.0, 10, and 30 μM) with BD Gentest UltraPool 150 donor human liver microsomes (all from the same liver pool, lot no. 38289; BD Biosciences, Oxford, UK) in the presence of CYP enzyme marker substrates and NADPH (Sigma Aldrich, Dorset, UK), followed by monitoring of the rate of formation of CYP isoform‐specific metabolites. Enzyme marker substrates, at or below their KM (the concentration of substrate that permits the enzyme to achieve half the maximum rate of reaction) were assessed in 2 mixtures: the first, assessed by AstraZeneca and incubated for 10 minutes at 37°C, contained 80 μM bupropion (for CYP2B6), 2 μM amodiaquine (for CYP2C8), and 25 μM nifedipine (for CYP3A4/5) (Sigma Aldrich, Dorset, UK); the second, assessed by Pharmaron (Beijing, China) and incubated for 5 minutes at 37°C, contained 30 μM phenacetin (for CYP1A2; Jince Analysis Technology Co Ltd, Tianjin, China), 10 μM diclofenac (for CYP2C9; Sigma Aldrich, Shanghai, China), 35 μM (S)‐mephenytoin (for CYP2C19; Toronto Research Chemicals, Toronto, Canada), 5 μM bufuralol (for CYP2D6; Sigma Aldrich, Shanghai, China), and 3 μM midazolam (for CYP3A4/5; International Laboratory USA, South San Francisco, California). CYP isoform‐selective reversible inhibitors were used as positive controls: thiotepa (0.3‐100 μM), quercetin (0.6‐200 μM), ketoconazole (0.001‐0.5 μM), α‐naphthoflavone (0.0017‐0.5 μM), sulfaphenazole (0.033‐10 μM), benzylnirvanol (0.055‐16.7 μM), and quinidine (0.0017‐0.5 μM) (all from Sigma Aldrich, Dorset, UK).
The formation of CYP isoform‐specific metabolites was measured under linear conditions for time and protein concentration. Following substrate incubation, the reaction was quenched with 10% trichloroacetic acid (containing 40 nM verapamil internal standard; Sigma Aldrich, Dorset, UK) and centrifuged for 15 minutes at 3000 rpm. The supernatant was analyzed for the concentrations of marker substrates by high‐performance liquid chromatography followed by tandem mass spectrometric detection using a Thermo TSQ Quantum Ultra mass spectrometer with an atmospheric pressure chemical ionization source in the multiple reaction‐monitoring (MRM) mode. Chromatography was performed using an ACE C18‐AR 75 × 2.1 mm (particle size 3 μm) or Thermo Scientific Hypersil Gold C18 50 × 2.0 mm (particle size 1.6 μm) column maintained at 50°C, using a gradient of 20% to 95% acetonitrile in water (0.1% formic acid) as the mobile phase. The MRM transitions for each metabolite were: hydroxybupropion, m/z 257.05 > 239.126; desethylamodiaquine, m/z 328.972 > 284.027; oxidized nifedipine, m/z 345.176 > 284.013; paracetamol, m/z 152.3 > 110.2; 4‐OH‐diclofenac, m/z 312.0 > 266.0; 4‐OH‐mephenytoin, m/z 235.1 > 150.1; 1‐OH‐bufuralol, m/z 278.4 > 186.0; 1‐OH‐midazolam, m/z 342.0 > 203.0; and m/z 455.2 > 303.1 for the verapamil internal standard.
Inhibition of each CYP isoform was measured as the percentage decrease in the activity of marker metabolite formation compared to noninhibited (dimethyl sulfoxide) controls. The mean of the enzyme activity (% of control) was plotted against the log of the inhibitor concentration and fitted to an IC50 (concentration required to inhibit metabolite formation by 50%) curve using Prism 5.0 (Graphpad, California). Representative IC50 curves for hydroxybupropion (CYP2B6) and oxidized nifedipine (CYP3A4/5) are shown in Supplementary Figure 1. A result of no inhibition was quoted if none was observed at the highest concentration tested (P < .05), based on a Student t‐test (test array = 100, 2 degrees of freedom, 2‐sample unequal equivalence). If inhibition was only observed at the highest concentration (P < .05), the IC50 was quoted as >30 μM.
In Vitro Assessment of CYP mRNA Induction by Tenapanor
Induction of CYP mRNA expression by tenapanor in vitro was assessed by Xenoblis (Saint‐Grégoire, France). The potential of tenapanor to induce CYP1A2, CYP2B6, or CYP3A4 was investigated by measurement of mRNA levels after incubation of human HepaRG cell line cultures (Biopredic International, Rennes, France) with tenapanor at concentrations of 0.0001 to 50 μM for 24 hours. In addition, HepaRG cell cultures were exposed for the same period to rifampicin (0.02‐20 μM), omeprazole (0.21‐50 μM), 6‐[4‐chlorophenyl]imidazo[2,1‐b][1,3]thiazole‐5‐carbaldehyde O‐[3,4‐dichlorobenzyl]oxime; 0.001‐1 μM (CITCO), and phenobarbital (8.2‐2000 μM) (Sigma Aldrich, Saint‐Quentin‐Fallavier, France) as positive controls, as well as to solvent vehicle controls. Four experiments were performed, each evaluating a range of tenapanor concentrations, with each concentration tested in triplicate.
CYP mRNA levels were assessed by quantitative real‐time polymerase chain reaction (Applied Biosystem 7900HT Fast Real‐Time PCR System, Life Technologies SAS, Villebon‐sur‐Yvette, France) relative to GAPDH, a housekeeping gene, using SYBR® Green validated primers for CYP1A2, CYP2B6, and CYP3A4.18 CYP mRNA fold induction was calculated as the ratio between the mRNA expression of induced cells and control cells incubated with vehicle only.
Clinical Study in Healthy Volunteers
Study Participants
Healthy volunteers aged 18 to 50 years were eligible to participate in this study designed to evaluate the effect of tenapanor on the pharmacokinetics of midazolam in humans (ClinicalTrials.gov identifier: NCT02140268). The key exclusion criteria were: history or presence of any condition known to interfere with the absorption, distribution, metabolism, or excretion of drugs; use of drugs or substances with enzyme‐inducing properties within 4 weeks prior to the first administration of study drug; loose stools (defined as a Bristol Stool Form Scale19 score of 6 or 7) for 2 or more days in the week before study drug administration; use of medications or supplements known to affect stool consistency and/or gastrointestinal motility, including fiber supplements, antidiarrheals, prokinetic drugs, enemas, probiotics, or salt or electrolyte supplements containing sodium, potassium, chloride, or bicarbonate formulations during the past 7 days before study drug administration. Women could not be pregnant, and if they were of childbearing potential, they were required to employ an effective method of contraception. Men were also required to use effective contraception.
All volunteers provided written informed consent. The protocol and informed consent form for this study were approved by MidLands Institutional Review Board (Overland Park, Kansas) before the study began. The study was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonisation, and Good Clinical Practice guidelines.
Study Design
This was a phase 1, open‐label, fixed‐sequence study conducted at a single site in the United States (Quintiles, Overland Park, Kansas). The study consisted of a screening visit, 2 residential treatment periods with an intervening nonresidential period, and a follow‐up visit (Figure 1).
Figure 1.
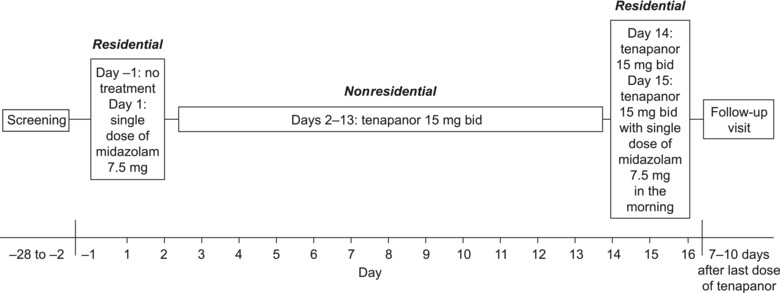
Design of the healthy volunteer study. bid, twice daily.
All volunteers received a single oral dose of midazolam 7.5 mg (syrup formulation) on day 1 (hereafter referred to as midazolam) followed by a tablet of tenapanor 15 mg twice daily from day 2 to day 15. On day 15, a single, oral dose of midazolam 7.5 mg was taken together with the morning dose of tenapanor (hereafter referred to as midazolam + tenapanor). Volunteers were screened up to 4 weeks before day −1 (ie, the day before the first treatment period) and were then resident in the study center for days −1 and 1 before being discharged at the beginning of day 2. Midazolam was administered 5 to 10 minutes before breakfast on day 1. Days 2 through 13 were nonresidential, during which the volunteers were expected to take tenapanor twice daily just before breakfast and dinner. Volunteers returned to the study center for days 14 to 16; tenapanor was given twice daily just before breakfast and dinner on days 14 and 15, and midazolam was administered 5 to 10 minutes before breakfast on day 15. Volunteers were discharged from the study center on day 16 after all of the safety assessments and sample collections were complete and then visited the study center for follow‐up 7 to 10 days after day 15.
Blood samples were taken predose and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 15, and 24 hours postdose for pharmacokinetic analysis of midazolam (and its 1‐OH and 4‐OH metabolites) on days 1 and 15. Blood samples for measurement of plasma tenapanor concentrations were collected on day 15 at predose and at 1, 2, and 4 hours post–morning dose. Safety assessments included monitoring of adverse events (AEs), vital signs, electrocardiograms, clinical laboratory evaluations, and physical examinations.
The 7.5‐mg dose of midazolam was chosen because it is below the maximum permitted oral dose (10‐15 mg) to yield an exposure and sedation margin in healthy volunteers. The tenapanor dose of 15 mg twice daily was chosen because it lies within the dose range likely to be used in later clinical trials of the drug. Importantly, this dose was also intended to ensure that any effect of tenapanor on midazolam metabolism would be caused by CYP inhibition/induction and not by other effects previously observed with high‐dose tenapanor treatment, such as diarrhea (also undesirable in a healthy volunteer study).
Pharmacokinetic Analyses
Plasma concentrations of tenapanor were measured by Covance Laboratories Inc. (Madison, Wisconsin). Human plasma samples containing tenapanor and a deuterium‐labeled analogue of tenapanor (d8‐tenapanor) as the internal standard were processed by liquid‐liquid extraction. After evaporation under nitrogen, the reconstituted samples were injected and analyzed by reversed‐phase high‐performance liquid chromatography using a Phenomenex Synergi Hydro‐RP 75 × 2.0 mm (particle size 4 μm) column (Phenomenex, Torrance, California) maintained at 35°C, with a gradient of 31% to 60% acetonitrile in water (0.1% formic acid) as the mobile phase. The extract was nebulized using heated nitrogen in electrospray positive ionization mode (Sciex API 5500, Applied Biosystems, Foster City, California). The ionized compounds were detected using liquid chromatography with tandem mass spectrometry (MRM transitions: tenapanor, m/z 573.0 > 502.1; d8‐tenapanor, m/z 577.4 > 504.0). The lower and upper limits of quantification were 0.5 ng/mL and 100 ng/mL, respectively, using a 100‐μL aliquot of human K2‐EDTA plasma. The accuracy and precision of quality‐control standards of tenapanor were determined at concentrations of 0.5, 1.5, 12, and 75 ng/mL. Inter‐run accuracy and precision were in the ranges of 100.1% to 109.2% and 3.3% to 7.7%, respectively.
Samples for determination of midazolam, 1‐OH‐midazolam, and 4‐OH‐midazolam concentration in plasma were analyzed by Covance Laboratories Inc. (Madison, Wisconsin). Midazolam, 1‐OH‐midazolam, 4‐OH‐midazolam, and the deuterated internal standards were extracted from samples using liquid‐liquid extraction. After evaporation under nitrogen, the residue was reconstituted and analyzed by reversed‐phase high‐performance liquid chromatography using a Waters Xbridge C18 50 × 2.1 mm (particle size 5 μm) column (Waters, Milford, Massachusetts) maintained at 30°C, using a gradient of 45% to 95% methanol in 30 mM ammonium formate (0.1% formic acid) as the mobile phase. The extract was nebulized using heated nitrogen in electrospray positive ionization mode, and the ionized compounds were detected by liquid chromatography with tandem mass spectrometry (MRM transitions: midazolam, m/z 326.3 > 291.3; 1‐OH‐midazolam, m/z 342.2 > 203.1; 4‐OH‐midazolam, m/z 342.2 > 234.2; d4‐midazolam, m/z 330.3 > 295.1; d4‐1‐OH‐midazolam, m/z 346.3 > 203.1). The lower and upper limits of quantification were 0.1 ng/mL and 100 ng/mL, respectively, using a 100‐μL aliquot of human K2‐EDTA plasma. The accuracy and precision of quality‐control standards of midazolam, 1‐OH‐midazolam, and 4‐OH‐midazolam were determined at concentrations of 0.1, 0.3, 5, and 75 ng/mL. Inter‐run accuracy and precision were in the ranges of 95.3% to 106.0% and 2.2% to 12.4%, respectively.
The bioanalytical methods were validated prior to sample analysis, and all study samples were analyzed within the known stability period of 278 days (tenapanor) or 150 days (midazolam and its metabolites). At a minimum, each analytical run included a calibration curve, a matrix blank, a control 0 sample (matrix blank containing internal standard), a reagent blank, and duplicate quality control samples at 3 concentrations within the calibration range. Both methods also demonstrated selectivity in the presence of coadministered drug.
Pharmacokinetic parameters were derived for midazolam and its metabolites on days 1 and 15 using standard noncompartmental methods with Phoenix® WinNonlin® 6.3 (Certara, St. Louis, Missouri). The pharmacokinetic parameters determined were as follows: the maximum concentration in plasma (Cmax); the area under the concentration‐time curve in plasma from time 0 (predose) to the time of the last quantifiable concentration (AUC0‐t), calculated by linear up/log down trapezoidal summation; the area under the concentration‐time curve in plasma from time 0 (predose) extrapolated to infinite time (AUC), calculated by linear up/log down trapezoidal summation and extrapolated to infinity by addition of the last quantifiable concentration (Ct) divided by the apparent elimination rate constant (λz; ie, AUC = AUC0‐t + Ct/λz).
Statistical Analyses
Statistical analyses were performed using Statistical Analysis System (SAS®, Cary, North Carolina) software, version 9.4. Natural log transformations of Cmax, AUC, and AUC0‐t for midazolam and its metabolites were separately analyzed using a mixed‐effects analysis of variance model, with a fixed effect for treatment and random effect for participant. The point estimate and 90%CI for the difference between treatments (midazolam + tenapanor and midazolam alone) were constructed. The point estimate and 90%CIs were then exponentially back‐transformed to provide point and CI estimates for the ratio of interest ([midazolam + tenapanor]/midazolam alone).
If there were no influence of tenapanor on the pharmacokinetics of midazolam, a standard deviation of at most 0.5 for the change in log‐transformed pharmacokinetic variables, and evaluable data from 24 participants, the probability of a 2‐sided 90%CI for the ratio of (midazolam + tenapanor)/midazolam completely contained within 70% to 143% was 90%. Therefore, up to 28 volunteers were required to be enrolled in order to obtain 24 evaluable participants.
Results
In Vitro Assessment of CYP Inhibition by Tenapanor
Tenapanor reversibly inhibited 5 of the 7 CYPs tested, being most potent against CYP3A4/5 with an IC50 of 0.402 μM for midazolam and 0.680 μM for nifedipine (Table 1). No IC50 could be determined for CYP2C19; however, inhibition was observed at the highest concentration tested (30 μM, mean 32.9% inhibition). No evidence of inhibition of CYP1A2 was observed over the concentration range tested.
Table 1.
In Vitro Assessment of Cytochrome P450 Inhibition by Tenapanor With Model Substrates
| CYP | Substrate, Concentration | Metabolite | Tenapanor IC50, μM (n = 3) |
|---|---|---|---|
| 1A2 | phenacetin, 30 μM | paracetamol | NI |
| 2B6 | bupropion, 80 μM | hydroxybupropion | 15.4 ± 7.5 |
| 2C8 | amodiaquine, 2 μM | desethylamodiaquine | 10.4 ± 3.0 |
| 2C9 | diclofenac, 10 μM | 4‐OH‐diclofenac | 14.2 ± 3.2 |
| 2C19 | S‐mephenytoin, 35 μM | 4‐OH‐mephenytoin | >30a (32.9 ± 1.9%) |
| 2D6 | bufuralol, 5 μM | 1‐OH‐bufuralol | 3.26 ± 0.54 |
| 3A4/5 | midazolam, 3 μM | 1‐OH‐midazolam | 0.402 ± 0.032 |
| 3A4/5 | nifedipine, 25 μM | oxidized nifedipine | 0.680 ± 0.157 |
CYP, cytochrome P450; IC50, half‐maximal inhibitory concentration; NI, no inhibition detected.
Data are presented as mean ± SD.
Statistically significant (P < .05) inhibition was observed at the highest concentration.
In Vitro Assessment of CYP mRNA Induction by Tenapanor
The positive control compounds were observed to induce at least a 13‐fold maximal increase in CYP1A2 (omeprazole, 32.0‐fold), CYP2B6 (CITCO, 13.2‐fold; phenobarbital, 17.0‐fold), and CYP3A4 (rifampicin, 18.4‐fold) mRNA levels relative to vehicle control. Tenapanor, at concentrations of up to 20 μM, did not induce CYP1A2 or CYP2B6 mRNA expression in any of the experiments (tenapanor was found to be cytotoxic to HepaRG cells at 50 μM but not at 20 μM). Four experiments were conducted to assess the effects of tenapanor on CYP3A4 mRNA. In 3 experiments tenapanor was not observed to influence CYP3A4 mRNA levels. In 1 experiment tenapanor induced CYP3A4 mRNA expression by up to 5.85‐fold, which was 16.8% of the positive control response (Figure 2).
Figure 2.
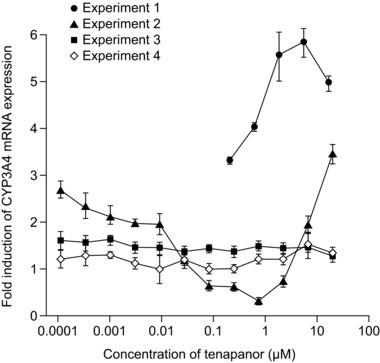
Fold induction of cytochrome P450 3A4 (CYP3A4) mRNA by tenapanor. Data are presented as mean ± SD. Experiments are independent replicates.
Clinical Study in Healthy Volunteers
Study Participants
A total of 28 volunteers (14 men) were enrolled. The mean ± SD [range] age was 29 ± 9 [18‐48] years. The mean ± SD [range] body mass index was 25.4 ± 3.2 [18.8‐29.8] kg/m2. Twenty‐six volunteers completed the study: 2 volunteers discontinued the study due to AEs.
Pharmacokinetic Evaluation
Midazolam distributed rapidly in plasma, and mean concentrations decreased in a monoexponential manner from 1 hour postdose (Figure 3a). Plasma midazolam exposure was similar when midazolam was given alone and when given concurrently with tenapanor (geometric least‐squares mean ratio [90%CI] [midazolam + tenapanor]/midazolam: AUC 107% [101% to 113%]; AUC0‐t 106% [100% to 113%]; Cmax 104% [89.6% to 122%]; Table 2). Both midazolam metabolites rapidly appeared and distributed in plasma, and then their mean concentrations decreased in a monoexponential manner in both treatment groups (Figure 3b and 3c). Plasma concentration profiles for 1‐OH‐midazolam and 4‐OH‐midazolam were also similar when midazolam was given alone and concurrently with tenapanor (Table 2). Plasma concentrations of tenapanor were below the lower limit of quantification (0.5 ng/mL) in all samples taken.
Figure 3.
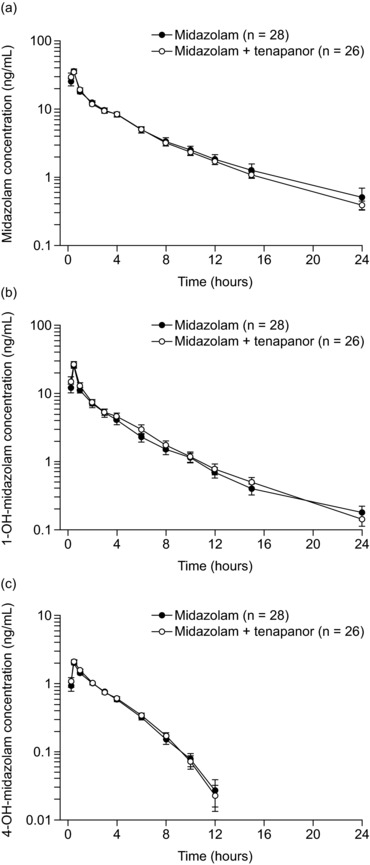
Plasma concentrations of midazolam (a) and its metabolites 1‐OH‐midazolam (b) and 4‐OH‐midazolam (c) in healthy volunteers over time following administration with and without tenapanor. Data are presented as arithmetic mean (SD). Values below the lower limit of quantification (0.1 ng/mL) were included in calculations as 0 ng/mL. Midazolam: single oral dose of midazolam 7.5 mg syrup, day 1. Midazolam + tenapanor: concurrent single oral dose of midazolam 7.5 mg and tenapanor 15‐mg tablet in the morning, day 15.
Table 2.
Summary of Pharmacokinetic Parameters of Midazolam and Its Metabolites Administered With and Without Tenapanor in Healthy Volunteers
| Parameter (Units) | Statistic | Midazolam (N = 28) | Midazolam + Tenapanor (N = 26) | Ratioa (90%CI), % |
|---|---|---|---|---|
| Midazolam | ||||
| AUC (ng·h/mL) | n | 27 | 26 | |
| Arithmetic mean (SD) | 96.2 (28.3) | 103 (30.9) | ||
| Geometric mean (GCV%) | 92.5 (29.2) | 98.5 (28.8) | 107 (101‐113) | |
| AUC0‐t (ng·h/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 101 (51.2) | 98.9 (28.9) | ||
| Geometric mean (GCV%) | 93.9 (37.9) | 95.2 (28.2) | 106 (100‐113) | |
| Cmax (ng/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 39.5 (15.4) | 40.8 (14.2) | ||
| Geometric mean (GCV%) | 36.8 (40.7) | 38.3 (38.4) | 104 (89.6‐122) | |
| tmax (h) | n | 28 | 26 | |
| Median (range) | 0.50 (0.25‐0.55) | 0.50 (0.25‐0.55) | ||
| t1/2 (h) | n | 28 | 26 | |
| Arithmetic mean (SD) | 5.43 (1.90) | 5.53 (1.49) | ||
| Geometric mean (GCV%) | 5.15 (34.4) | 5.33 (28.5) | ||
| 1‐OH‐midazolam | ||||
| AUC (ng·h/mL) | n | 28 | 25 | |
| Arithmetic mean (SD) | 54.1 (31.6) | 60.6 (32.9) | ||
| Geometric mean (GCV%) | 48.8 (43.6) | 55.0 (43.5) | 110 (103‐116) | |
| AUC0‐t (ng·h/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 52.1 (29.7) | 58.2 (31.9) | ||
| Geometric mean (GCV%) | 47.2 (43.1) | 52.7 (43.8) | 110 (104‐116) | |
| Cmax (ng/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 26.6 (12.0) | 28.0 (12.9) | ||
| Geometric mean (GCV%) | 24.1 (47.8) | 25.2 (51.3) | 103 (87.2‐122) | |
| tmax (h) | n | 28 | 26 | |
| Median (range) | 0.50 (0.25‐0.55) | 0.50 (0.25‐0.55) | ||
| t1/2 (h) | n | 28 | 25 | |
| Arithmetic mean (SD) | 5.14 (2.22) | 5.01 (1.66) | ||
| Geometric mean (GCV%) | 4.64 (50.0) | 4.73 (37.1) | ||
| 4‐OH‐midazolam | ||||
| AUC (ng·h/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 6.14 (1.56) | 6.32 (1.54) | ||
| Geometric mean (GCV%) | 5.96 (25.3) | 6.14 (24.3) | 105 (98.7‐111) | |
| AUC0‐t (ng·h/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 5.65 (1.50) | 5.87 (1.47) | ||
| Geometric mean (GCV%) | 5.47 (26.9) | 5.69 (25.3) | 106 (99.1‐113) | |
| Cmax (ng/mL) | n | 28 | 26 | |
| Arithmetic mean (SD) | 2.11 (0.757) | 2.20 (0.649) | ||
| Geometric mean (GCV%) | 1.99 (37.6) | 2.10 (33.8) | 105 (90.5‐122) | |
| tmax (h) | n | 28 | 26 | |
| Median (range) | 0.50 (0.25‐1.02) | 0.50 (0.25‐2.00) | ||
| t1/2 (h) | n | 28 | 26 | |
| Arithmetic mean (SD) | 2.48 (0.820) | 2.36 (0.581) | ||
| Geometric mean (GCV%) | 2.37 (30.2) | 2.30 (22.0) |
AUC, area under the plasma concentration‐time curve from 0 to infinity; AUC0‐t, area under the plasma concentration‐time curve from 0 (predose) to the last quantifiable concentration; Cmax, maximum observed concentration in plasma; GCV%, geometric coefficient of variation; tmax, time to Cmax; t1/2, apparent terminal half‐life.
Ratio is (midazolam + tenapanor)/tenapanor, based on geometric least‐squares means.
Safety and Tolerability
No serious AEs or deaths were reported in the study. The numbers of volunteers reporting at least 1 AE while receiving midazolam (27/28) and midazolam + tenapanor (26/26) were comparable, and fewer volunteers reported at least 1 AE on the days when tenapanor was taken alone (9/27). The most commonly reported AE in the study was somnolence, reported by all participants (28/28); this affected 27/28 volunteers taking midazolam alone and 26/26 volunteers taking midazolam + tenapanor. Of the 9 volunteers who reported AEs while receiving tenapanor alone, 5 reported gastrointestinal disorders (abnormal gastrointestinal sounds, n = 2; abdominal pain, n = 1; defecation urgency, n = 1; hypertrophy of tongue papillae, n = 1). The only nongastrointestinal AE to be reported by at least 2 volunteers taking tenapanor alone was dizziness (n = 2). Two volunteers discontinued the study prematurely owing to AEs; 1 participant withdrew after administration of midazolam on day 1 with an AE of headache, which resolved on the same day; a second participant withdrew on day 14 after the morning dose of tenapanor with an AE of increased white blood cell count, which was ongoing at the end of the study and was not considered to be related to midazolam or tenapanor by the investigator. There were no clinically relevant findings in terms of laboratory values, vital signs, ECG results, or physical examinations.
Discussion
Tenapanor is a small‐molecule inhibitor of NHE3 that acts locally in the gut to reduce absorption of sodium and phosphate. It is currently in phase 3 clinical development for the treatment of patients with IBS‐C as well as for the control of serum phosphate in patients with CKD on dialysis. Here, we investigated the potential for interactions between tenapanor and drugs that are metabolized by various enzymes of the CYP family, leading to a clinical study of the impact of tenapanor on the metabolism of midazolam, a model substrate of CYP3A4, in healthy volunteers. Our results suggest that tenapanor is unlikely to inhibit or induce CYP3A4 to a clinically meaningful extent in humans.
The results of our in vitro assays in human liver microsomes indicated that tenapanor could inhibit CYP3A4/5 but was unlikely to inhibit other CYPs to a clinically relevant degree. The results of a further in vitro assay evaluating CYP mRNA induction in the human HepaRG cell line were inconclusive, and the assay did not test protein expression or metabolic function, but there was a suggestion in 1 experiment that tenapanor may induce CYP3A4 mRNA expression to a small extent. Further assessment was therefore warranted in a human drug‐drug interaction study, designed in line with appropriate guidelines,16, 17 in which the ability of tenapanor to inhibit or induce CYP3A4 in vivo was investigated by evaluation of the pharmacokinetics of the model substrate midazolam when administered with and without tenapanor. We found that exposure of midazolam was comparable whether it was administered alone or in combination with tenapanor, with AUC, AUC0‐t, and Cmax ratios within the bioequivalence range of 70% to 143% for all analytes tested. Tenapanor was confirmed to have minimal systemic availability, consistent with other phase 1 and 2 studies of the drug,4, 13 and there were no unexpected safety findings.
Midazolam, a short‐acting benzodiazepine, is widely used as a regulatory standard for evaluation of potential drug interactions with CYP3A4.16, 17 The somnolence reported by all volunteers during midazolam treatment was consistent with its effects as a central nervous system depressant. Because midazolam is a sensitive CYP3A4 probe substrate, the finding that coadministered tenapanor does not alter its pharmacokinetics may also be generalized to other CYP3A4 substrates. This suggests that tenapanor can be coadministered with drugs primarily metabolized by CYP3A4 without negatively impacting on their exposure and without the need for dose adjustments.
Both the FDA and European Medicines Agency guidelines for drug interaction studies recommend that the systemic exposure of the inhibiting/inducing compound should be the exposure obtained with the highest generally recommended dose, maximizing the possibility of demonstrating an interaction, should it exist.16, 17 The dose of tenapanor used in our study was 15 mg twice daily. This is in the dose range being tested for the treatment of hyperphosphatemia in patients with CKD on dialysis.11 However, this dose is about 3‐fold lower than the highest dose of tenapanor recently evaluated for treatment of IBS‐C, 50 mg twice daily,8 and that is being investigated in 2 phase 3 clinical studies in IBS‐C.9, 10 Higher levels of CYP3A4 inhibition and induction would have been expected if tenapanor 50 mg twice daily had been tested in this study, as other drug‐drug interactions are generally known to be dose proportional.20, 21 However, even if inhibition of CYP3A4 by tenapanor is dose proportional, the level of inhibition seen with tenapanor 50 mg twice daily (ie, an approximately 3‐fold higher dose than was tested here) would result in about a 23% increase in AUC compared with the observed 7% increase for 15 mg twice daily. Both levels of interaction are anticipated to be clinically irrelevant. Therefore, a 15‐mg twice‐daily dose of tenapanor was expected to be sufficient to evaluate for clinically meaningful CYP3A4 inhibition or induction in this initial study. We thus believe that the findings of this study are also applicable to higher doses of tenapanor, such as those tested in IBS‐C.
The design of the healthy volunteer study, with 14 days of repeated dosing with tenapanor before administration of midazolam (ie, before the midazolam + tenapanor treatment), made it possible to look at both inhibition and induction of CYP3A4 and to assess the net influence on midazolam when it was coadministered with tenapanor. The 14‐day duration of tenapanor pretreatment was selected based on the half‐life of the CYP3A4 enzyme, to ensure that enough time had elapsed to detect any impact on CYP3A4.
In summary, the results of our in vitro studies into the effects of tenapanor on CYP‐mediated metabolism warranted a clinical investigation of the impact of tenapanor on CYP3A4 but found no evidence of inhibition of other CYPs by tenapanor. The subsequent assessment of midazolam pharmacokinetics with and without tenapanor in healthy volunteers indicated that tenapanor does not have a clinically relevant impact on CYP3A4 in humans. This suggests that tenapanor can be coadministered with CYP3A4‐metabolized drugs without impacting on their exposure and that clinically significant drug‐drug interactions are unlikely to occur.
Declaration of Conflicting Interests
H.R., M.E., and S.J. are employees of, and have ownership interest in, AstraZeneca. B.S., M.A., and M.K. are employees of AstraZeneca. D.P.R. is an employee of, and has ownership interest in, Ardelyx Inc.
Funding
This study was funded by AstraZeneca.
Supporting information
Supplementary Information
Acknowledgments
We thank the volunteers of the clinical study, the study center, and the clinical team, including the Principal Investigator Dr Eleanor Lisbon (Quintiles, Overland Park, Kansas), as well as the personnel involved in performing in vitro assays. We thank Maria Heijer, MSc, from AstraZeneca Gothenburg, Mölndal, Sweden and Brian Dayton, PhD, from Covance Laboratories Inc. (Madison, Wisconsin) for providing details of analytical methods for the healthy volunteer study. Medical writing support was provided by Richard Claes, PhD, and Steven Inglis, PhD, from PharmaGenesis London, London, UK, and was funded by AstraZeneca Gothenburg, Mölndal, Sweden and Ardelyx Inc., Fremont, California.
References
- 1. Broere N, Chen M, Cinar A, et al. Defective jejunal and colonic salt absorption and altered Na+/H+ exchanger 3 (NHE3) activity in NHE regulatory factor 1 (NHERF1) adaptor protein‐deficient mice. Pflugers Arch. 2009;457:1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Orlowski J, Kandasamy RA, Shull GE. Molecular cloning of putative members of the Na/H exchanger gene family. cDNA cloning, deduced amino acid sequence, and mRNA tissue expression of the rat Na/H exchanger NHE‐1 and two structurally related proteins. J Biol Chem. 1992;267:9331–9339. [PubMed] [Google Scholar]
- 3. Tse CM, Brant SR, Walker MS, Pouyssegur J, Donowitz M. Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney‐specific Na+/H+ exchanger isoform (NHE‐3). J Biol Chem. 1992;267:9340–9346. [PubMed] [Google Scholar]
- 4. Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014;6:227ra36. [DOI] [PubMed] [Google Scholar]
- 5. Johansson S, Rosenbaum DP, Knutsson M, Leonsson‐Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2016; doi:10.1007/s10157-016-1302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium‐hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol. 2014;26:1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosenbaum D, Johansson S, Carlsson B, et al. Tenapanor, a minimally absorbed NHE3 inhibitor, reduces dietary phosphorus absorption in healthy volunteers. J Am Soc Nephrol. 2014;25:72A. [Google Scholar]
- 8. Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor treatment of patients with constipation-predominant irritable bowel syndrome: a phase 2, randomized, placebo-controlled efficacy and safety trial. Am J Gastroenterol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. ClinicalTrials.gov. A 12‐week study with a 4‐week randomized withdrawal period to evaluate the efficacy and safety of tenapanor for the treatment of IBS‐C (T3MPO‐1). https://clinicaltrials.gov/ct2/show/NCT02621892. Accessed July 11, 2016.
- 10. ClinicalTrials.gov. A 26‐week study to evaluate the efficacy and safety of tenapanor in IBS‐C (T3MPO‐2). https://clinicaltrials.gov/ct2/show/NCT02686138. Accessed June 1, 2016.
- 11. Block GA, Rosenbaum DP, Leonsson‐Zachrisson M, et al. Effect of tenapanor on serum phosphate in patients receiving hemodialysis [published online ahead of print 2017]. J Am Soc Nephrol. doi:10.1681/ASN.2016080855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. ClinicalTrials.gov. An 8‐week study to evaluate tenapanor in the treatment of hyperphosphatemia in end‐stage renal disease patients on hemodialysis (ESRD‐HD). https://clinicaltrials.gov/ct2/show/NCT02675998. Accessed June 28, 2016.
- 13. Block GA, Rosenbaum DP, Leonsson‐Zachrisson M, et al. Effect of tenapanor on interdialytic weight gain in patients on hemodialysis. Clin J Am Soc Nephrol. 2016;11:1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thelen K, Dressman JB. Cytochrome P450‐mediated metabolism in the human gut wall. J Pharm Pharmacol. 2009;61:541–558. [DOI] [PubMed] [Google Scholar]
- 15. Zhang QY, Dunbar D, Ostrowska A, Zeisloft S, Yang J, Kaminsky LS. Characterization of human small intestinal cytochromes P‐450. Drug Metab Dispos. 1999;27:804–809. [PubMed] [Google Scholar]
- 16. U.S. Food and Drug Administration . Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed April 30, 2015.
- 17. European Medicines Agency . Guideline on the investigation of drug interactions. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed April 30, 2015.
- 18. Girault I, Rougier N, Chesné C, et al. Simultaneous measurement of 23 isoforms from the human cytochrome P450 families 1 to 3 by quantitative reverse transcriptase‐polymerase chain reaction. Drug Metab Dispos. 2005;33:1803–1810. [DOI] [PubMed] [Google Scholar]
- 19. Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924. [DOI] [PubMed] [Google Scholar]
- 20. Baldan N, Rigotti P, Furian L, et al. Co‐administration of sirolimus alters tacrolimus pharmacokinetics in a dose‐dependent manner in adult renal transplant recipients. Pharmacol Res. 2006;54:181–185. [DOI] [PubMed] [Google Scholar]
- 21. Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Dose‐dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab Dispos. 2011;39:1977–1986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information