

Abstract
This study tests the hypothesis that the prevalence of severe clinical manifestations in Gaucher disease type 1 (GD1) patients at the time of treatment initiation has changed since alglucerase/imiglucerase enzyme replacement therapy (ERT) was approved in the United States (US) in 1991. US alglucerase/imiglucerase‐treated GD1 patients from the International Collaborative Gaucher Group Gaucher Registry clinicaltrials.gov NCT00358943 were stratified by age at ERT initiation (<18, 18 to <50, ≥50 years), era of ERT initiation (1991‐1995, 1996‐2000, 2001‐2005, 2006‐2009), and splenectomy status pre‐ERT. Prevalence of splenectomy decreased dramatically across the eras among all age groups. Bone manifestations were more prevalent in splenectomized patients than non‐splenectomized patients in all age groups. Prevalence of bone manifestations differed across eras in certain age groups: non‐splenectomized patients had a lower prevalence of ischemic bone events (pediatric patients) and bone crisis (pediatric patients and adults 18 to <50 years) in later eras; splenectomized adult (18 to <50 years) patients had a lower prevalence of ischemic bone events and bone crisis in later eras. Over two decades after the introduction of ERT, the prevalence of splenectomy and associated skeletal complications has declined dramatically. Concomitantly, the interval between diagnosis and initiation of ERT has decreased, most strikingly in pediatric patients who have the most severe disease. Together, these findings suggest that since the introduction of alglucerase/imiglucerase ERT, optimal standard of care has become established in the US to prevent destructive complications of GD1.
1. INTRODUCTION
In Gaucher disease (GD) biallelic mutations in the glucocerebrosidase gene result in defective lysosomal acid β‐glucosidase (glucocerebrosidase, EC 3.2.1.21) that causes widespread accumulation of macrophages engorged with glucosylceramide‐laden lysosomes and system‐wide pathophysiology mediated by accumulating sphingolipids.1 The clinical effects are multisystemic with heterogeneous manifestations and natural history.2, 3 Symptomatic presentation of GD can occur at any age with variable combinations of cytopenia, hepatosplenomegaly, diverse patterns of skeletal disease, and growth retardation, and in the most severe cases, neurodegenerative disease. The vast majority of cases are patients with GD type 1 (GD1) who lack the early‐onset neurological disease that characterizes type 2 and type 3 disease.3
GD1 is a chronic progressive disease.4 The natural history of GD1 prior to the availability of an effective treatment was that of a devastating disease associated with a high incidence of splenectomy, bleeding complications, liver disease, painful and disabling skeletal disease due to fragility fractures, bone crisis and avascular osteonecrosis (AVN), pulmonary hypertension, malignancies, and premature death.5, 6, 7, 8, 9, 10 Only symptomatic care was available in the form of blood transfusions and joint replacement surgery. Although splenectomy was frequently performed for symptomatic relief (pressure symptoms and cytopenia), it resulted in accelerated disease in bone and in other organs as well as life‐threatening complications such as pulmonary hypertension.5, 10, 11
Following proof of principle studies12 and a pivotal clinical trial in 1991,13 the United States (US) Food and Drug Administration (FDA) approved alglucerase (mannose‐terminated, human placental acid β‐glucosidase, Ceredase®, Sanofi Genzyme, Cambridge, MA), the first macrophage‐targeted enzyme replacement therapy (ERT) for GD1. In 1994, following a clinical trial showing clinical equivalency of alglucerase and a recombinant version of mannose‐terminated glucocerebrosidase,14 imiglucerase (Cerezyme®, Sanofi Genzyme, Cambridge, MA) was approved for treatment of GD1. ERT with mannose‐terminated acid β‐glucosidase (imiglucerase and others) is the current standard of care for GD1; imiglucerase has been shown to reverse many disease manifestations, including some bone pathologies,15, 16, 17 and to reduce the risk of AVN.18, 19
Since ERT became the standard of care in the 1990s, there appears to be a noticeable decline in the frequency of severe phenotypes typically encountered in the pre‐ERT era. The purpose of this analysis is to confirm these clinical observations through a descriptive analysis of the disease manifestations of GD1 patients at the initiation of alglucerase/imiglucerase treatment during the two decades following the approval of alglucerase and imiglucerase. The International Collaborative Gaucher Group (ICGG) Gaucher Registry (clinicaltrials.gov NCT00358943), initiated in 1991, offers an excellent instrument to assess temporal changes in the impact of the availability of ERT. Over the past quarter‐century, the ICGG Gaucher Registry has captured phenotypic, genotypic, natural history and treatment response data in patients with GD from around the world. More than 6000 GD patients from over 60 countries have been enrolled in the Registry.20
We analyzed clinical characteristics of symptomatic GD1 patients at the initiation of alglucerase/imiglucerase ERT in four approximately 5‐year intervals over the first two decades after approval of alglucerase and imiglucerase, from 1991 to 2009. We focused in particular on whether the prevalence of splenectomy at the time of treatment initiation was declining, as this procedure has been associated with rapid progression to severe skeletal, pulmonary and hepatic manifestations as well as increased cancer risk.2, 5, 11, 19, 21
2. METHODS
2.1. The ICGG Gaucher Registry
The ICGG Gaucher Registry is a voluntary, global, observational database, which is governed by an international group of experts (ICGG Gaucher Registry Board of Advisors), with operational support provided by Sanofi Genzyme. It is open to all GD patients around the world regardless of their treatment status or choice of treatment. Clinical assessments and care of patients enrolled in the Registry are determined by their treating physicians. Treating physicians voluntarily provide data on assessments currently used to monitor the clinical manifestations, disease progression of GD and response to therapy.
2.2. Patient selection
As of March 04, 2016, GD1 patients were selected from the ICGG Gaucher Registry based on the following criteria: (1) enrolled from clinics and physician practices in the US, (2) began imiglucerase (“imiglucerase” from here forward will refer to both alglucerase and imiglucerase treatment) ERT between January 1, 1991 and June 24, 2009 (inclusive), (3) had a known date of GD1 diagnosis and of imiglucerase initiation, (4) had reported splenectomy status and (5) if splenectomized, had a known date of splenectomy. The sample was limited to US patients as imiglucerase treatment first became available in the US. The rationale for restricting the study to US patients was that this dataset provided the longest potential time window of treatment initiation for analysis. Also, focusing on a single country minimizes confounders with respect to variable clinical practice and phenotypes in different parts of the world, for example, indications and timing of starting treatment and genotypes.
2.3. Study design
Patients were classified into three age cohorts based upon their age at treatment initiation: pediatric patients <18 years of age, adult patients 18 to <50 years of age, adult patients ≥50 years of age. Patients were also stratified by splenectomy status and date of imiglucerase treatment initiation.
Four approximately equal eras based on date of treatment initiation were defined beginning in 1991, the year that alglucerase became commercially available in the US. Era 1 was defined as 1991 through 1995, Era 2 as 1996 through 2000, Era 3 as 2001 through 2005, and Era 4 as 2006 through 2009. Eras are of equal length (5 years) with the exception of Era 4, which is 4 years due to the shortage of imiglucerase that occurred beginning June 25, 2009.
This study was conducted according to the principles of the Declaration of Helsinki (2013).22 Informed consent was obtained from all subjects. The data are reported in accordance with STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines.23
2.4. Demographics
Demographic variables included sex, age at GD1 diagnosis, year of GD1 diagnosis, age at imiglucerase treatment initiation, time interval between diagnosis and initiation of imiglucerase, splenectomy status, year of splenectomy, and genotype (N370S homozygous, N370S heterozygous, and other). Genotype was not available for all patients since it was not an inclusion criterion for the current analysis. Splenectomized patients included those with either total or partial splenectomy.
2.5. Clinical parameters
Clinical parameters analyzed included bone, visceral and hematologic manifestations as entered by the reporting sites. Bone manifestations included bone pain, acute bone crisis, AVN, infarction, fractures and hip replacement. Bone pain according to the Registry guidance was defined as present if the patient reported bone pain as occurring in the month before the medical visit. Bone crisis was defined as stated in the Registry guidance, as acute pain requiring immobilization, narcotics, and accompanied by one or more of the following: periosteal elevation, elevated white blood cell count, fever and/or debilitation of >3 days. Information on AVN, infarction, and fracture was collected primarily from skeletal radiology exam reports, and secondarily from medical history at the time of enrollment in the ICGG Gaucher Registry. Information on hip replacement was collected from reports of medically significant surgical procedures. AVN is defined as bone necrosis resulting from infarction in a sub‐articular (ie, joint) location. Infarction is defined as an area of necrosis at other sites, (ie, medullary infarction). An ischemic bone event was defined as present if the patient was reported as having either AVN or infarction. Baseline was defined as any occurrence of bone crisis, bone pain, ischemic bone events, fracture, or hip replacement prior to treatment initiation or up to 6 weeks from the date of treatment initiation.
Visceral parameters included liver and spleen volumes expressed as multiples of normal (MN) (2.5% of body weight for liver; 0.2% of body weight for spleen) as described previously.18 Splenomegaly was classified categorically as Mild or None (≤5 MN), Moderate (>5 to ≤15 MN), and Severe (>15 MN). Hepatomegaly was defined categorically as Mild or None (≤1.25 MN), Moderate (>1.25 to ≤ 2.5 MN), and Severe (>2.5 MN). Baseline was defined as the data point closest to initiation of treatment with imiglucerase using a window of no more than −6 months to +6 weeks (inclusive) from initiation of treatment for liver and spleen volumes.
Hematological parameters included hemoglobin concentration (g/dL) and platelet count (×103/mm3). Anemia was defined categorically based on hemoglobin concentration according to age and gender norms: <12 g/dL for males older than 12 years; <11 g/dL for females older than 12 years; <10.5 g/dL for children ages >2 to 12 years; <9.5 g/dL for children ages 6 months to 2 years; <10.1 g/dL for children younger than 6 months of age. As defined in the Registry protocol, severity of thrombocytopenia was classified categorically according to the platelet count: Mild or None (≥120 × 103/mm3), Moderate (60‐<120 × 103/mm3) or Severe (<60 × 103/mm3). Baseline was defined as the data point closest to initiation of treatment with imiglucerase using a window of no more than −8 weeks to +2 weeks (inclusive) from initiation of treatment for hemoglobin and platelet count.
2.6. Statistical analyses
Descriptive analyses for each age cohort by splenectomy status at the time of treatment initiation and by era were conducted for baseline demographics. For patients who had been splenectomized prior to treatment initiation, summary statistics were calculated for age at splenectomy, calendar year of splenectomy, and whether they were splenectomized prior to 1991 or during/after 1991. Patients splenectomized on or after the date of treatment initiation were considered to have an intact spleen at baseline. Within each age cohort, the chi‐square test was used to compare splenectomy status at treatment initiation and genotype frequency across the eras, and the Kruskal‐Wallis test was used to compare age at diagnosis, age at treatment initiation, and the time interval between diagnosis and initiation of imiglucerase across the eras.
Collapsing data from all four eras together, the prevalence of bone crisis, bone pain, and ischemic bone events was compared between splenectomized and non‐splenectomized patients at baseline within each age group using the chi‐square test or Fisher's exact test (for strata with small sample sizes). Within categories of age group and splenectomy status at initiation of imiglucerase, the chi‐square test (or Fisher's exact test, when appropriate) was used to test differences in prevalence across the eras of each bone manifestation at baseline.
Hematologic endpoints (hemoglobin concentration, platelet count) and liver volume at baseline were analyzed using analysis of variance (ANOVA) to compare mean values across the eras, stratified by age cohort and splenectomy status. Spleen volume for patients with intact spleens at treatment initiation was also analyzed using ANOVA to compare mean values across the eras stratified by age cohort.
All hypothesis tests were two‐tailed with an alpha level of 0.05. Statistical analyses were conducted using the SAS software version 9.2 (Cary, North Carolina).
3. RESULTS
3.1. Patients
As of March 04, 2016, 1427 (69.0%) of 2069 US GD1 patients in the ICGG Gaucher Registry met all inclusion and exclusion criteria. Most excluded patients were excluded because they were never reported as treated (n = 327); 178 patients were excluded because they did not start imiglucerase within the specified time windows; 99 were excluded because of missing a GD1 diagnosis date; 12 were excluded because the first treatment was unknown; five were excluded because the first treatment was not imiglucerase; five patients were excluded for unknown imiglucerase initiation date; two patients were excluded for conflicting treatment initiation dates; 13 were excluded for missing date of splenectomy (if splenectomized) and one patient was excluded for missing splenectomy status.
The distributions of sex, age at GD1 diagnosis, age at imiglucerase initiation, splenectomy status before treatment initiation, and genotype by age cohort and treatment initiation era are presented in Table 1. Mean age at diagnosis differed significantly across the eras for each of the three age groups (P < .05 for pediatric patients; P < .0001 for adults 18 to <50 years; P < .05 for adults ≥50 years) as did age at initiation of treatment for pediatric patients (P < .0001) (Table 1). Genotype frequency across the eras was significantly different for the adults 18 to <50 years (P < .0001). There appears to be a greater frequency of the N370S homozygous genotype in later eras for this age group (Table 1). No statistically significant differences in genotype were present in the other two age groups.
Table 1.
Patient characteristics by age group and imiglucerase initiation era
| Imiglucerase Initiation Eras | |||||
|---|---|---|---|---|---|
| Statistic | 1991–1995 | 1996–2000 | 2001–2005 | 2006–2009 | |
| Total number of patientsa | N | 694 | 345 | 265 | 123 |
| <18 Years of Age at Imiglucerase Initiation | |||||
| Total Patientsa | N | 172 | 109 | 80 | 38 |
| Sex | |||||
| Male | n (%) | 80 (46.5) | 53 (48.6) | 46 (57.5) | 16 (42.1) |
| Female | n (%) | 92 (53.5) | 56 (51.4) | 34 (42.5) | 22 (57.9) |
| Age at GD1 diagnosis (years) | Mean (SD) | 5.5 (3.57) | 6.0 (4.18) | 7.1 (4.34) | 6.2 (4.54) |
|
Median (25th, 75th) |
4.9 (3.0, 7.3) |
5.0 (2.8, 8.6) |
6.2 (3.7, 9.9) |
5.3 (2.3, 9.9) |
|
| Min, Max | 0.0, 15.9 | 0.0, 16.3 | 0.8, 17.2 | 0.1, 16.8 | |
| Age at imiglucerase initiation (years) | Mean (SD) | 9.9 (4.49) | 7.4 (4.72) | 7.9 (4.23) | 7.8 (4.84) |
|
Median (25th, 75th) |
10.1 (6.0, 13.7) |
5.9 (3.7, 10.3) |
6.6 (4.6, 11.1) |
5.7 (4.1, 11.3) |
|
| Min, Max | 0.7, 17.9 | 0.4, 17.8 | 1.7, 17.5 | 1.5, 17.5 | |
| Splenectomized prior to date of imiglucerase initiation | n (%) | 34 (19.8) | 3 (2.8) | 1 (1.3) | 0 |
| Genotypeb | n | 137 | 100 | 72 | 36 |
| N370S/N370S | n (%) | 21 (15.3) | 12 (12.0) | 10 (13.9) | 5 (13.9) |
| N370S/Other | n (%) | 81 (59.1) | 66 (66.0) | 47 (65.3) | 18 (50.0) |
| Other/Other | n (%) | 35 (25.5) | 22 (22.0) | 15 (20.8) | 13 (36.1) |
| 18 to <50 years age at imiglucerase initiation | |||||
| Total Patientsa | N | 370 | 156 | 120 | 43 |
| Sex | |||||
| Male | n (%) | 163 (44.1) | 74 (47.4) | 61 (50.8) | 19 (44.2) |
| Female | n (%) | 207 (55.9) | 82 (52.6) | 59 (49.2) | 24 (55.8) |
| Age at GD1 diagnosis (years) | Mean (SD) | 18.7 (12.28) | 25.5 (12.70) | 28.4 (12.44) | 27.0 (12.84) |
|
Median (25th, 75th) |
17.8 (7.5, 28.0) |
26.8 (17.3, 34.7) |
29.4 (21.0, 38.2) |
26.8 (17.8, 38.2) |
|
| Min, Max | 0.0, 49.5 | 0.0, 49.4 | 1.4, 49.7 | 4.2, 49.4 | |
| Age at imiglucerase initiation (years) | Mean (SD) | 34.9 (8.88) | 34.7 (9.24) | 35.8 (8.52) | 36.7 (8.00) |
|
Median (25th, 75th) |
35.7 (26.9, 42.3) |
34.1 (27.3, 42.8) |
37.2 (29.4, 42.0) |
38.4 (30.6, 42.6) |
|
| Min, Max | 18.2, 50.0 | 18.1, 49.7 | 18.2, 49.8 | 18.1, 50.0 | |
| Splenectomized prior to date of imiglucerase initiation | n (%) | 158 (42.7) | 30 (19.2) | 19 (15.8) | 7 (16.3) |
| Genotypeb | n | 272 | 140 | 112 | 36 |
| N370S/N370S | n (%) | 75 (27.6) | 71 (50.7) | 59 (52.7) | 15 (41.7) |
| N370S/Other | n (%) | 168 (61.8) | 60 (42.9) | 49 (43.8) | 18 (50.0) |
| Other/Other | n (%) | 29 (10.7) | 9 (6.4) | 4 (3.6) | 3 (8.3) |
| ≥50 years of age at imiglucerase initiation | |||||
| Total Patientsa | N | 152 | 80 | 65 | 42 |
| Sex | |||||
| Male | n (%) | 75 (49.3) | 42 (52.5) | 34 (52.3) | 25 (59.5) |
| Female | n (%) | 77 (50.7) | 38 (47.5) | 31 (47.7) | 17 (40.5) |
| Age at GD1 diagnosis (years) | Mean (SD) | 40.2 (21.40) | 44.8 (18.42) | 48.6 (17.67) | 46.3 (20.60) |
|
Median (25th, 75th) |
38.3 (24.9, 55.5) |
47.2 (29.3, 58.2) |
52.5 (39.9, 59.3) |
51.1 (28.2, 63.9) |
|
| Min, Max | 0.5, 81.5 | 5.9, 74.8 | 4.5, 81.8 | 4.9, 78.9 | |
| Age at imiglucerase initiation (years) | Mean (SD) | 62.0 (9.36) | 61.8 (8.59) | 62.6 (8.48) | 63.5 (7.92) |
|
Median (25th, 75th) |
60.1 (54.2, 68.3) |
60.4 (54.4, 69.5) |
61.8 (56.5, 68.1) |
65.2 (57.6, 69.0) |
|
| Min, Max | 50.0, 84.2 | 50.1, 85.0 | 50.1, 84.1 | 50.0, 82.0 | |
| Splenectomized prior to date of imiglucerase initiation | n (%) | 67 (44.1) | 22 (27.5) | 11 (16.9) | 8 (19.0) |
| Genotypeb | n | 104 | 70 | 58 | 37 |
| N370S/N370S | n (%) | 59 (56.7) | 47 (67.1) | 43 (74.1) | 23 (62.2) |
| N370S/Other | n (%) | 40 (38.5) | 19 (27.1) | 12 (20.7) | 12 (32.4) |
| Other/Other | n (%) | 5 (4.8) | 4 (5.7) | 3 (5.2) | 2 (5.4) |
“Total Patients” includes all patients who met inclusion criteria for analysis.
Sample sizes are not the same as for other variables because genotype was not part of the inclusion criteria for the study and not all patients had a genotype recorded in the Registry.
3.2. Prevalence of splenectomy
Of the 1427 patients starting ERT across all 4 eras, 360 (25.2%) patients had undergone splenectomy prior to the initiation of imiglucerase (Table 1). Among the patients who had undergone splenectomy (379), the majority (360 patients, 95%) were splenectomized prior to treatment. The reduction in splenectomy prevalence pre‐ERT treatment was striking. Both adult groups who started ERT in Era 1 had a high prevalence of splenectomy, with 43% of adults 18 to <50 years and 44% of adults ≥50 years having been splenectomized pre‐ERT (Table 1). In pediatric patients, approximately 20% of patients who started ERT in Era 1 were splenectomized (Table 1). Compared to Era 1, patients who started ERT in subsequent Eras 2, 3, and 4, had significantly lower prevalence of splenectomy at the time of initiation of treatment (Table 1 and Figure 1). The trend was most marked in the pediatric population in whom splenectomy at the time of treatment initiation virtually disappeared in Era 4 relative to Era 1 (0% vs. 19.8%, respectively). The majority of the splenectomized adult patients, even those who began ERT in the later eras, had been splenectomized prior to 1991 (Supporting Information Table S1).
Figure 1.
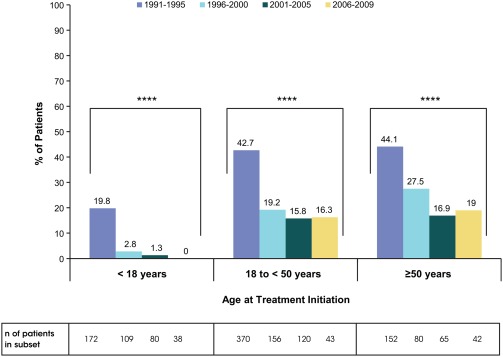
The proportion of GD1 patients who were splenectomized prior to imiglucerase initiation by age group and era of imiglucerase initiation. ****P < .0001. P‐values are from chi‐square test. Abbreviations: GD1, Gaucher disease type 1
3.3. Bone manifestations
In each age group, there was a highly significant excess of key manifestations of bone disease (bone crisis, ischemic bone events, and bone pain) in splenectomized patients compared to patients with intact spleen (P < .0001, Supporting Information Figure S1) with data collapsed across all 4 treatment initiation eras. Further stratification of patients with and without spleen by treatment era revealed a significantly lower prevalence of bone crisis in pediatric patients with intact spleen with each successive era of ERT (P < .01) (Figure 2A). We also found a decreasing prevalence of bone crisis in splenectomized (P < .05) and non‐splenectomized (P < .05) adults 18 to <50 years of age in later eras than in the first era. There were no significant differences in adults ≥50 years of age across eras.
Figure 2.
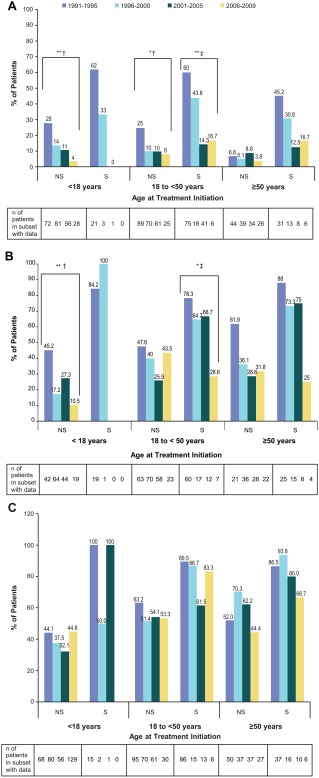
Prevalence of bone manifestations of GD1 by splenectomy status, age group, and era of imiglucerase initiation: Panel A) Bone crisis; Panel B) Ischemic bone events; Panel C) Bone pain. *P < .05; **P < .01; † P‐values are from chi ‐square test; ‡ P ‐values are from Fisher's exact test. Strata without a corresponding P‐value indicate that P > .05. Abbreviations: GD1, Gaucher disease type 1; NS, non‐splenectomized; S, splenectomized
The results for ischemic bone events parallel those for bone crisis, with a lower prevalence of patients with ischemic bone events in later eras (Figure 2B). Decreasing prevalence of ischemic bone events with each successive era is significant in pediatric patients with intact spleens (P < .01) and splenectomized adults 18 to <50 years of age (P < .05). Very few patients who initiated therapy in childhood have been splenectomized and the proportion of splenectomized patients declines in later eras for adult patients (Table 1), necessitating caution when interpreting results involving small numbers of patients.
In general, bone pain was reported by a large proportion of patients by the time of treatment initiation with no significant differences in prevalence of bone pain across the eras in any age cohort (Figure 2C). Data on prevalence of fractures and hip replacement by era, age group, and splenectomy status were too sparse for meaningful interpretation (data not shown).
3.4. Indicators of splenic and hepatic involvement
Splenomegaly and hepatomegaly were evident prior to treatment initiation in all eras (Supporting Information Table S1). Spleen volumes differed across eras, with significantly lower mean volumes observed in later eras for children and adults 18 to <50 years (P < .01) (Supporting Information Figure S2 Panel A). For hepatomegaly, the difference across the eras did not reach significance for any group except the intact spleen adults 18 to <50 years of age cohort (P < .01, Supporting Information Figure S2 Panel B).
In pediatric patients, >94% of non‐splenectomized patients had moderate or severe splenomegaly in each era and >95% had moderate or severe hepatomegaly in each era (Supporting Information Table S1). All splenectomized pediatric patients with reported liver volumes (n = 15) had moderate or severe hepatomegaly. In adults 18 to <50 years of age, greater than 70% (range: 70% to 89%) of spleen‐intact patients had moderate or severe splenomegaly in each era and greater than 44% (range 45% to 81%) had moderate or severe hepatomegaly; greater than 66% (range 67% to 95%) of splenectomized patients had moderate or severe hepatomegaly. In adults ≥50 years of age, greater than 42% (range 43% to 68%) of spleen‐intact patients had moderate or severe splenomegaly in each era; 15% to 39% had moderate or severe hepatomegaly. The prevalence of splenectomized patients who had moderate or severe hepatomegaly in this age group ranged from 20% to 100% across the eras, with only two splenectomized patients representing the era where the prevalence was 100%. In general, there was less visceral disease in the adults ≥50 years.
3.5. Indicators of hematological disease
Overall there were no clinically meaningful differences in hemoglobin concentrations or platelet counts across the eras. Significant P‐values (P < .05) were observed for hemoglobin concentration among non‐splenectomized and splenectomized adults 18 to <50 years, with slightly higher concentrations in later eras, and for platelet counts among non‐splenectomized pediatric patients, with no apparent trend across eras (Supporting Information Figure S2 panels C and D, respectively). Anemia ranged from 18.8% to 61.1% across the eras and age groups with no discernable trend (Supporting Information Table S2). Thrombocytopenia tended to be in the mild to moderate range; pediatric patients had the lowest prevalence of severe thrombocytopenia (Supporting Information Table S2).
3.6. Time interval between GD1 diagnosis and initiation of imiglucerase
The median time interval between GD1 diagnosis and initiation of treatment differed across eras (P < .0001 for pediatric patients and adults 18 to <50 years and P < .01 for adults ≥50 years), with the longest interval observed in Era 1 in all age groups (Figure 3).
Figure 3.
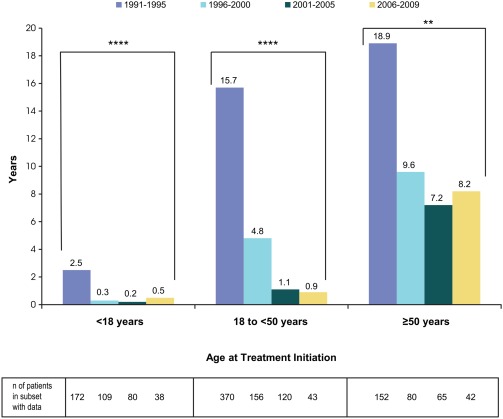
Median interval between age at diagnosis and age at imiglucerase initiation by age group and era of imiglucerase initiation. **P < .01; ****P < .0001. P‐values were derived from Kruskal‐Wallis test
4. DISCUSSION
Until the introduction of alglucerase/imiglucerase ERT in the early 1990s, individual cohort studies of GD1 depicted generally devastating disease with high rates of splenectomies, disabling skeletal complications and premature deaths.8, 10, 24 During two decades of ERT, clinicians have observed a remarkable transformation in the presenting phenotype of GD1. Our study is the first formal assessment of the changing spectrum of disease manifestations at the time of treatment initiation in the era of ERT. We focused our study on the US as imiglucerase ERT was first approved here, and we hoped our study cohort would represent a generally uniform clinical practice with respect to indications for starting treatment and dose.
During the first 5 years after approval of ERT, the patient cohort largely represented the accumulation of patients in advanced stages of GD1 with a high prevalence of splenectomies, disabling skeletal complications, and a long interval between diagnosis and initiation of ERT. During the subsequent 5‐year periods, imiglucerase ERT became established as the standard of care. Splenectomies and disabling skeletal complications declined steadily in patients starting therapy. Eras 2 (1996–2000) and 3 (2001–2005) represent the adoption of ERT in clinical practice when increasing numbers of physicians, especially hematologists, were becoming more familiar with the efficacy and safety of imiglucerase. Educational efforts with physicians during this time period focused on improving the understanding of the natural progression of GD1, the benefits of ERT and the evidence of maximal effect on AVN risk reduction through early initiation of ERT. Hence by Era 4 (2006–2009), the backlog of severely affected patients with advanced destructive complications associated with splenectomy and GD1 was significantly depleted. Patients in the most recent era are symptomatic patients who commenced ERT generally with an intact spleen and prior to the development of irreversible skeletal complications. Importantly, even among splenectomized patients, the prevalence of pretreatment bone crisis and ischemic bone events have decreased significantly relative to Era 1.
Our most clinically significant finding is the markedly diminished role of splenectomy in the management of GD1 since the introduction of ERT in 1991. In all age groups, the vast majority of pre‐ERT splenectomies were performed prior to 1991 (Supporting Information Table S1). In the pediatric group, where the time interval between diagnosis and initiation of ERT showed a striking closure across the eras, splenectomies were completely eliminated by Era 4. We saw a similar trend in the reduction in splenectomies in the adult cohorts, but a significant proportion of adult patients had undergone splenectomies sometime prior to ERT treatment even in Era 4. The majority of the splenectomies in these adult patients were performed many years prior to the commencement of ERT. In adults 18 to <50 years the median calendar year of splenectomy in each of the eras ranged from 1974 to 1990 and in adults ≥50 years from 1966 to 1981 (Supporting Information Table S1). Although the ICGG Gaucher Registry does not capture indications for splenectomies, the collective experience of the authors suggest they were most likely performed for massive splenomegaly, pressure symptoms and cytopenia, with some patients having the procedure for misdiagnosis of hematological malignancy.
A higher frequency of signature bone events (bone crisis, ischemic bone events, and bone pain) in splenectomized patients compared to non‐splenectomized patients was observed in our large cohort of US ICGG Gaucher Registry patients in keeping with smaller single‐center studies.2, 5, 6, 19, 25 Without GD1‐specific treatment, splenectomy removes a reservoir organ for the storage of the primary substrate glucosylceramide and secondary pathogenic substrates such as glucosylsphingosine,26 leading to accelerated accumulation of Gaucher cells at other sites, which in turn accelerates bone disease,5, 25 liver disease,27 pulmonary hypertension,28 marrow disease causing bone marrow failure,29 and immune alterations and comorbidities.30 Thus, a consensus conference of leading Gaucher disease physician experts have determined that a major goal in management of GD1 is to prevent splenectomy through timely initiation of ERT.11, 31 With widely available, non‐invasive, reliable testing for GD, diagnostic splenectomy is strongly contraindicated and palliative total splenectomy is not recommended except under life‐threatening circumstances such as uncontrollable bleeding or splenic rupture.32 The concept that treatment instituted after splenectomy may avert postsplenectomy complications is questionable in light of studies showing that postsplenectomy patients treated with ERT continue nevertheless to suffer new ischemic bone events.19, 33 It is therefore gratifying to confirm that with the advent of ERT, the number of splenectomized GD1 patients initiating treatment has decreased dramatically. Our study suggests management of GD1 patients in the US is making steady progress towards timely initiation of ERT to prevent the need for splenectomy. The reduced need for splenectomy, in the era of ERT has also been reported in a study of 99 Dutch patients.11 Further studies should examine the temporal trends with respect to splenectomy in GD1 and define the rare indications for splenectomy in the era of ERT.
A statistically significant decrease in the prevalence of bone crisis and ischemic bone events was observed across the eras in spleen‐intact pediatric patients and a similar trend occurred in some adult patients as well (Figure 2A,B). This important finding underscores that splenectomy is not the only determinant of bone crises and ischemic bone events. Bone manifestations can occur in the setting of intact spleen and their prevalence may be reduced by early initiation of ERT.19 Consistent with the latter concept, we found that the median interval between the age at diagnosis and the age at beginning imiglucerase treatment decreased over the eras (Figure 3). This effect was observed most clearly in pediatric patients who began ERT almost simultaneously with diagnosis in later eras relative to earlier eras when there was significant delay between diagnosis and treatment initiation. This salutary development of timely institution of treatment should decrease the risk of AVN,19 enhance bone development and growth in children, prevent delayed puberty,34, 35, 36 and increase the years of improved quality of life for patients with GD1.37
One of the most interesting findings of our study was that a history of bone pain remained constant across the eras although the prevalence of splenectomies and irreversible skeletal complications declined markedly in the new generation of patients. This highlights the importance of considering Gaucher disease in the evaluation of patients with unexplained recurrent or chronic bone pain.
The general lack of change from 1991 to 2009 in average pretreatment hemoglobin concentration and platelet count is not surprising in view of previously published Registry data showing that severe anemia and thrombocytopenia are uncommon. Only about 40% of GD1 patients are anemic at all prior to starting treatment, with no linear correlation between platelet count and magnitude of splenomegaly.21, 38 Because of insufficient data, the frequency of clinically significant bleeding, with or without blood transfusions, could not be analyzed.
GBA gene genotype frequencies in patient cohorts showed the expected distribution in Era 1, with a lower prevalence of N370S homozygosity in pediatric patients (15%), rising to 28% in adults 18 to < 50 years and 57% in adults ≥50 years (Table 1), and with reciprocal trends for N370S/other genotypes. The comparison of genotype frequencies across the eras within each age group did not find any significant differences with the exception of the adults 18 to <50 years of age in whom there is an increased prevalence of N370S homozygosity in later eras. While generally N370S homozygosity is associated with milder disease, several studies have shown many of these patients present with severe disease manifestations even in childhood.39, 40, 41, 42 Our study does not address the issue of N370S homozygous or other patients identified through newborn screening and whether there is role for ERT in the prevention of disease manifestations. Our study is entirely focused on patients with variable degrees of symptomatic disease with visceral, hematological and skeletal manifestations including bone pain.
Current treatment algorithms and third party payor guidelines largely presuppose that patients will have advanced disease. These guidelines may need to be revisited as evidence accrues that early treatment before the onset of irreversible manifestations may improve long‐term, patient‐centered outcomes and may prevent the irreversible morbidity that characterized GD1 patients in the pre‐ERT era at great cost to the quality and quantity of their lives.18, 19, 34 Whether early institution of treatment can avert some of the late‐onset complications of GD1 and reduce the incidence of hematologic malignancies such as multiple myeloma, for which Gaucher patients have a 5–60‐fold increased risk,43, 44 needs further study.
Although our study is one of largest cohorts reported, the study has some limitations common to any observational registry study. These analyses were restricted to US patients for the reasons described above. Cultural, economic, and healthcare accessibility differences in other countries may affect the generalizability of these results to other regions. Patients enrolled in the Registry may not be representative of the patient population as a whole due to selection biases related to the centers that choose to participate. Centers that contribute to the Registry are well informed with respect to GD1 and ERT. Treatment initiation for GD1 may be more delayed for patients receiving care in other settings. Assessments, lab techniques, and imaging are not necessarily standardized across reporting centers. Attrition of patients and missing data tend to accrue over time and may not necessarily be random. Classification of patients as GD1 was ascertained by the treating physician as is the standard practice. Some patients may have been misclassified as GD1 when they were actually GD3, but we believe if such instances occurred, they are rare. Also as previously noted, the small number of splenectomized patients in the later eras, particularly in the pediatric age cohort, limits interpretation of some results.
Our results confirm the hypothesis that the current phenotype for GD1 at the time of treatment initiation is characterized by a markedly lower prevalence of splenectomy and a lower frequency of destructive bone complications, relative to the period when ERT first became commercially available in the US. This is particularly evident in pediatric patients and adult patients younger than 50 years of age. This transformation in phenotype at treatment initiation suggests a need to revisit treatment guidelines and goals.
5. DISCLOSURES
PKM is a member of the ICCG North American Advisory Board. He is supported by a grant from the National Institutes of Arthritis, Musculoskeletal and Skin Diseases AR 65932 and Center of Excellence Grant in Clinical Translational Research from Sanofi Genzyme. He has received research support, honoraria and consulting fees from Sanofi Genzyme.
HA is a member of the ICGG North American Advisory Board and has received honoraria and travel support from Sanofi Genzyme for advisory board activities.
MB is a member of ICGG North American Advisory Board and has received honoraria and travel support from Sanofi Genzyme for advisory boards and lectures. She has receive honoraria and travel reimbursements from Shire, Alexion, Recordati, and Alnylam.
JB is a Sanofi Genzyme employee.
TAB has received honoraria, travel reimbursements from Sanofi Genzyme and is a member of the ICGG North American Registry Advisory Board, and a researcher for the ENCORE study; he has received honoraria, travel reimbursements from Shire.
JC has received consulting fees for participation in advisory boards and data safety monitoring boards from Sanofi Genzyme, Alexion and honoraria for speaking engagements from Sanofi Genzyme, The National Gaucher Foundation, and the Fabry Support and Interest Group, and is an investigator in clinical trials sponsored by Sanofi Genzyme, Shire, BioMarin, Amicus.
PKaplan has received unrestricted educational grants from Sanofi Genzyme, fellowship grants for Genetic Counselors and honoraria from Sanofi Genzyme, compensation as Chairperson of Safety Review Committee for Elelyso Registry from Pfizer Inc. and as Chairperson of Safety Steering Committee from Synageva‐Alexion.
AK is a member of the ICGG Advisory Board and has received honoraria and travel support for advisory board activities, and research grants, speaker?s fees and travel grants from Sanofi Genzyme; consulting fees, speaker fees, travel and research grants from Actelion; research and travel grants, speaker and consultant fees from Cytonet LLC; research grants, speaker?s fees and travel awards from Shire HGT; consulting fees, travel awards and research grants from Alexion; consulting fees, travel grants and research grants from Biomarin; consulting fees, travel grants and research grants from Horizon.
PKishnani has received research/grant support from Genzyme Corporation (Sanofi Aventis), Valerion Therapeutics, Shire Pharmaceuticals, Roche, Pfizer, Alexion, NIH, FDA, and PCORI; consulting fees and honoraria from Genzyme Corporation (Sanofi Aventis), Shire Pharmaceuticals, Alexion Pharmaceuticals, Amicus Therapeutics and Roche and is a member of the Pompe and Gaucher Disease Registry Advisory Board for Sanofi Genzyme; Scientific Advisory Board for Shire Pharmaceuticals; and Registry Board Member for Alexion Pharmaceuticals. She also serves on the Data Safety Monitoring Board for PTC Therapeutics.
EK is a member of the North American ICGG Advisory board for Sanofi Genzyme and is a consultant for SIFAP study sponsored by Shire Human Gene Therapies.
BR has received honoraria from Sanofi Genzyme as a member of the ICCG North American Advisory Board.
RCS is a member of the ICGG North American Advisory Board and has received fees from Sanofi Genzyme for advisory board activities. He has also received research grants from Sanofi Genzyme and fees as a member of the advisory board for Fabry disease.
NW has received research support, consultation fees, honoraria and travel reimbursement for attending advisory boards or for speaking from Sanofi Genzyme, Shire HGT, and Pfizer Corporation. He is a member of the North American and International Advisory Boards for the ICCG Gaucher Registry.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Table 1.
Supporting Information Table 2.
ACKNOWLEDGMENTS
The authors would like to acknowledge all the patients with Gaucher disease, their physicians and health care personnel who submit data to the ICGG Gaucher Registry and the Gaucher Registry support team at Sanofi Genzyme. Laura Croal, PhD and Jennifer Ibrahim, MD, Sanofi Genzyme employees, provided editorial support. Eva Stamenovic of Sanofi Genzyme provided statistical programming support. Writing assistance provided by Shelton Panak, PhD, an independent contractor, was funded by Sanofi Genzyme.
Mistry PK, Batista JL, Andersson HC, et al. Transformation in pretreatment manifestations of Gaucher disease type 1 during two decades of alglucerase/imiglucerase enzyme replacement therapy in the International Collaborative Gaucher Group (ICGG) Gaucher registry. Am J Hematol. 2017;92:929–939. https://doi.org/10.1002/ajh.24801
REFERENCES
- 1. Mistry PK, Cappellini MD, Lukina E, et al. A reappraisal of Gaucher disease‐diagnosis and disease management algorithms. Am J Hematol. 2011;86:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mistry PK, Belmatoug N, Vom Dahl S, Giugliani R. Understanding the natural history of Gaucher disease. Am J Hematol. 2015;90(Suppl 1):S6–11. [DOI] [PubMed] [Google Scholar]
- 3. Grabowski GA. Gaucher disease and other storage disorders. Hematol Am Soc Hematol Educ Progr. 2012;2012:13–18. [DOI] [PubMed] [Google Scholar]
- 4. Maaswinkel‐Mooij P, Hollak C, van Eysden‐Plaisier M, Prins M, Aerts H, Poll R. The natural course of Gaucher disease in The Netherlands: implications for monitoring of disease manifestations. J Inherit Metab Dis. 2000;23:77–82. [DOI] [PubMed] [Google Scholar]
- 5. Fleshner PR, Aufses AH, Jr. , Grabowski GA, Elias R. A 27‐year experience with splenectomy for Gaucher's disease. Am J Surg. 1991;161:69–75. [DOI] [PubMed] [Google Scholar]
- 6. Beighton P, Goldblatt J, Sacks S. Bone involvement in Gaucher disease. Prog Clin Biol Res. 1982;95:107–129. [PubMed] [Google Scholar]
- 7. Kolodny EH, Ullman MD, Mankin HJ, Raghavan SS, Topol J, Sullivan JL. Phenotypic manifestations of Gaucher disease: clinical features in 48 biochemically verified type 1 patients and comment on type 2 patients. Prog Clin Biol Res. 1982;95:33–65. [PubMed] [Google Scholar]
- 8. Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res. 1982;95:177–217. [PubMed] [Google Scholar]
- 9. Wenstrup RJ, Roca‐Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol. 2002;75(Suppl 1):A2–12. [DOI] [PubMed] [Google Scholar]
- 10. Weinreb NJ, Barbouth DS, Lee RE. Causes of death in 184 patients with type 1 Gaucher disease from the United States who were never treated with enzyme replacement therapy. Blood Cells, Mol Dis. 2016. doi:10.1016/j.bcmd.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 11. Cox TM, Aerts JM, Belmatoug N, et al. Management of non‐neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis. 2008;31:319–336. [DOI] [PubMed] [Google Scholar]
- 12. Furbish FS, Steer CJ, Krett NL, Barranger JA. Uptake and distribution of placental glucocerebrosidase in rat hepatic cells and effects of sequential deglycosylation. Biochim Biophys Acta. 1981;673:425–434. [DOI] [PubMed] [Google Scholar]
- 13. Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency–macrophage‐targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324:1464–1470. [DOI] [PubMed] [Google Scholar]
- 14. Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122:33–39. [DOI] [PubMed] [Google Scholar]
- 15. Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113:112–119. [DOI] [PubMed] [Google Scholar]
- 16. Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis. 2011;46:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Fost M, Hollak CE, Groener JE, et al. Superior effects of high‐dose enzyme replacement therapy in type 1 Gaucher disease on bone marrow involvement and chitotriosidase levels: a 2‐center retrospective analysis. Blood. 2006;108:830–835. [DOI] [PubMed] [Google Scholar]
- 18. Charrow J, Scott CR. Long‐term treatment outcomes in Gaucher disease. Am J Hematol. 2015;90(Suppl 1):S19–S24. [DOI] [PubMed] [Google Scholar]
- 19. Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol. 2009;147:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weinreb NJ, Goldblatt J, Villalobos J, et al. Long‐term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J Inherit Metab Dis. 2013;36:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lo SM, Liu J, Chen F, et al. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long‐term outcomes of therapy. J Inherit Metab Dis. 2011;34:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. [DOI] [PubMed] [Google Scholar]
- 23. von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. Int J Surg. 2014;12:1495–1499. [DOI] [PubMed] [Google Scholar]
- 24. Zimran A, Kay A, Gelbart T, et al. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine (Baltimore). 1992;71:337–353. [PubMed] [Google Scholar]
- 25. Rose JS, Grabowski GA, Barnett SH, Desnick RJ. Accelerated skeletal deterioration after splenectomy in Gaucher type 1 disease. AJR Am J Roentgenol. 1982;139:1202–1204. [DOI] [PubMed] [Google Scholar]
- 26. Murugesan V, Chuang WL, Liu J, et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am J Hematol. 2016;91:1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bohte AE, van Dussen L, Akkerman EM, et al. Liver fibrosis in type I Gaucher disease: magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS One. 2013;8:e57507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher's disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77:91–98. [DOI] [PubMed] [Google Scholar]
- 29. Mistry PK, Davies S, Corfield A, Dixon AK, Cox TM. Successful treatment of bone marrow failure in Gaucher's disease with low‐dose modified glucocerebrosidase. Q J Med. 1992;83:541–546. [PubMed] [Google Scholar]
- 30. Sønder SU, Limgala RP, Ivanova MM, et al. Persistent immune alterations and comorbidities in splenectomized patients with Gaucher disease. Blood Cells Mol Dis. 2016;59:8–15. [DOI] [PubMed] [Google Scholar]
- 31. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41:4–14. [DOI] [PubMed] [Google Scholar]
- 32. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists‐oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82:697–701. [DOI] [PubMed] [Google Scholar]
- 33. Drelichman G, Fernandez Escobar N, Basack N, et al. Skeletal involvement in Gaucher disease: An observational multicenter study of prognostic factors in the Argentine Gaucher disease patients. Am J Hematol. 2016;91:E448–E453. [DOI] [PubMed] [Google Scholar]
- 34. Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172:447–458. [DOI] [PubMed] [Google Scholar]
- 35. Andersson H, Kaplan P, Kacena K, Yee J. Eight‐year clinical outcomes of long‐term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122:1182–1190. [DOI] [PubMed] [Google Scholar]
- 36. Kaplan P, Mazur A, Manor O, et al. Acceleration of retarded growth in children with Gaucher disease after treatment with alglucerase. J Pediatr. 1996;129:149–153. [DOI] [PubMed] [Google Scholar]
- 37. Weinreb N, Barranger J, Packman S, et al. Imiglucerase (Cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin Genet. 2007;71:576–588. [DOI] [PubMed] [Google Scholar]
- 38. Hollak CE, Belmatoug N, Cole JA, et al. Characteristics of type I Gaucher disease associated with persistent thrombocytopenia after treatment with imiglucerase for 4–5 years. Br J Haematol. 2012;158:528–538. [DOI] [PubMed] [Google Scholar]
- 39. Balwani M, Fuerstman L, Kornreich R, Edelmann L, Desnick RJ. Type 1 Gaucher disease: significant disease manifestations in “asymptomatic” homozygotes. Arch Intern Med. 2010;170:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160:603–608. [DOI] [PubMed] [Google Scholar]
- 41. Taddei TH, Kacena KA, Yang M, et al. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am J Hematol. 2009;84:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fairley C, Zimran A, Phillips M, et al. Phenotypic heterogeneity of N370S homozygotes with type I Gaucher disease: an analysis of 798 patients from the ICGG Gaucher Registry. J Inherit Metab Dis. 2008;31:738–744. [DOI] [PubMed] [Google Scholar]
- 43. Rosenbloom BE, Weinreb NJ, Zimran A, Kacena KA, Charrow J, Ward E. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood. 2005;105:4569–4572. [DOI] [PubMed] [Google Scholar]
- 44. de Fost M, Vom Dahl S, Weverling GJ, et al. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol Dis. 2006;36:53–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Table 1.
Supporting Information Table 2.