

Abstract
Inflammatory bowel diseases (IBD) are key risk factors for the development of colorectal cancer, but the mechanisms that link intestinal inflammation with carcinogenesis are insufficiently understood. Card9 is a myeloid cell‐specific signaling protein that regulates inflammatory responses downstream of various pattern recognition receptors and which cooperates with the inflammasomes for IL‐1β production. Because polymorphisms in Card9 were recurrently associated with human IBD, we investigated the function of Card9 in a colitis‐associated cancer (CAC) model. Card9−/− mice develop smaller, less proliferative and less dysplastic tumors compared to their littermates and in the regenerating mucosa we detected dramatically impaired IL‐1β generation and defective IL‐1β controlled IL‐22 production from group 3 innate lymphoid cells. Consistent with the key role of immune‐derived IL‐22 in activating STAT3 signaling during normal and pathological intestinal epithelial cell (IEC) proliferation, Card9−/− mice also exhibit impaired tumor cell intrinsic STAT3 activation. Our results imply a Card9‐controlled, ILC3‐mediated mechanism regulating healthy and malignant IEC proliferation and demonstrates a role of Card9‐mediated innate immunity in inflammation‐associated carcinogenesis.
Keywords: Card9, Colitis‐associated‐cancer, Innate lymphoid cells, Interleukin‐1β, Interleukin‐22
Introduction
Inflammatory bowel diseases (IBDs) such as Crohn's disease and ulcerative colitis are relapsing and remitting disorders, which are most prevalent in the Western world. The chronic inflammatory state within the intestine results in a debilitating disorder and in addition predisposes to the development of colitis‐associated cancer (CAC), which is a major cause of death in IBD patients 1. Data from epidemiologic and genetic studies indicate a deregulated innate immune response against intestinal microbes in a genetically susceptible host as the underlying cause of IBD, but the signals and immune mechanisms that promote CAC growth are still insufficiently defined. Candidate gene searches of innate immune pathways in IBD identified associations of Card9 gene polymorphisms (rs10870077) with Crohn´s disease and ulcerative colitis 2, confirmed by genome‐wide association studies 3, 4, 5. The Card9 gene encodes for a cytoplasmic adaptor protein, which is selectively expressed in myeloid‐derived innate immune cells 6, 7. It belongs to a small family of Caspase recruitment domain (CARD) containing proteins that also includes the lymphocyte specific member Card11 (Carma1), and which assemble immune signaling complexes for context‐specific NF‐κB and Mitogen‐activated protein kinases (MAPK) activation 7, 8, 9. The Card9 module cooperates with inflammasomes in antigen presenting cells (APCs) 7, to induce the generation of pro‐IL‐1β, which is subsequently cleaved into bioactive IL‐1β. It also regulates the production of other cytokines including IL‐6, which all together orchestrate the differentiation of activated T cells towards Th‐17 immune responses during systemic or local fungal or bacterial infection 10, 11, 12, 13, 14, 15. However, the pathophysiological importance of these pathways in inflammatory diseases is still ill defined and their role in inflammation‐associated cancer is unknown.
Multiple pattern recognition receptors (PRRs) of the C‐type lectin receptor (CLR) family including the anti‐fungal receptor Dectin‐1 require Card9 for the activation of innate immunity. Card9‐signaling is also responsive to sterile cell death and to the stimulation of nucleotide‐oligomerization domain (Nod) 2 or cytoplasmic viral nucleic acid sensors such as Rig‐I or Rad50 7, 13. Interestingly, polymorphisms in Dectin‐1 and in Nod2 are similar to Card9 polymorphisms associated with human IBD 5, 16, 17, indicating that these signaling pathways play important roles in intestinal mucosal immunity. In line with these findings, Card9‐signaling has been implicated in intestinal immune responses and the maintenance of homeostasis after epithelial injury and bacterial infection in mice 14. Therefore, we here investigated the function of Card9‐mediated innate immunity in inflammation‐associated colon carcinogenesis. We report that Card9‐signaling drives the production of IL‐1β within the damaged intestine and regulates the subsequent generation of IL‐22 by group3 innate lymphoid cells, which promotes tumorigenesis via STAT3 activation within the transformed epithelium.
Results
Reduced colitis‐associated tumor growth in Card9‐deficient mice
To study the role of Card9 in CAC, we injected Card9−/− mice with one dose of the genotoxic carcinogen Azoxymethane (AOM) to induce mutations in intestinal epithelial cells (IECs), followed by three cycles of feeding with the detergent Dextran Sodium Sulfate (DSS) (Fig. 1A). DSS‐treatment causes epithelial injury and subsequent colonic inflammation that drives intestinal epithelial dysplasia after AOM treatment 18, 19, 20. We assessed colitis severity by daily weighing the mice. During the first round of DSS‐treatment, Card9−/− mice lost less weight compared to their wild type littermates (Fig. 1A) while no differences in systemic inflammatory TNF, IL‐6, IL‐2 and IL‐1β responses were detected during the acute phase of colitis (Fig. 1B). However, during the second and third cycle of DSS treatment, Card9−/− mice displayed a higher loss of body weight (Fig. 1A) with finally increased systemic TNF, IL‐6, IL‐2 and IL‐1β concentrations (Fig. 1B) and a delayed and insufficient recovery from the DSS challenge (Fig. 1A). Consistently and in line with previously published results 14, 21, Card9‐deficient mice showed a delayed recovery following prolonged DSS administration in an acute colitis model (data not shown). About 40% of all Card9−/− mice had to be sacrificed before day 68 of the experiment (Fig. 1C). Sacrificed Card9−/− mice had dilated colons without macroscopically visible polyps and were filled entirely with luminal content indicative of a paralyzed colonic muscle layer, which can occur as a life‐threatening complication of severe intestinal inflammation.
Figure 1.
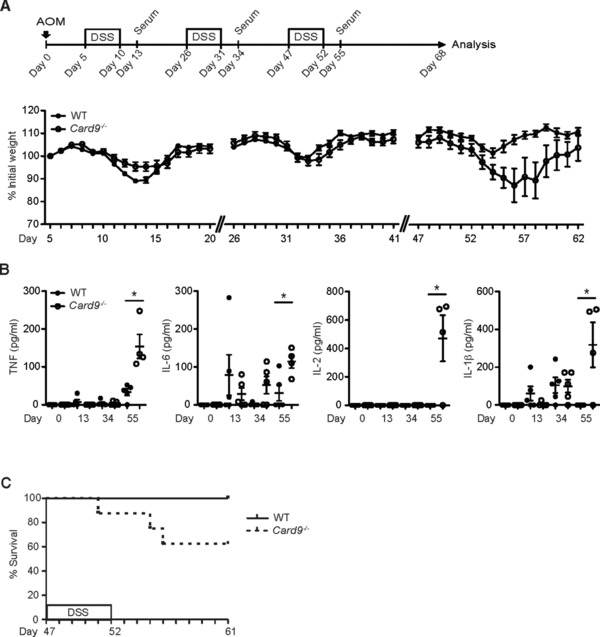
Increased susceptibility to chronic colitis in Card9−/− mice. Mice were injected with a single dose of AOM, followed by three 5‐day cycles of feeding with DSS, interrupted by periods of normal drinking water. Serum was collected at the indicated time points. (A) The body weight of WT (n = 10) and Card9−/− (n = 8) mice was monitored daily. Data points reflect the average weight per group in percent of initial weight ± S.E.M. (B) Systemic cytokine levels, including TNF, IL‐6, IL‐2 and IL‐1β, were determined in the serum of WT and Card9−/− mice at the indicated time points by cytometric bead array Each symbol represents an individual mouse. Data from one experiment are presented. Small horizontal lines indicate the mean, error bars indicate the S.E.M. * p < 0.05, Student's t‐test. (C) Kaplan–Meier survival curve of WT (n = 10) and Card9−/− (n = 8) mice during the third DSS cycle. Statistical survival analysis was performed using the log‐rank test (p < 0.05). (A, C) Data are from single experiments representative of three independent experiments.
At day 68 after initial AOM treatment, we sacrificed all remaining animals for detailed analysis. As expected, the WT animals developed multiple large polyps in their distal colons 18, 19, 20, but the colonic polyps in Card9−/‐ mice appeared macroscopically smaller in size. Histological analysis revealed a hyperplastic and regenerating epithelium in WT mice, whereas Card9−/− mice showed broad areas of epithelial abrasion with ulcers, edema and mucosal infiltration with Ly6G+ neutrophils (Supporting Information Fig. 1A), which was also observed in the animals that were prematurely sacrificed (data not shown), consistent with defects in epithelial regeneration. To test whether mucosal barrier defects contribute to epithelial damage in Card9‐deficient mice, we stained for acidic and neutral mucins in the inflamed epithelia and found an equal distribution between WT and Card9−/− animals (Supporting Information Fig. 1B).
To characterize the tumor growth in a quantitative manner, we performed serial colon sections for histology. In Card9‐deficient animals we detected a significant reduction in the size of polyps and a lower degree of dysplasia characterized by less elongated and less crowded nuclei with more preserved polarity and crypt architecture in comparison to WT mice (Fig. 2A and B). Yet, the total number of neoplasms per mouse did not differ between WT and Card9−/− mice (Fig. 2C), indicating that Card9 signaling is not important for the AOM‐mediated tumor initiation, but required for signals that promote tumor growth. To test whether Card9 signaling would control the proliferation of normal and transformed intestinal epithelial cells, we performed immunohistochemical analysis of BrdU incorporation into IECs. We observed significantly reduced numbers of BrdU‐positive, proliferating cells per crypt within the tumors and also in the untransformed epithelia of Card9−/‐ mice (Fig. 2D). Because an increased susceptibility to epithelial cell apoptosis could additionally contribute to the observed phenotype, we also studied caspase‐3 activation within the epithelium. While we detected an increase in the frequency of apoptotic cells in the inflamed epithelia of Card9−/‐ mice compared to WT mice (Fig. 2E left), active caspase‐3 staining did not differ in the tumor regions (Fig. 2E right). Together, this first set of in vivo experiments reveals that the innate immune adapter Card9 is required to deliver signals during intestinal inflammation that positively control the regeneration of epithelial cells after injury and which in addition drive the growth of transformed IECs during colon tumor progression.
Figure 2.
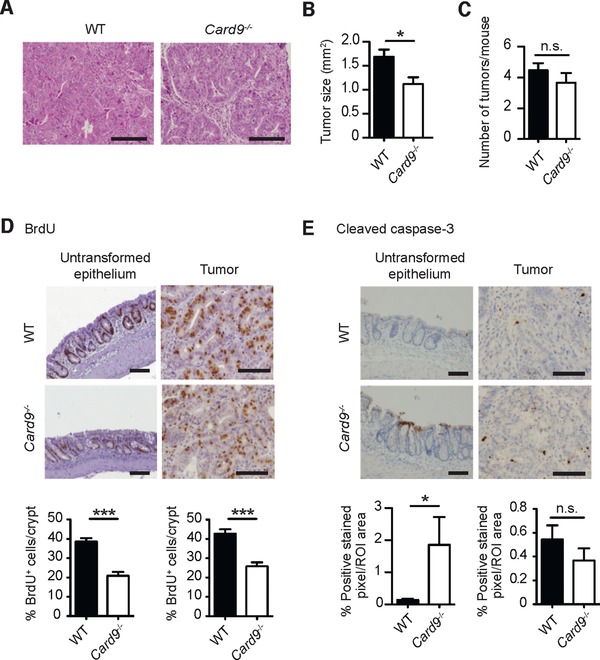
Reduced colitis‐associated tumor growth in Card9−/− mice. CAC was induced in WT and Card9−/− mice as described in Fig. 1 using the AOM/DSS model. (A) Representative H&E stains of colonic polyps from AOM/DSS‐treated WT and Card9−/− mice at day 68. (B) The size of the tumors in WT (n = 67) and Card9−/− (n = 33) mice were analyzed histologically at day 68. (C) Number of tumors per mouse in WT (n = 15) and Card9−/− (n = 10) mice were determined histologically. (D) Representative images of BrdU‐incorporation into IECs of the inflamed untransformed epithelium (left) or tumor tissue (right), detected by IHC in the colon of AOM/DSS‐treated WT and Card9−/− mice. BrdU‐incorporation is also quantified as the percentage of BrdU‐positive cells per crypt (bottom). (E) Active cleaved caspase‐3 IHC of the inflamed untransformed epithelium (left) or tumor tissue (right) in the colon of AOM/DSS‐treated WT and Card9−/− mice. The percentage of positive stained pixel/ region of interest (ROI) in the epithelium or tumor tissue is depicted (bottom). (A, D, E) Representative data of at least two independent experiments are shown. (B, C) Data of two independent experiments are pooled and presented as mean + S.E.M of (B, C). * p < 0.05 and *** p < 0.001, Student's t‐test; n.s., not significant; Scale bars represent 100 μm.
Card9 controls the injury‐induced IL‐1β response within the colonic mucosa
To study the mechanisms by which Card9 would regulate transformed IEC growth, we analyzed the expression of factors that mediate tissue regeneration after acute DSS challenge. The production of IEC‐derived growth factors, including TFF3, KGF and IL‐18, which are all required for tissue regeneration 22, 23, 24, 25, 26 did not differ between DSS‐treated WT and Card9−/− mice (Fig. 3A). This finding is in line with the fact that Card9 is not expressed within IECs but specifically in leukocytes as indicated in previous studies 7, 9, 27, 28.
Figure 3.
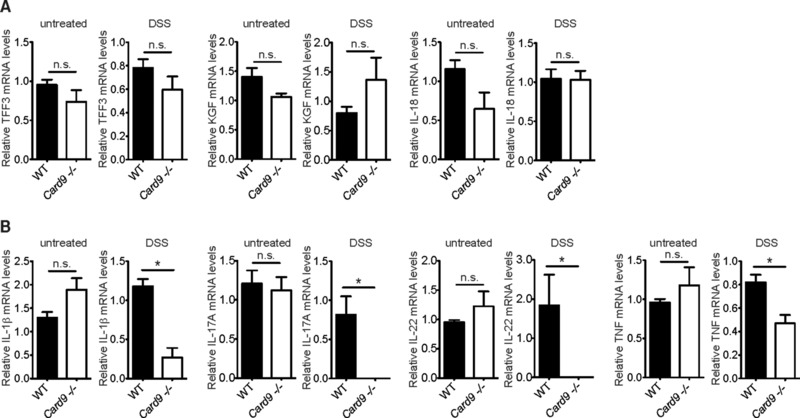
Impaired pro‐inflammatory cytokine expression in Card9‐deficient lamina propria leukocytes during colitis. Acute colitis was induced in WT and Card9−/− mice by feeding with DSS for 7 days, followed by 5 days of recovery. (A) Relative expression of Trefoil factor 3 (TFF3), Keratinocyte growth factor (KGF), and IL‐18 mRNA was measured by quantitative PCR and normalized to β‐actin transcript levels in colon tissues of untreated WT (n = 5) and Card9−/− (n = 5), or acute DSS‐treated WT (n = 4) and Card9−/− (n = 4) mice at day 12 of the experiment (DSS). (B) IL‐1β, IL‐17A, IL‐22, and TNF transcript levels in lamina propria leucocytes of untreated WT (n = 5) and Card9−/− (n = 5), or acute DSS‐treated WT (n = 4) and Card9−/− (n = 5) mice at day 12 of the experiment (DSS) were analyzed by qPCR as described above. The data of one representative experiment are shown as the mean + S.E.M of the indicated numbers of samples. * p < 0.05, Student's t‐test; n.s., not significant.
Next, we investigated the leukocyte intrinsic expression of cytokines that drive or induce tumor‐promoting Th17 cell and IL‐22‐mediated immune responses. Intriguingly, IL‐1β mRNA expression within leukocytes isolated from the colonic lamina propria was almost completely abolished in mice lacking Card9 (Fig. 3B). Moreover, we also observed a reduced IL‐1β protein production in ex vivo colon cultures from AOM/DSS‐treated Card9‐deficient mice at day 68 of the experiment (Supporting Information Fig. 2). In addition, and consistent with the function of Card9 signaling and IL‐1β production in promoting Th17 cell differentiation 10, 15, 29, 30, 31, 32, 33, the expression of IL‐17A within lamina propria leukocytes was also almost completely abolished in Card9−/− animals (Fig. 3B).
IL‐1β does not only regulate Th17‐responses, but also maintains and enhances the production of IL‐22 by innate lymphoid cells 34, 35, 36, 37, which is a key growth factor for intestinal epithelial cells 38 in inflammatory disease wound healing and tumorigenesis 39. Consistent with a lack in IL‐1β production during the recovery phase after colitis, Card9−/− mice expressed significantly less IL‐22 in lamina propria leukocytes (Fig. 3B). To test whether elevated TNF contributes to increased DSS‐mediated colitis and colonic epithelial cell death in Card9−/− animals, we also analyzed TNF expression during acute DSS colitis. However, TNF expression was also significantly reduced in lamina propria leukocytes of DSS treated Card9−/− mice (Fig. 3B).
Card9 signals regulate IL‐22 production from type 3 innate lymphoid cells
IL‐22‐producing lymphoid cells migrate to and are activated in the lamina propria during inflammation. They are also frequently detected in human colorectal cancer and play a tumor‐promoting role in the disease 39, 40. IL‐22 signals through its heterodimeric receptor, which is exclusively expressed on non‐immune cells and which induces IEC proliferation during tissue repair. IL‐22 signaling can promote malignancy in tumor cells and the neutralization of IL‐22 can directly reduce the level of dysplasia and tumor growth 39. Because the predominant source of IL‐22 in the intestinal mucosa are type 3 innate lymphoid cells (ILC3) 37, we investigated IL‐22‐production by ILC3s within the intestines of Card9−/− mice using flow cytometry. We also studied IL‐17 or IFN‐γ generation by ILC3s, or IFN‐γ production by ILC1s, which constitute together with ILC3s the main ILC subsets within the gastro‐intestinal tract.
Interestingly, the frequency of IL‐22‐producing ILC3s did not differ between wild‐type and Card9‐deficient mice under homeostatic conditions (Fig. 4A). Yet, during the regeneration phase after DSS challenge, the percentage of IL‐22‐producing ILC3s was significantly reduced in Card9−/− mice compared to the wild‐type specifically within the colon (Fig. 4A), but not in the small intestine (Fig. 4B). The production of IL‐17 or IFN‐γ by ILC3s, or IFN‐γ by ILC1s 41, was also not impaired by the absence of Card9 (Fig. 4C–G), indicating that Card9 specifically regulates IL‐22 production by ILC3s during colonic inflammation.
Figure 4.
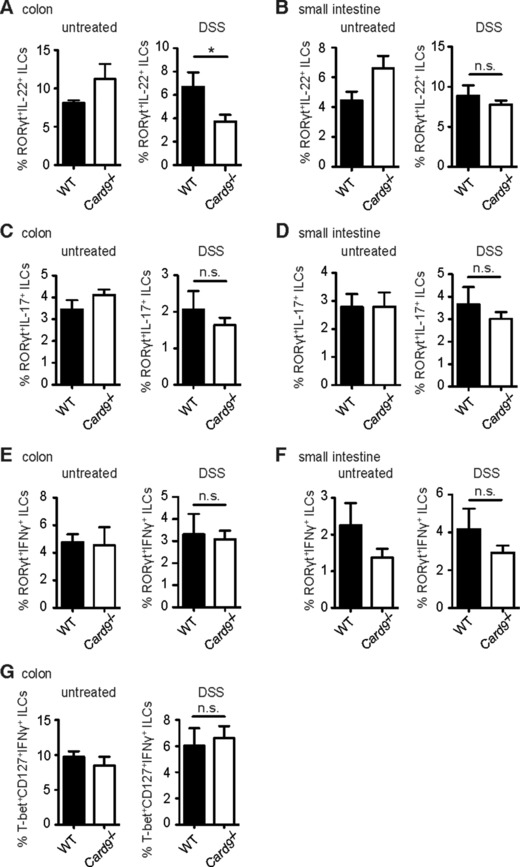
Reduced IL‐22 production by ILC3s in response to acute DSS treatment in the colon of Card9−/− mice. Intracellular cytokine levels in ILC subsets were determined by flow cytometry during the recovery phase of acute DSS‐induced colitis. (A‐G) Lamina propria leukocytes were isolated from the colon (A, C, E, G) and small intestine (B, D, F) of untreated WT (n = 4) and Card9−/− (n = 2) mice (untreated), or acute DSS‐treated WT (n = 5) and Card9−/− (n = 7) mice during the recovery phase at day 12 (DSS). Intracellular cytokine levels within ILCs were determined by flow cytometry. (A, B) Percentages of RORγt+IL‐22+ ILCs in the colon (A) and small intestine (B). (C, D) Relative numbers of RORγt+IL‐17+ ILCs in the colon (C) and small intestine (D). (E, F) Percentages of RORγt+IFN‐γ+ ILCs in the colon (E) and small intestine (F). (G) Percentages of T‐bet+CD127+IFN‐γ+ ILCs in the colon. Data of one single experiment are shown as the mean + s.e.m of the indicated number of samples. *p < 0.05, Student's t‐test; n.s., not significant.
The defective IL‐22 generation in Card9−/− mice could be due to an ILC3‐intrinsic signaling defect, or caused by the lack of external signals, which are induced via Card9. We therefore investigated the expression of Card9 within ILCs. Yet, similar to other lymphoid lineage cells including conventional NK cells 42, ILC2s and ILC3s express predominantly the homologue Card11 (Fig. 5A), indicating that Card9 has no critical function within the ILC compartment. Important for ILC‐responses in the mucosa are instructive cytokines generated by MHCIIhi myeloid cells including Notch2‐dependent CD103+CD11b+ dendritic cells 43. Card9−/− mice exhibit regular frequencies of MHCIIhi myeloid cells and normal percentages of all major intestinal DC subsets within the lamina propria and in the mesenteric lymph nodes, including CD103−CD11b+, CD103+CD11b−, and CD103+CD11b+ DCs (Fig. 5B, C and Supporting Information Fig. 3A), further indicating that pure developmental APC defects are unlikely to account for the defective IL‐22 response in ILC3s. Because myeloid derived IL‐1β controls the production of IL‐22 by ILCs together with IL‐23 34, 35, 37, we next investigated IL‐23 expression in Card9‐deficient leukocytes during DSS colitis. Yet, in contrast to defective IL‐1β production (see above, Fig. 3B and Supporting Information Fig. 2), IL‐23 is regularly expressed in Card9‐deficient lamina propria leukocytes (Fig. 5D). Together, these data suggest that the Card9‐mediated generation of IL‐1β by myeloid cells within the inflamed tissue secondarily drives the production of IL‐22 by Card11 expressing ILCs. To test this model, we isolated lamina propria leukocytes from Card9−/‐ mice and stimulated these cells with recombinant IL‐1β and IL‐23 in vitro (Fig. 5E and Supporting Information Fig. 3B). Supplementation of exogenous IL‐1β and IL‐23 was indeed sufficient to induce the regular generation of IL‐22 in ILCs from Card9‐deficient mice. Thus, the ILCs in Card9−/− animals are not intrinsically defective in IL‐22 production, indicating that the failure to provide myeloid‐derived IL‐1β is a cause for the defective IL‐22 production in Card9‐deficient mice.
Figure 5.
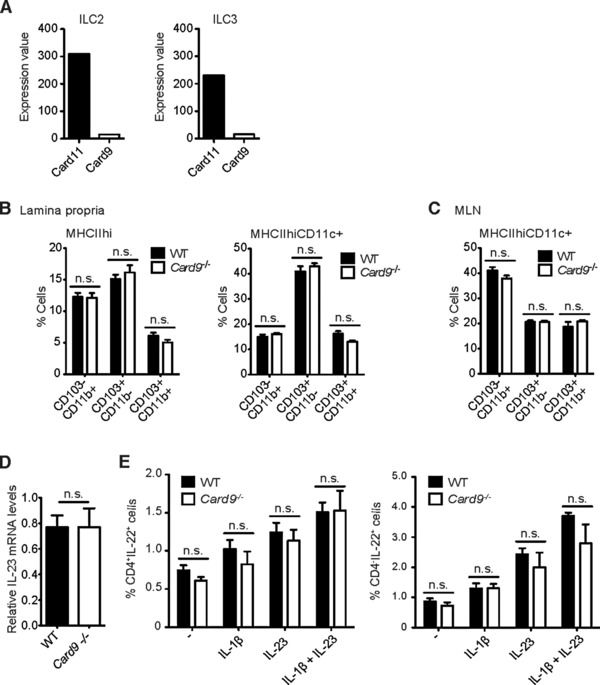
Normal ILC function and DC development in the intestine of Card9‐deficient mice. Leukocytes were isolated from the intestinal lamina propria and mesenteric lymph nodes of untreated WT and Card9−/− mice. DC subsets as well as ILC function were analyzed by flow cytometry. (A) Relative expression values of Card9 and Card11 in intestinal FACS‐purified type 2 and type 3 ILC subsets from untreated WT mice were determined by microarray analysis (n = 5). (B, C) Percentages of MHCIIhi (left) and MHCIIhiCD11c+ (right) DCs subdivided into CD103−CD11b+, CD103+CD11b−, and CD103+CD11b+ DCs in the colonic lamina propria (B) and mesenteric lymph nodes (MLN) (C) of untreated WT (n = 5) and Card9−/− (n = 5) mice were determined by flow cytometry. (D) Relative IL‐23 mRNA expression normalized to β‐actin transcript levels in colonic lamina propria leucocytes from acute DSS‐treated WT (n = 4) and Card9−/− (n = 4) mice at day 12. (E) Percentages of IL‐22‐producing CD4+ (left) or CD4− (right) ILCs that were isolated from the mesenteric lymph nodes (MLN) of untreated WT (n = 3) and Card9−/− (n = 3) mice. MLN cells were either left untreated or restimulated in vitro with IL‐1β, IL‐23 or a combination of IL‐1β and IL‐23 and analyzed by flow cytometry. (B–E) Data from one single experiment are shown as the mean + s.e.m of the indicated number of samples. n.s., not significant, Student's t‐test.
Reduced tumor cell intrinsic STAT3 signaling in Card9‐deficient animals
Binding of IL‐22 to the IL‐22‐receptor on IECs activates JAK/STAT signaling, mainly STAT3, by inducing its phosphorylation. Subsequently, activated STAT3 is essential to induce the expression of a plethora of target genes within the IEC compartment that mediate mitogenic and regenerative activities both under physiological and pathogenic conditions 39, 44, 45. Consistently, mice with an IEC‐specific deletion of STAT3 recover later from colitis with epithelial abrasion and develop smaller tumors in CAC‐models similar to Card9 deficient animals 44, 46. Because activated STAT3 is responsible for most IL‐22 responses, we investigated the activity of IL‐22‐receptor signaling in the colonic crypt cells of tumors from AOM/DSS‐treated Card9−/− mice by measuring phosphorylated STAT3 levels. Consistent with reduced IL‐22‐production in Card9−/− animals, we observed also a significantly lower percentage of phospho‐STAT3 positive crypt cells in their tumors in comparison to WT animals (Fig. 6A and B). Other cytokines and growth factors such as IL‐6, IL‐11 and VEGF are also potent activators of STAT3‐signaling in IECs. We therefore analyzed the expression of these cytokines in colonic tissues from AOM/DSS‐treated WT and Card9 −/‐ mice, but detected no statistically significant differences (Supporting Information Fig. 4A–C). Together, these results indicate that the failure to fully activate STAT3 signaling within IECs accounts for the reduced tumor growth and defective epithelial cell regeneration in Card9−/− mice.
Figure 6.
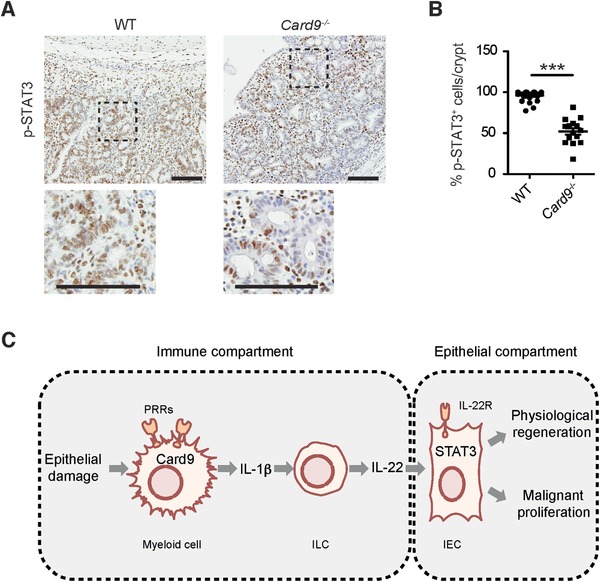
Reduced STAT3 activation in IECs within colonic tumors of Card9−/− mice and proposed mechanistic model for Card9 function in CAC. (A) Representative images of phospho‐STAT3 IHC on colon sections from AOM/DSS‐treated WT and Card9−/− mice at day 68. Scale bars represent 100 μm. (B) Percentage of phospho‐STAT3 positive crypt cells/crypt within tumors from WT (n = 10) and Card9−/− (n = 5) mice. Each dot represents one crypt within a tumor as evaluated by histology. (C) Simplified model for Card9 function in intestinal epithelial regeneration and malignancy. For details see main text. (B) Data are shown as the mean + S.E.M of the indicated number of samples and have been pooled from two experiments. *** p < 0.001, Student's t‐test.
Discussion
In our study we demonstrate a previously unrecognized role for Card9 in the regulation of intestinal immune responses and in the promotion of colitis‐associated cancer. The impaired production of IL‐1β in Card9‐deficient mice after inflammatory challenge is associated with a defective induction of a major regenerative and tumor‐promoting pathway in the intestine that operates through IL‐22 signaling and the STAT3 pathway. Based on our findings, we propose the following model for Card9 function during colitis‐associated cancer development (see also Fig. 6C): Upon intestinal damage and loss of epithelial integrity, microbes and/or damage‐associated danger signals are sensed by Card9‐coupled CLRs on myeloid cells including Dectin‐1, Dectin‐2, or Mincle, or intracellular PRRs such as Nod2 or nucleic acid sensors. These receptors activate the Card9 signaling module, which cooperates with the inflammasomes to induce IL‐1β generation and presumably other cytokines by myeloid cells. Together these factors induce, enhance and maintain local ILC activity presumably together with local metabolites such as aryl hydrocarbon receptor (AHR) ligands. These Card9‐controlled pathways drive in particular local ILC3‐mediated IL‐22 production for subsequent STAT3 activation within IECs to promote both normal epithelial regeneration and malignant CAC growth. Besides ILC3s, other cell types such as TH22, Th17, γδT, NK, or NKT cells in principle also produce IL‐22 and thus might contribute to local IL‐22 levels in a Card9‐dependent manner 21.
While we identified the production of IL‐22 to be impaired in Card9−/− mice, the phenotype of Card9‐deficient animals in the model of colitis‐associated cancer is distinct from that of Il‐22−/‐ mice. While IL‐22‐deficient mice suffer from exacerbated colitis during DSS‐treatment, the severe colitis in these animals promotes tumor growth 47. Unlike Il‐22−/‐ mice, Card9−/− mice do not only lack IL‐22, but they are also impaired in the production of several other inflammatory factors including IL‐1β, TNFα and IL‐17. Thus, it is highly likely that such factors would overcome a pure IL‐22 deficiency and drive inflammation‐mediated tumor growth in the Il‐22−/− mice. Moreover, as we could not detect any defects in the innate cytokine IL‐23 in leukocytes from the intestine of Card9−/− mice, we propose that the impaired induction of intestinal IL‐22, IL‐17 and TNF in these animals is caused by attenuated IL‐1β‐signaling, which was shown to be an inducer of IL‐22, IL‐17 and TNF expression 10, 15, 35, 37, 48, 49. Consistently, former studies have also implicated a role for the Th17 cell cytokine IL‐17 in the promotion of tumor growth in the intestine 50. Nevertheless, only the acute neutralization of IL‐22 with antibodies was able to reduce dysplasia and colorectal cancer growth, while a blockage of IL‐17 only inhibited some parameters of intestinal inflammation including splenic mass 39 demonstrating a predominant role of IL‐22 in the maintenance of the disease 39. Thus, we hypothesize that despite the significant reduction of IL‐17 in lamina propria leukocytes from the intestine of Card9−/− mice, the impairment in IL‐22 production is the major cause for the reduced dysplasia and retarded tumor growth.
During the preparation of this manuscript, an independent report has also demonstrated that Card9−/− mice exhibit a reduction in the percentage of IL‐22+ cells in the lamina propria of the colon after DSS treatment, resulting in defective healing and increased susceptibility to colitis 21. These findings are consistent with our data and further support our concept. In addition, while Lamas et al. did not study the function of these mechanisms during inflammation‐triggered oncogenesis, they could elegantly demonstrate a critical function for Card9 in the control of the composition of the gut microbiome. In their study, the altered microbiome in Card9−/− mice is impaired in the metabolism of tryptophan into indole derivates, which act as AHR ligands and promote local IL‐22 production. We continuously co‐housed Card9‐deficient mice and littermate controls from birth on, to minimize effects of genotype on the microbiota. Nevertheless, based on the current study by Lamas et al. 21, it is conceivable that decreased levels of bacteria that have tryptophan‐catabolizing functions in Card9−/− mouse microbiota might additionally affect tumor growth through IL‐22 regulation. Furthermore, other Card9‐mediated pro‐inflammatory mediators are likely to contribute to tumor pathogenesis. Additional investigations are required to precisely dissect the individual contribution of the different Card9 effector mechanisms including inflammatory cytokines that regulate IL‐22 and collaborate in the promotion of colitis‐associated cancer. Because a growing number of studies have identified Card9 polymorphisms in IBD patients, and IBD is a strong risk factor for colorectal cancer development [2, 3, 4, 5, 51], dysfunctional Card9‐regulated immune pathways could contribute to CAC in humans and could as such constitute a target for therapeutic intervention. Moreover, given that the pathogenic role of aberrant STAT3‐signaling in multiple tumor types is well established 52 and because the IL‐22 receptor is also expressed by other cell types including hepatocytes, lung epithelial cells or keratinocytes 53, Card9‐induced pathways that induce IL‐22 signaling may also impact on other adeno‐ or squamous cell carcinomas.
Materials and methods
Mice, induction of CAC, and acute colitis
Card9−/− mice were generated by our laboratory and described earlier 8. All mice were kept under specific pathogen free conditions. Card9−/− and WT mice were cohoused for at least 3 weeks before and throughout the experiments to minimize variations in the endogenous microbiota.. We therefore used only female mice in all experiments. Azoxymethane (Sigma) was injected intraperitoneally (i.p.) once at 10 mg/kg, followed by three cycles of 3.5% Dextran sulfate sodium salt (MP Biomedicals) in the drinking water for five continuous days at days 5, 26, and 47. For acute DSS colitis, mice were given DSS for 7 days in the drinking water followed by 5 days of water. Mice suffering from weight loss higher than 20% of their initial weight or showing indication of illness were euthanized by cervical dislocation. In order to monitor proliferation of IECs, mice were injected intraperitoneally with Bromodeoxyuridine (BrdU) (Sigma) at 75 mg/kg 12 h prior to sacrifice. For systemic cytokine analysis, blood was collected by submandibular bleeding. All procedures were reviewed and approved by the Regierung von Oberbayern.
Histological analysis
Colons were prepared as ‘Swiss rolls’ and formalin‐fixed for 24 h. For histopathological analysis 2 μm‐thick serial sections (every 200 μm) of paraffin‐embedded colons were subjected to Hematoxylin & Eosin (H&E) staining according to standard procedures. BrdU‐incorporation was detected using anti‐bromodeoxyuridine antibody (AbD Serotec; MCA2060) according to standard immunohistochemical procedures. Automated immunohistochemical staining for cleaved caspase‐3 (Cell Signalling, #9661, Asp175), Ly6G (BD Pharmingen, 551459, monoclonal (1A8)) and phospho‐STAT3 (Cell Signaling, #9145, Tyr705) was performed using a BondMax staining machine (Leica). Hematoxylin was used for counterstaining. Images were acquired using an Olympus SZX12 microscope equipped with a JVC digital camera and using the image processing software cellSens. For tumor quantification and tumor sizes, H&E‐stained slides of colonic serial sections were scanned using a SCN400 Slide scanner (Leica) and analyzed using TissueIA image analysis software (Slidepath, Leica). The area of every single tumor was measured on the slide with the biggest tumor diameter of all serial sections from a specific mouse. The loss of cellular integrity during apoptosis required quantification of apoptosis by measuring the positive stained pixel in confined region of interest (ROI) areas discriminating epithelial and tumor regions for each mouse. BrdU incorporation and STAT3 activation in IECs was evaluated by counting at least five crypts per tumor or epithelium field in three fields/mouse of at least four animals per group. Researchers were blinded to sample identity for histological analysis.
Cytokine quantification
Blood was obtained on days 13, 34, and 55 of the chronic AOM/DSS regimen and stored for 30 min on ice in order to allow clotting. After 10 min centrifugation at 4°C and 2000 g the serum supernatant was collected and stored at ‐80°C until analysis. Cytokine levels were quantified by cytometric bead array (CBA; BD Biosciences) according to the manufacturer's instructions.
Gene expression analysis
Total RNA was isolated with Trizol (Invitrogen) followed by further purification using the RNeasy Mini Kit (Qiagen) and transcribed into cDNA using Superscript II reverse transcriptase (Invitrogen). The specific primer pairs were: IL‐1β, 5′‐tgtaatgaaagacggcacacc‐3′ and 5′‐tcttctttgggtattgcttgg‐3′; IL‐17A, 5’‐ctccagaaggccctcagactac‐3’ and 5’‐ agctttccctccgcattgacacag‐3’; IL‐18, 5’‐tccttgaagttgacgcaaga‐3’ and 5’‐tccagcatcaggacaaagaa‐3’; IL‐22, 5’‐tttcctgaccaaactcagca‐3’ and 5’‐tctggatgttctggtcgtca‐3’; IL‐23a, 5’‐tccctactaggactcagccaac‐3’ and 5’‐tgggcatctgttgggtct‐3’; TFF3, 5’‐gcaccatacattggcttgg‐3’ and 5’‐agagccctctggctaatgct‐3’; KGF (fgf7) 5’‐ccctttgattgccacaattc‐3’ and 5’‐ttgacaaacgaggcaaagtg‐3’; TNF, 5′‐tcttctcattcctgcttgtgg‐3′ and 5′‐ggtctgggccatagaactga‐3′; IL6, 5′‐gctaccaaactggatataatcagga‐3′ and 5′‐ccaggtagctatggtactccagaa‐3′; IL‐11, 5′‐gcaggtggtccttccctaa‐3′ and 5′‐aggcgagacatcaagagctg‐3′; Vegfa, 5′‐aatgctttctccgctctgaa‐3′ and 5′‐gcttcctacagcacagcaga‐3′and β‐actin, 5′‐agacctctatgccaacacag‐3′ and 5′‐tcgtactcctgcttgctgat‐3′. The qPCR Core kit for SYBR Green I (Eurogentec) and a LightCycler® 480 Real‐Time PCR System were used as indicated by the manufacturer. Relative target mRNA expression was calculated as the ratio of the real‐time PCR signal of the particular target mRNA to that of the β‐actin mRNA and normalized to a WT unstimulated control.
Ex vivo colon culture
After AOM/DSS‐treatment, colons from WT and Card9−/− mice were removed and cultured ex vivo as described by Zenewicz et al. 54. On day 3, supernatants were harvested and stored at ‐80°C until analysis of cytokine concentrations by cytometric bead array (BD).
Isolation of lamina propria leukocytes and FACS staining for cytokine production in ILCs
The isolation of lamina propria leukocytes followed by immunofluorescence staining and flow cytometric analysis has been previously described 55. Fluorophore‐conjugated antibodies for flow cytometry and reactive to mouse antigens were purchased from eBioscience unless otherwise indicated: CD3 (145‐2C11), CD19 (1D3), CD45 (30F11), CD127 (A7R34), IL‐22 (1H8PWSR), IL‐17A (TC11‐18H10; BD Biosciences), RORγt (B2D), IFN‐γ(XMG1.2), and T‐bet (eBIO 4B10).
Card gene expression in ILC2 and ILC3 subsets
Microarrays for the expression of Card9 and Card11 in ILC3 and ILC2 subsets were performed as previously described by Hoyler et al. 56.
Isolation of mesenteric lymph node cells, ex vivo stimulation and flow cytometric analysis of IL‐22 production
MLNs were dissected and incubated in HBSS (Ca2+; Mg2+) supplemented with 2% FCS, 2 mg/mL Collagenase D (Roche) and 0.12 mg/mL DNase I (Roche) for 30 min at 37°C. The cell suspension was incubated for 7 min with red blood cell lysis solution (eBioscience) on ice (1 mL/mouse) and plated in complete RPMI (RPMI supplemented with 10% FCS, 1% L‐Glutamine, 1% Penicillin/Streptomycin and 0.1% 2‐Mercaptoethanol, all from Gibco) at a cell concentration of 2 × 106/mL on a 96‐well plate (0.2 mL/well) in duplicates. MLN cells were stimulated for 4 h at 37°C in the presence of recombinant mouse IL‐1β (PreproTech) at a concentration of 10 ng/mL, or/and recombinant mouse IL‐23 (RnD) at a concentration of 10 ng/mL, and 3 μg/mL Brefeldin A (Sigma) or left untreated. Following stimulation, cell suspensions were stained with the following fluorophore‐conjugated antibodies for flow cytometry: B220 (RA3‐6B2), CD3 (145‐2C11), CD4 (GK1.5; eBioscience and BD Biosciences), CD45 (30‐F11), CD90.2 (53‐2.1), IL‐22 (1H8PWSR), NK1.1 (PK136). All antibodies and fixable viability dyes (eFluor 506, eFluor 660) were purchased from eBioscience unless otherwise indicated. Intracellular IL‐22 expression by ILCs following restimulation was assessed by gating on B220−CD3−NK1.1−CD45+CD90.2+ cells and further gating on CD4+ and CD4− ILCs using a FACSCanto II (BD) and FACSDiva (BD) and FlowJo Software.
Statistical analysis
Data were analyzed and graphed using Excel (Microsoft Office) and Prism (GraphPad Software). For comparison between two groups two‐tailed Student's t‐test was used. Survival data were analyzed using the log‐rank test. The level of significance was defined as * p < 0.05 and *** p < 0.001.
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Abbreviations
- CAC
Colitis‐associated cancer
- IEC
Intestinal Epithelial cell
- ILC
Innate lymphoid cell
- DSS
Dextran sodium sulfate
- AOM
Azoxymethane
- WT
wild‐type
Supporting information
Supporting Information Figure 1. Enhanced epithelial damage and inflammation in the colon of Card9‐deficient mice during chronic colitis.
Supporting Information Figure 2. Impaired IL‐1β production in colons of Card9‐/‐ mice in the AOM/DSS colitis‐associated cancer model.
Supporting Information Figure 3. Representative FACS blots and gating strategies.
Supporting Information Figure 4. Expression of STAT3 activators in the colon of AOM/DSS‐treated mice.
Acknowledgements
We would like to thank Daniel Kull, Annette Feuchtinger, Ruth Hillermann, and Michaela Diamanti for their technical expertise and assistance. We would like to thank Axel Walch for his expertise in histopathology. The work was supported by research grants from the Helmholtz Alliance Preclinical Comprehensive Cancer Center (PCCC) to J.R. and M.H., the DFG (SFB 1054/B01 and RU 695/6‐1) to J.R., the "Stiftung Experimentelle Biomedizin" (Hofschneider Foundation) to M.H., and the ERC (grant 322865 to JR, grant 311377 to A.D., and LiverCancerMech to M.H.). H.B. and S.R. designed and performed experiments, analyzed data and wrote the manuscript. K.P. and E.K. designed and performed experiments and analyzed data. R.H. performed experiments. M.H., A.D., and F.G. designed experiments. J.R. designed the study, analyzed data, and wrote the manuscript.
References
- 1. Beaugerie, L. and Itzkowitz, S. H. , Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015. 372: 1441–1452. [DOI] [PubMed] [Google Scholar]
- 2. Zhernakova, A. , Festen, E. M. , Franke, L. , Trynka, G. , van Diemen, C. C. , Monsuur, A. J. , Bevova, M. et al., Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 2008. 82: 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Franke, A. , McGovern, D. P. , Barrett, J. C. , Wang, K. , Radford‐Smith, G. L. , Ahmad, T. , Lees, C. W. et al., Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 2010. 42: 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGovern, D. P. , Gardet, A. , Torkvist, L. , Goyette, P. , Essers, J. , Taylor, K. D. , Neale, B. M. et al., Genome‐wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010. 42: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rivas, M. A. , Beaudoin, M. , Gardet, A. , Stevens, C. , Sharma, Y. , Zhang, C. K. , Boucher, G. et al., Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011. 43: 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsu, Y. M. , Zhang, Y. , You, Y. , Wang, D. , Li, H. , Duramad, O. , Qin, X. F. et al., The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 2007. 8: 198–205. [DOI] [PubMed] [Google Scholar]
- 7. Roth, S. and Ruland, J. , Caspase recruitment domain‐containing protein 9 signaling in innate immunity and inflammation. Trends Immunol. 2013. 34: 243–250. [DOI] [PubMed] [Google Scholar]
- 8. Gross, O. , Gewies, A. , Finger, K. , Schafer, M. , Sparwasser, T. , Peschel, C. , Forster, I. et al., Card9 controls a non‐TLR signalling pathway for innate anti‐fungal immunity. Nature 2006. 442: 651–656. [DOI] [PubMed] [Google Scholar]
- 9. Hara, H. , Ishihara, C. , Takeuchi, A. , Imanishi, T. , Xue, L. , Morris, S. W. , Inui, M. et al., The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM‐associated and Toll‐like receptors. Nat. Immunol. 2007. 8: 619–629. [DOI] [PubMed] [Google Scholar]
- 10. Acosta‐Rodriguez, E. V. , Napolitani, G. , Lanzavecchia, A. and Sallusto, F. , Interleukins 1beta and 6 but not transforming growth factor‐beta are essential for the differentiation of interleukin 17‐producing human T helper cells. Nat. Immunol. 2007. 8: 942–949. [DOI] [PubMed] [Google Scholar]
- 11. Gross, O. , Poeck, H. , Bscheider, M. , Dostert, C. , Hannesschlager, N. , Endres, S. , Hartmann, G. et al., Syk kinase signalling couples to the Nlrp3 inflammasome for anti‐fungal host defence. Nature 2009. 459: 433–436. [DOI] [PubMed] [Google Scholar]
- 12. Poeck, H. , Bscheider, M. , Gross, O. , Finger, K. , Roth, S. , Rebsamen, M. , Hannesschlager, N. et al., Recognition of RNA virus by RIG‐I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010. 11: 63–69. [DOI] [PubMed] [Google Scholar]
- 13. Roth, S. , Rottach, A. , Lotz‐Havla, A. S. , Laux, V. , Muschaweckh, A. , Gersting, S. W. , Muntau, A. C. et al., Rad50‐CARD9 interactions link cytosolic DNA sensing to IL‐1beta production. Nat. Immunol. 2014. 15: 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sokol, H. , Conway, K. L. , Zhang, M. , Choi, M. , Morin, B. , Cao, Z. , Villablanca, E. J. et al., Card9 mediates intestinal epithelial cell restitution, T‐helper 17 responses, and control of bacterial infection in mice. Gastroenterology 2013. 145: 591–601 e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sutton, C. , Brereton, C. , Keogh, B. , Mills, K. H. and Lavelle, E. C. , A crucial role for interleukin (IL)‐1 in the induction of IL‐17‐producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006. 203: 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hugot, J. P. , Chamaillard, M. , Zouali, H. , Lesage, S. , Cezard, J. P. , Belaiche, J. , Almer, S. et al., Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 2001. 411: 599–603. [DOI] [PubMed] [Google Scholar]
- 17. Iliev, I. D. , Funari, V. A. , Taylor, K. D. , Nguyen, Q. , Reyes, C. N. , Strom, S. P. , Brown, J. et al., Interactions between commensal fungi and the C‐type lectin receptor Dectin‐1 influence colitis. Science 2012. 336: 1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neufert, C. , Becker, C. and Neurath, M. F. , An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation‐driven tumor progression. Nat. Protoc. 2007. 2: 1998–2004. [DOI] [PubMed] [Google Scholar]
- 19. Okayasu, I. , Ohkusa, T. , Kajiura, K. , Kanno, J. and Sakamoto, S. , Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut 1996. 39: 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka, T. , Kohno, H. , Suzuki, R. , Yamada, Y. , Sugie, S. and Mori, H. , A novel inflammation‐related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003. 94: 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamas, B. , Richard, M. L. , Leducq, V. , Pham, H. P. , Michel, M. L. , Da Costa, G. , Bridonneau, C. et al., CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016. 22: 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Byrne, F. R. , Farrell, C. L. , Aranda, R. , Rex, K. L. , Scully, S. , Brown, H. L. , Flores, S. A. et al., rHuKGF ameliorates symptoms in DSS and CD4(+)CD45RB(Hi) T cell transfer mouse models of inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver. Physiol. 2002. 282: G690–701. [DOI] [PubMed] [Google Scholar]
- 23. Holmkvist, P. , Pool, L. , Hagerbrand, K. , Agace, W. W. and Rivollier, A. , IL‐18Ralpha‐deficient CD4(+) T cells induce intestinal inflammation in the CD45RB(hi) transfer model of colitis despite impaired innate responsiveness. Eur. J. Immunol. 2016. 46: 1371–1382. [DOI] [PubMed] [Google Scholar]
- 24. Izcue, A. , Coombes, J. L. and Powrie, F. , Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009. 27: 313–338. [DOI] [PubMed] [Google Scholar]
- 25. Salcedo, R. , Worschech, A. , Cardone, M. , Jones, Y. , Gyulai, Z. , Dai, R. M. , Wang, E. et al., MyD88‐mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J. Exp. Med. 2010. 207: 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taupin, D. and Podolsky, D. K. , Trefoil factors: initiators of mucosal healing. Nat. Rev. Mol. Cell Biol. 2003. 4: 721–732. [DOI] [PubMed] [Google Scholar]
- 27. Bertin, J. , Guo, Y. , Wang, L. , Srinivasula, S. M. , Jacobson, M. D. , Poyet, J. L. , Merriam, S. et al., CARD9 is a novel caspase recruitment domain‐containing protein that interacts with BCL10/CLAP and activates NF‐kappa B. J. Biol. Chem. 2000. 275: 41082–41086. [DOI] [PubMed] [Google Scholar]
- 28. Glocker, E. O. , Hennigs, A. , Nabavi, M. , Schaffer, A. A. , Woellner, C. , Salzer, U. , Pfeifer, D. et al., A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009. 361: 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chung, Y. , Chang, S. H. , Martinez, G. J. , Yang, X. O. , Nurieva, R. , Kang, H. S. , Ma, L. et al., Critical regulation of early Th17 cell differentiation by interleukin‐1 signaling. Immunity 2009. 30: 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dorhoi, A. , Desel, C. , Yeremeev, V. , Pradl, L. , Brinkmann, V. , Mollenkopf, H. J. , Hanke, K. et al., The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 2010. 207: 777–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LeibundGut‐Landmann, S. , Gross, O. , Robinson, M. J. , Osorio, F. , Slack, E. C. , Tsoni, S. V. , Schweighoffer, E. et al., Syk‐ and CARD9‐dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007. 8: 630–638. [DOI] [PubMed] [Google Scholar]
- 32. Schoenen, H. , Bodendorfer, B. , Hitchens, K. , Manzanero, S. , Werninghaus, K. , Nimmerjahn, F. , Agger, E. M. et al., Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose‐dibehenate. J. Immunol. 2010. 184: 2756–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Werninghaus, K. , Babiak, A. , Gross, O. , Holscher, C. , Dietrich, H. , Agger, E. M. , Mages, J. et al., Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma‐Syk‐Card9‐dependent innate immune activation. J. Exp. Med. 2009. 206: 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cella, M. , Otero, K. and Colonna, M. , Expansion of human NK‐22 cells with IL‐7, IL‐2, and IL‐1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. U. S. A. 2010. 107: 10961–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hughes, T. , Becknell, B. , Freud, A. G. , McClory, S. , Briercheck, E. , Yu, J. , Mao, C. et al., Interleukin‐1beta selectively expands and sustains interleukin‐22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 2010. 32: 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montaldo, E. , Juelke, K. and Romagnani, C. , Group 3 innate lymphoid cells (ILC3s): Origin, differentiation, and plasticity in humans and mice. Eur. J. Immunol. 2015. 45: 2171–2182. [DOI] [PubMed] [Google Scholar]
- 37. Sonnenberg, G. F. , Fouser, L. A. and Artis, D. , Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL‐22. Nat. Immunol. 2011. 12: 383–390.21502992 [Google Scholar]
- 38. Sugimoto, K. , Ogawa, A. , Mizoguchi, E. , Shimomura, Y. , Andoh, A. , Bhan, A. K. , Blumberg, R. S. et al., IL‐22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 2008. 118: 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kirchberger, S. , Royston, D. J. , Boulard, O. , Thornton, E. , Franchini, F. , Szabady, R. L. , Harrison, O. et al., Innate lymphoid cells sustain colon cancer through production of interleukin‐22 in a mouse model. J. Exp. Med. 2013. 210: 917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lim, C. and Savan, R. , The role of the IL‐22/IL‐22R1 axis in cancer. Cytokine Growth Factor Rev. 2014. 25: 257–271. [DOI] [PubMed] [Google Scholar]
- 41. Spits, H. , Artis, D. , Colonna, M. , Diefenbach, A. , Di Santo, J. P. , Eberl, G. , Koyasu, S. et al., Innate lymphoid cells–a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013. 13: 145–149. [DOI] [PubMed] [Google Scholar]
- 42. Hara, H. and Saito, T. , CARD9 versus CARMA1 in innate and adaptive immunity. Trends Immunol. 2009. 30: 234–242. [DOI] [PubMed] [Google Scholar]
- 43. Satpathy, A. T. , Briseno, C. G. , Lee, J. S. , Ng, D. , Manieri, N. A. , Kc, W. , Wu, X. et al., Notch2‐dependent classical dendritic cells orchestrate intestinal immunity to attaching‐and‐effacing bacterial pathogens. Nat. Immunol. 2013. 14: 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grivennikov, S. , Karin, E. , Terzic, J. , Mucida, D. , Yu, G. Y. , Vallabhapurapu, S. , Scheller, J. et al., IL‐6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis‐associated cancer. Cancer Cell 2009. 15: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pickert, G. , Neufert, C. , Leppkes, M. , Zheng, Y. , Wittkopf, N. , Warntjen, M. , Lehr, H. A. et al., STAT3 links IL‐22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009. 206: 1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bollrath, J. , Phesse, T. J. , von Burstin, V. A. , Putoczki, T. , Bennecke, M. , Bateman, T. , Nebelsiek, T. et al., gp130‐mediated Stat3 activation in enterocytes regulates cell survival and cell‐cycle progression during colitis‐associated tumorigenesis. Cancer Cell 2009. 15: 91–102. [DOI] [PubMed] [Google Scholar]
- 47. Huber, S. , Gagliani, N. , Zenewicz, L. A. , Huber, F. J. , Bosurgi, L. , Hu, B. , Hedl, M. et al., IL‐22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012. 491: 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cella, M. , Fuchs, A. , Vermi, W. , Facchetti, F. , Otero, K. , Lennerz, J. K. , Doherty, J. M. et al., A human natural killer cell subset provides an innate source of IL‐22 for mucosal immunity. Nature 2009. 457: 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ikejima, T. , Okusawa, S. , Ghezzi, P. , van der Meer, J. W. and Dinarello, C. A. , Interleukin‐1 induces tumor necrosis factor (TNF) in human peripheral blood mononuclear cells in vitro and a circulating TNF‐like activity in rabbits. J. Infect. Dis. 1990. 162: 215–223. [DOI] [PubMed] [Google Scholar]
- 50. Grivennikov, S. I. , Wang, K. , Mucida, D. , Stewart, C. A. , Schnabl, B. , Jauch, D. , Taniguchi, K. et al., Adenoma‐linked barrier defects and microbial products drive IL‐23/IL‐17‐mediated tumour growth. Nature 2012. 491: 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lees, C. W. , Barrett, J. C. , Parkes, M. and Satsangi, J. , New IBD genetics: common pathways with other diseases. Gut 2011. 60: 1739–1753. [DOI] [PubMed] [Google Scholar]
- 52. Bollrath, J. and Greten, F. R. , IKK/NF‐kappaB and STAT3 pathways: central signalling hubs in inflammation‐mediated tumour promotion and metastasis. EMBO Rep. 2009. 10: 1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Witte, E. , Witte, K. , Warszawska, K. , Sabat, R. and Wolk, K. , Interleukin‐22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010. 21: 365–379. [DOI] [PubMed] [Google Scholar]
- 54. Zenewicz, L. A. , Yancopoulos, G. D. , Valenzuela, D. M. , Murphy, A. J. , Stevens, S. and Flavell, R. A. , Innate and adaptive interleukin‐22 protects mice from inflammatory bowel disease. Immunity 2008. 29: 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kiss, E. A. , Vonarbourg, C. , Kopfmann, S. , Hobeika, E. , Finke, D. , Esser, C. and Diefenbach, A. , Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011. 334: 1561–1565. [DOI] [PubMed] [Google Scholar]
- 56. Hoyler, T. , Klose, C. S. , Souabni, A. , Turqueti‐Neves, A. , Pfeifer, D. , Rawlins, E. L. , Voehringer, D. et al., The transcription factor GATA‐3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012. 37: 634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1. Enhanced epithelial damage and inflammation in the colon of Card9‐deficient mice during chronic colitis.
Supporting Information Figure 2. Impaired IL‐1β production in colons of Card9‐/‐ mice in the AOM/DSS colitis‐associated cancer model.
Supporting Information Figure 3. Representative FACS blots and gating strategies.
Supporting Information Figure 4. Expression of STAT3 activators in the colon of AOM/DSS‐treated mice.